推荐厂家
暂无
暂无
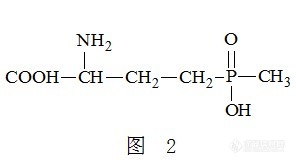
关于”新版GB 2763 食品安全国家标准 规定茶叶中限量农残草甘膦和草铵膦项目“的检测研究 一、研究意义及现状 随着新版GB 2763 食品安全国家标准的不断更新及发布实施,草甘膦和草铵膦已被明确列为茶叶中农药残留强检(必检)项目,草甘膦在茶叶中的限量为1mg/kg,草铵膦在茶叶中的限量为0.5mg/kg。同时,草甘膦和草铵膦也成为中国茶叶出口国外的检测项目(来源于中华人民共和国商务部),且已成为越来越严的限量指标。 文献(2013年农药行业预测和草甘膦市场机遇分析,杨益军,农药市场信息,2013.03)报道,除草剂草甘膦因其高效、广谱、低毒等特性使其被广泛应用,未来需求量也将大幅增加。但草甘膦的使用容易使植物产生抗性(IARC国际研究机构发布报告称草甘膦很可能对人类致癌),而草铵膦可克服该缺陷,现已有学者(草铵膦、百草枯、草甘膦对非耕地杂草的防效比较,凌进,农药,2014年第53卷第8期,613-615)对草铵膦和草甘膦的除草性能进行了研究,确证了草铵膦代替草甘膦的可行性。 因草甘膦和草铵膦为广谱除草剂,被广泛应用于农业、林业及园艺的栽培。我国作为农业大国,其茶叶产量世界第一、出口量世界第二,草甘膦和草铵膦的生产和使用量都位居世界前列(草甘膦 草铵膦及其代谢产物的检测方法,李小娟、周信康、孟品佳,公共安全中的化学问题研究进展)。同时,我单位对西南茶叶原料主产区进行了初步调研,进一步确认茶农使用草甘膦和草铵膦农药的现状。 随着草甘膦和草铵膦除草剂使用量的日益增大,使其常被发现存在于环境水样、土壤及植物中,这样长期积累会引起环境污染,从而对人类健康造成严重威胁。草甘膦和草铵膦结构类似,且均含有膦酸基、羟基、氨基,是极强的两性化合物,易溶于水,难挥发。鉴于草甘膦和草铵膦特殊的物化性质和茶叶基质自身的复杂性,无论国内外,茶叶中草甘膦和草铵膦同时检测的标准还未见发布。 目前,可用于检测草甘膦和草铵膦农药残留量的主要方法有液相色谱法,柱前衍生后气相色谱法、气相色谱-质谱法及液相色谱-质谱/质谱法。 快速发展起来的超高效液相色谱-质谱联用技术,具有检测灵敏度高、适用范围广、分析速度快和能有效排除复杂基质产生的干扰等优点,当今已成为检测型实验室检测农残的首选。然而,若采用液质质直接测定草甘膦和草铵膦,则仪器响应较低,无法满足茶叶中草甘膦和草铵膦农药残留量检测的要求。 近两年来,已有研究文献陆续发表,用柱前衍生-液相色谱串联质谱法检测。笔者结合其文献研究结果,对茶叶中草甘膦和草铵膦农药残留量的检测方法系统地进行研究,采用9-芴甲氧羰酰氯(FMOC-Cl)作为常用衍生剂,在硼酸盐缓冲盐溶液的条件下,能与草甘膦和草铵膦的提取液发生衍生反应,形成衍生产物,衍生产物注入UPLC进行色谱洗脱分离,采用串联质谱探测响应信号,外标法直接快速定量茶叶中的草甘膦和草铵膦的含量。二、液质质检测分析原理 质谱原理是先将物质离子化,按离子的质荷比分离,然后测量各种离子谱峰的强度而实现分析目的的一种分析方法。液质联用是将色谱的分离能力与质谱强大的定性功能结合起来,实现对复杂混合物更准确的定量和定性分析,简化样品的前处理流程,使样品分析更简便。主要针对不挥发性、极性、热不稳定、大分子量等化合物的分析测定。液质联用检测技术灵敏度高,且串联质谱(三重四级杆)定性准确,可有效杜绝微量甚至痕量物质分析时的假阳性现象,常用于目标物质的痕量分析。 采用柱前衍生-液相色谱串联质谱法检测茶叶中的草甘膦和草铵膦有以下优势: 1)、灵敏度高、线性好、检出限低(可达ng/mL级及其以下); 2)、定量结果准确、稳定、重复性好; 3)、实验操作简单、步骤少、耗时短、分析速度快、检测效率高; 4)、实验试剂无污染、无毒、安全; 5)、有效减弱基质对目标物检测的影响。三、茶叶中草甘膦和草铵膦农药残留量检测的前处理试验 茶叶经GB/T8303磨碎、过筛制得待测茶样(发酵茶应先低温去除水分,使样品易于磨碎); 准确称取已磨碎处理过的茶样1g(精确至0.001g)置于80mL具盖离心管中,加入10mL水,涡旋混匀静置,加入2mL二氯甲烷,混匀,超声提取或回旋振荡10min,低速离心机4500r/min离心5min,取上清液,制得提取上清液; 注意:若茶样为新采摘的鲜叶,则称取约5g鲜叶于研钵中,加入30mL水,研磨约10min,将其转入离心管中,用10mL水洗涤研钵后转移至离心管,重复洗涤一次,再次加入10mL二氯甲烷于离心管,均质至混匀,4500r/min离心10min,取上清液,制得提取上清液; 将提取上清液用净化柱CAX、C18,以及活性炭小柱等进行比对试验,确定以C18小柱净化提取液,制得提取净化液; 通过缓冲液浓度、衍生液浓度、衍生液用量、缓冲液用量、净化液用量,衍生时间等条件试验,得出最优衍生试验参数为缓冲液浓度为50g/L,衍生液浓度20g/L,衍生液用量:缓冲液用量:净化液用量的体积比为1:1:1,衍生时间为约3h,衍生液过0.22μm的有机滤膜后进样。四、茶叶中草甘膦和草铵膦农药残留量检测的衍生机理 茶叶中草甘膦和草铵膦农药残留量的衍生机理为:在硼酸钠缓冲盐溶液条件下,草甘膦(分子结构如图1所示)和草铵膦(分子结构如图2所示)中R-NH-R’的-H被FMOC-Cl(分子结构如图3所示)中的FMOC-取代,生成 http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414494663_01_0_3.png,得到衍生目标产物草甘膦衍生物和草铵膦衍生物。 其中,草甘膦分子结构图,见图1;草铵膦分子结构图,见图2;9-芴甲氧羰酰氯(FMOC-Cl)分子结构图,见图3;草甘膦和草铵膦与9-芴甲氧羰酰氯(FMOC-Cl)的衍生机理图,见图4。http://ng1.17img.cn/bbsfiles/images/2017/10/2015070414424276_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414430242_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2015070414432007_01_2275853_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507061123_553629_2275853_3.png五、茶叶中草甘膦、草铵膦农残衍生物在质谱中的裂解机理1、茶叶中草甘膦农残衍生物在质谱中的裂解机理 通过对草甘膦衍生物在串联质谱中的裂解机理进行系统的分析研究,可探索出草甘膦衍生物的裂解机理为:首先草甘膦衍生物裂解为离
[font=宋体][size=14px][/size][/font][font='微软雅黑','sans-serif']你好,想问一下老师[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]跑乙酰甲胺磷单标的时候不出峰,跑有机磷混标的时候乙酰甲胺磷可以出峰,换新的一支标准品也是同样的情况,想请教一下老师是哪里有问题,谢谢[/font][font='微软雅黑','sans-serif']求助问题来自微信群。[/font][font=宋体][size=14px][/size][/font]
食品中甲胺磷和乙酰甲胺磷农药残留量的测定方法1.适用范围本方法适用于谷物、蔬菜和植物油中甲胺磷和乙酰甲胺磷的残留量分析,其最小检出限分别为7.79×10-12g和1.79×10-11g。2.原理概要含有机磷的样品在富氢焰上燃烧,以HPO碎片的形式,放射出波长526nm的特征光,这种特征光通过滤光片选择后,由光电倍增管接收,转换成电信号,经微电流放大器放大后,被记录下来,样品的峰高与标准品的峰高相比,计算出样品相当的含量。3.主要试剂和仪器3.1.主要试剂丙酮;二氯甲烷:重蒸;无水硫酸钠;活性炭:用3mol/L盐酸浸泡过夜,抽滤,用水洗至中性,在120℃下烘干备用;甲胺磷(methamidophos):≥99%;乙酰甲胺磷(acephate):≥99%;甲胺磷和乙酰甲胺磷标准溶液的配制:分别准确称取甲胺磷和乙酰甲胺磷的标准品,用丙酮分别制成0.1mg/mL的标准储备液。使用时用丙酮稀释配制成单一品种的标准使用液(1mg/mL)和混合标准工作液(每个品种浓度为1mg/mL)。贮藏于冰箱中。3.2.仪器气相色谱仪:具有火焰光度检测器;电动振荡器;K-D浓缩器或旋转蒸发器;离心机。4.试样的制备取谷物实验样品经粉碎机粉碎,过20目筛后,制成谷物试样。取蔬菜实验样品洗净,晾干,去掉非食部分后剁碎或经组织捣碎机捣碎,制成蔬菜试样。5.过程简述5.1.提取和净化蔬菜:称取蔬菜试样10g,精确至0.001g,用无水硫酸钠(因蔬菜含水量不同而加入量不同,约50~80g)研磨呈干粉状,倒入具塞锥形瓶中,加入0.2~0.4g活性炭(根据蔬菜色素含量)及80mL丙酮,振摇0.5h,抽滤,滤液浓缩定容至5mL,待气相色谱分析。谷物:称取谷物试样10g,精确至0.001g,置于具塞锥形瓶中,加入40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。小麦:称取小麦试样10g,精确至0.001g,置于具塞锥形瓶中,加入0.2g活性炭及40mL丙酮,振摇1h,抽滤,浓缩,定容至5mL,待气相色谱分析。植物油:称取植物油试样5g,用45mL丙酮分次洗入50mL的离心管内,加入5mL水,混匀,在3 000r/min下离心5min,吸取上清液,下面油层再加10mL水和10mL丙酮,离心5min,吸取上清液,合并两次上清液,用K-D浓缩器浓缩近干,残渣和水加入40g无水硫酸钠,研磨呈干粉状,倒入具塞锥形瓶中,加入0.3g活性炭、60mL二氯甲烷,振荡0.5h,抽滤,定容至5mL,待气相色谱分析。5.2.色谱条件色谱柱:玻璃柱,内径3mm,长0.5m,内装2%dEGS/Chromosorb W AWdMCS,80~100mesh。气流:载气,氮气70mL/min,空气0.7kg/cm2,氢气1.2kg/cm2。温度:进样口200℃,柱温180℃。5.3.测定定性:以甲胺磷和乙酰甲胺磷农药标样的保留时间定性。定量:用外标法定量,以甲胺磷和乙酰甲胺磷农药已知浓度的标准样品溶液作外标物,按峰高定量。6.结果计算Xi=hi•Esi•V1hsi•V2•m式中:Xi——样品中i组分有机磷含量,mg/kg;Esi——注入标样中i组分有机磷的含量,ng;hi——样品的峰高,mm;hsi——标样中i组分的峰高,mm;V1——浓缩定容体积,mL;V2——注入色谱样品的体积,μL;m——样品的质量,g。7.方法的精密度添加回收试验中甲胺磷和乙酰甲胺磷的变异系数分别为2.36%和3.95%。8.甲胺磷和乙酰甲胺磷的保留时间在5.2的气相色谱条件下,甲胺磷的保留时间为0.9min,乙酰甲胺磷的保留时间为1.9min。9.来源:GB 14876—94






 400-877-2799
400-877-2799
 留言咨询
留言咨询
 400-860-5168转3372
留言咨询
400-860-5168转3372
留言咨询
 留言咨询
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP


