推荐厂家
暂无
暂无
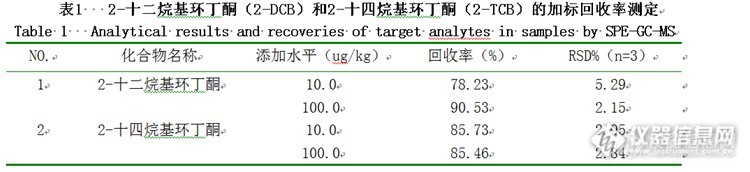
摘 要:建立固相萃取-气相色谱- 质谱联用(solid phase extraction with gas chromatography-mass spectrometry,SPE-GC-MS)法测定植物油中2-十二烷基环丁酮和2-十四烷基环丁酮。对影响分析物萃取效率的诸因素如洗脱溶剂等进行详细考察和优化。最佳萃取条件为0.5 g样品与5 mL乙腈混匀,经ProElut Silica (500 mg/3mL)固相萃取柱净化后,以GC-MS 进行测定,该方法对2-十二烷基环丁酮和2-十四烷基环丁酮的检出限为10μg/kg,线性范围为0.01~0.5μg/mL,线性相关系数分别为0.99938和0.99977,相对标准偏差(relative standard deviation,RSD)(n=3)小于6%。该方法成功应用于植物油中2-十二烷基环丁酮和2-十四烷基环丁酮的分析,加标回收的回收率为78%~91%。关键词:固相萃取;气相色谱-质谱;2-十二烷基环丁酮;2-十四烷基环丁酮;植物油 食品辐照作为对物质或食品进行加工处理的新型保藏技术,在国际上已逐渐被认可,但是在商业化应用、国际贸易以及辐照食品的市场监管方面,迫切需要有辐照食品鉴定检测方法。 经辐照后,在含脂食品中会产生特异性辐解产物2-烷基环丁酮(2-Alkylcyclobutanones ,2-ACBs),它是含脂辐照食品的特异性辐解产物,在未辐照的含脂食品中,至今还从未检测到此类化合物。在1990年, 2-ACBs 类化合物可作为检测含脂辐照食品的标志性化合物, 首次被报道,随后依据该结论制定了欧盟标准EN1785和GB\T 21926-2008 。2-ACBs由食品中的游离脂肪酸或甘油三酸酯的羰基氧失去一个电子,再经由重排过程生成,其过程如图1所示。http://ng1.17img.cn/bbsfiles/images/2015/07/201507091523_554630_2452211_3.png图1 经辐照后游离的脂肪酸转化为2-ACBs的示意图 在大多数食品中,棕榈酸、硬脂酸、油酸、亚油酸是主要的脂肪酸,而棕榈酸和硬脂酸是其中含量最高的饱和脂肪酸,其辐解物2-十二烷基环丁酮(2-dodecylcyclobutanone,2-DCB)和2-十四烷基环丁酮(2-tetradecylcyclobutanone,2-TCB)相对于其它2-ACBs较为稳定,因此一般作为检测含脂辐照食品的主要标志性化合物。目前对含脂辐照食品大多采用佛罗里硅土柱进行净化,但是该法的应用范围有限。本实验拟通过优化固相萃取(solidphase extraction,SPE)条件,采用气相色谱-质谱联用(gas chromatography-massspectrometry,GC-MS)技术测定植物油中2-十二烷基环丁酮和2-十四烷基环丁酮,为进一步缩短2-ACBs 萃取和分离时间、减少溶剂使用量、提高检测灵敏度以及扩大方法应用范围提供基础数据和理论依据。1 材料与方法1.1 材料、试剂与仪器GCMS-QP2010 气相色谱-质谱联用仪 日本岛津公司;DM-5MS 毛细管柱(30 m×0.25 mm,0.25 μm)迪马公司;XH-C 涡旋混合器 江苏金坛市盛威实验仪器;80-1 高速离心机 河南省予华仪器;OSB-2100 旋转蒸发仪 上海爱朗仪器有限公司;12孔固相萃取装置 迪马公司; ProElut Silica(500 mg/3mL)固相萃取柱 迪马公司。HSC-12B 氮吹仪天津市威仪科技发展有限公司;丙酮、二氯甲烷、乙酸乙酯乙腈、甲基叔丁基醚、正己烷(均为色谱纯)迪马公司。实验所用的植物油均购自当地市场。1.2 方法1.2.1 标准贮备液的制备称取一定量标准品,溶于正己烷溶剂中,配制成浓度为0.5 mg/mL的标准贮备液。再配制成质量浓度系列为0.01μg/mL、0.02μg/mL、0.05μg/mL、0.1μg/mL、0.2μg/mL、0.5μg/mL的标准工作溶液,备用。1.2.2 仪器分析条件气相色谱条件:色谱柱为DM-5MS (30.0m×250μm,0.25μm);载气He(99.995%);恒流,柱流速1.0mL/min;不分流,进样量1μL,进样口温度为260℃;起始温度80℃(保持1min),以15℃/min的速度升至150℃,再以8℃/min升温至200℃,再以20℃/min升温至260℃(保持5min)。质谱条件:EI源,离子源200℃,溶剂延迟为3min,选择离子监测模式(SIM),选择监测离子(m/z):69、84、98、112、125。1.2.3 样品的提取称取0.5 g样品于10 mL带塞试管中,加入5 mL乙腈,涡旋混合2 min,超声提取2 min,4000 rpm下离心2min,取上清液;下层油脂再用5 mL乙腈重复上述步骤,合并两次上清液。将得到的上清液在50℃下,氮吹近干,再慢慢挥干,再向氮吹瓶中加入2.5 mL正己烷复溶,待净化。1.2.4 样品的净化依次用5 mL甲基叔丁基醚,5mL正己烷缓慢通过ProElut Silica固相萃取柱,以达到润湿小柱,活化填料,除去干扰杂质的目的;再将1.2.3节方法制得的待净化液转移到ProElut Silica固相萃取柱中,流出液弃去;然后用5 mL正己烷淋洗,弃去流出液;再用10 mL甲基叔丁基醚:正己烷(1:99V:V)洗脱,用旋转蒸发瓶接收,直至洗脱液完全自然滴出。在50 ℃下,将收集到的洗脱液氮吹浓缩,然后用正己烷定容至1 mL后供GC-MS分析。2 结果与分析在固相萃取操作中,影响分析物峰面积的主要固相萃取因素有洗脱剂、洗脱体积、洗脱速率和上样速率。为了获得最佳分析结果,需要对其进行优化。2.1固相萃取条件的确定2.1.1 提取溶剂的选择2-十二烷基环丁酮(2-DCB)和2-十四烷基环丁酮(2-TCB)与脂肪酸的结构及其类似,故能溶于极性和中等极性的试剂中。分别用丙酮、二氯甲烷、甲基叔丁基醚、乙酸乙酯作为2-DCB 和2-TCB的提取溶剂。实验结果表明乙腈提取效果较好,再加以涡旋振荡后结合超声提高回收率。2.1.2 固相萃取柱的选择对于油脂类样品,采用固相萃取柱进行样品净化是必不可少的步骤。结合相应参考文献,本实验采用了硅胶、PSA、Florisil、Alumina等填料的固相萃取柱,结果表明对于植物油,硅胶柱相对于其他填料的固相萃取柱来说,2-DCB 和2-TCB回收率较高,添加回收率达到了80%-120%,满足分析检测的要求,且达到很好的净化效果。如图2所示http://ng1.17img.cn/bbsfiles/images/2015/07/201507091524_554631_2452211_3.pngA:标准品;B:空白样品;C:添加标品的样品图2 植物油空白样品及其添加样品的总离子流图2.1.3 淋洗曲线的建立固相萃取技术最重要的目的在于通过固相萃取柱将目标化合物与主要干扰物分开,从而实现净化的目的。在此过程中应非常注意选择合适的洗脱溶剂。样品处理过程是先用正己烷将其中的中性化合物除去,参照Horvarovich 等报道,用硅胶柱分离样品中的2-DCB和2-TCB,选用弱极性的甲基叔丁基醚(methyl-t-butyl ether,TBME)/正己烷(V/V)混合溶剂将稍强极性的2-DCB 和2-TCB洗脱下来。由于样品基质与文献不一样,淋洗液与洗脱液的选择也会不一样。因次需要考察正己烷以及其与甲基叔丁基醚不同比列的混合液作为洗脱液时2-DCB和2-TCB的回收率。选用5根ProElut Silica固相萃取柱,取0%、0.5%、1%、2%、5%不同浓度的甲基叔丁基醚:正己烷(V/
为了达到某些需求,很多时候单一的溶剂并不能满足挥发速率等方面的要求,故产生了对混合溶剂的需求,然而现在的一些混合溶剂的气味很大,如中干水、洗网水、硝基稀料等,他们大多是甲苯、环己酮、丁酮等混合而成,如何才能将这种混合溶剂的气味降低呢,还请各位多多指教!不胜感激!
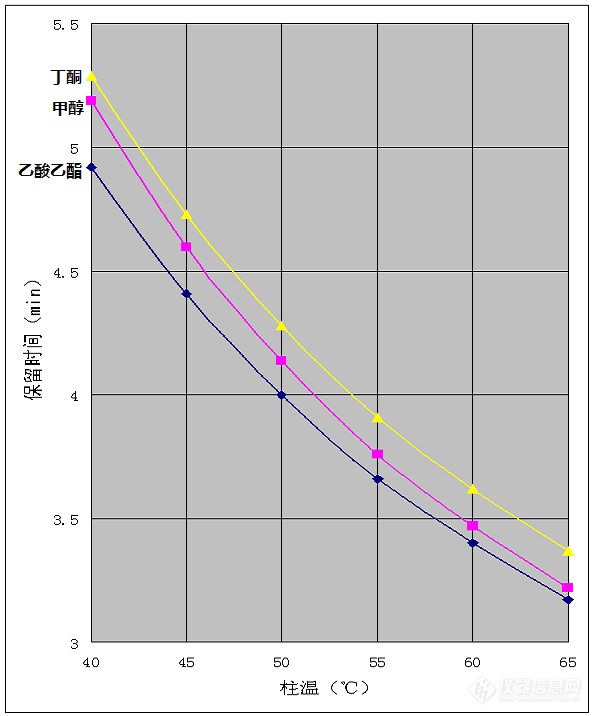
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。





 400-860-5168转3986
400-860-5168转3986
 留言咨询
留言咨询
 400-860-5168转2141
留言咨询
400-860-5168转2141
留言咨询
 400-860-5168转4637
留言咨询
400-860-5168转4637
留言咨询










 我要推广仪器
我要推广仪器
 下载APP
下载APP