
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转3457
仪器信息网认证电话,请放心拨打

均苯并四咪唑(HBTM): 一种用于不对称酰基化的通用有机催化剂----阿拉丁试剂
简介作为我们不对称催化产品组合的一部分,阿拉丁以(R,产品货号:B120975)和(S,产品货号:B120963)对映体的形式提供异硫脲有机催化剂苯并四咪唑(HBTM)。这些化合物已广泛应用于低催化剂负载量的各种官能团的对映选择性转化和动力学拆分。代表性转化动力学拆分S-HBTM最初是由Birman及其同事开发作为酶促条件的一种替代而用于动力学中仲醇的拆分[1]。已报道了最多有122种不同的可选变量。HBTM催化剂已在各种温度范围内与多种有机溶剂一起被用于烯丙基、炔丙基和苄基仲醇的动力学拆分,反应通常在数小时内完成,在较低温度(0 - 55 ℃)下的甲苯、氯仿或叔戊醇中可达到最佳条件[1, 2]。HBTM同样可用于α-芳基链烷酸、α-芳氧基-链烷酸、α-烷氧基-链烷酸、α-卤代链烷酸和受保护的α-氨基酸的动力学拆分[3, 4]。已报道了最多有96种不同的可选变量。该转化通常是在低温下利用甲苯作为溶剂进行24小时反应来完成的。同样,有学者研究了利用HBTM对对映体富集的α-硫代链烷酸的形成进行了动态动力学拆分[4, 5]。该过程可选择多种不同的底物,并且具有高产率和高对映体纯度。对映选择性转化HBTM已被用于恶唑基碳酸酯的不对称羧基转移反应[6]。该过程以二氯甲烷作为溶剂,在低温下反应16小时,底物普适性较好,能以较高的产率和对映选择性得到相应的产物。有研究表明HBTM可用于不对称亲核体催化迈克尔-醛醇-β-内酰胺化(NCMAL)的串联反应[7]。该过程是在四氢呋喃/二氯甲烷中进行24小时的反应发生的,从而得到具有高非对映异构体比率、高产率和高对映纯度的产物。 绝对构型的测定HBTM是被选择用于竞争对映选择性转化(CEC)方法的催化剂,该方法可用于分配对映体富集的立体中心的绝对构型。利用HBTM的CEC方法已被报道用于仲醇,其中反应的转化率是通过1H NMR[8]和TLC进行测量的[9]。TLC分析也已被纳入为本科阶段的实验室实验课程中了[10]。该过程是在室温下进行的,需经历30分钟至数小时不等。以下助记符可用于识别使用R-HBTM和S-HBTM催化剂的仲醇的绝对构型[8-10]。利用HBTM的CEC方法已被报道用于确定恶唑烷酮、内酰胺和硫内酰胺的绝对构型[11]。该过程通常是在室温或50 oC条件下进行数小时后完成的。以下助记符可用于识别这些使用R-HBTM和S-HBTM催化剂的体系的绝对构型。 更多研究HBTM的亲核性和Lewis碱度已通过来自利用苯甲醛离子的一系列实验的速率和平衡常数而进行了研究[12]。此外,还对HBTM催化的仲醇酯化反应进行了详细的动力学分析,为该转化的催化循环提供了思路和想法[13]。参考文献1. Purification of Basic Compounds with Functionalized SilicaGel. Synfacts. 2008(10): 1115-1115. https://doi.org/10.1055/s-2008-10781982. Li X, Jiang H, Uffman EW, Guo L, Zhang Y, Yang X, Birman VB. 2012. Kinetic Resolution of Secondary Alcohols Using Amidine-Based Catalysts. J. Org. Chem.. 77(4): 1722-1737. https://doi.org/10.1021/jo202220x3. Yang X, Birman V. 2009. Homobenzotetramisole-Catalyzed Kinetic Resolution of α-Aryl-, α-Aryloxy-, and α-Arylthioalkanoic Acids. Adv. Synth. Catal.. 351(14-15): 2301-2304. https://doi.org/10.1002/adsc.2009004514. Yang X, Birman VB. 2011. Chem.–Eur. J.. 17, 11296.5. Yang X, Birman VB. 2011. Nonenzymatic Dynamic Kinetic Resolution of α-(Arylthio)- and α-(Alkylthio)alkanoic Acids. Angew. Chem. Int. Ed.. 50(24): 5553-5555. https://doi.org/10.1002/anie.2010078606. Joannesse C, Johnston C, Concellón C, Simal C, Philp D, Smith A. 2009. Isothiourea-Catalyzed Enantioselective Carboxy Group Transfer. Angewandte Chemie International Edition. 48(47): 8914-8918. https://doi.org/10.1002/anie.2009043337. Liu G, Shirley ME, Van KN, McFarlin RL, Romo D. 2013. Rapid assembly of complex cyclopentanes employing chiral, α, β-unsaturated acylammonium intermediates. Nature Chem. 5(12): 1049-1057. https://doi.org/10.1038/nchem.17888. Wagner AJ, David JG, Rychnovsky SD. 2011. Determination of Absolute Configuration Using Kinetic Resolution Catalysts. Org. Lett.. 13(16): 4470-4473. https://doi.org/10.1021/ol201902y9. Wagner AJ, Rychnovsky SD. 2013. Determination of Absolute Configuration of Secondary Alcohols Using Thin-Layer Chromatography. J. Org. Chem.. 78(9): 4594-4598. https://doi.org/10.1021/jo400432q10. Wagner AJ, Miller SM, Nguyen S, Lee GY, Rychnovsky SD, Link RD. 2013. J. Chem. Educ. in press..11. Perry MA, Trinidad JV, Rychnovsky SD. 2013. Absolute Configuration of Lactams and Oxazolidinones Using Kinetic Resolution Catalysts. Org. Lett.. 15(3): 472-475. https://doi.org/10.1021/ol303239t12. Maji B, Joannesse C, Nigst TA, Smith AD, Mayr H. 2011. Nucleophilicities and Lewis Basicities of Isothiourea Derivatives. J. Org. Chem.. 76(12): 5104-5112. https://doi.org/10.1021/jo200803x13. Wagner AJ, Rychnovsky SD. 2013. Kinetic Analysis of the HBTM-Catalyzed Esterification of an Enantiopure Secondary Alcohol. Org. Lett.. 15(21): 5504-5507. https://doi.org/10.1021/ol402643n阿拉丁提供相关产品,详情可查看:Homobenzotetramisole (HBTM): A General Organocatalyst for Asymmetric Acylations (aladdin-e.com)阿拉丁相关产品列表产品货号产品名称C120993 (-)二异松蒎基氯硼烷 ,60% in Hexane, ca. 1.7mol/LC104780(-)二异松蒎基氯硼烷 ,60% in Heptane,ca. 1.7mol/LC283118 氯氢化[2-[[(R)-[2-[(R)-[2-(4-吗啉基-κN4)乙基]氨基-κN]乙基]硫-κS]甲基]苯基-κC]铱(III) ,M282866 甲烷磺酸(2-二环己基膦-3,6-二甲氧基-2',4',6'-三异丙基-1,1'-联苯)(2'-甲胺基-1,1'-联苯-2-基)钯(II) ,≥98%B105983 O-叔丁基-L-苏氨酸 ,98%B119767 (1S,2S)-二(4-甲氧基苯)-1,2-乙二胺 ,98%B120844 N-苄基氯化辛可宁丁 ,98%B120854 N-苄基氯化辛可宁[手性相转移催化剂] ,99%B120963 (+)-苯并四咪唑 ,97%B120975 (-)-苯并四咪唑 ,98%B123140 (R)-[2,2′-双(二苯基膦)-1,1′-联萘]二氯化钌 ,95%B281649 3-[[[3,5-双(三氟甲基)苯基]氨基]-4-[[((1S,2S)-2-(1-吡咯烷基)环己基]氨基]-3-环丁烯-1,2-二酮 ,98%,99% eeC100499 L-(-)樟脑磺酸 ,99%C104776 (+)二异松蒎基氯硼烷 ,60% in Heptane,ca. 1.7mol/LC106038 (+)-10-樟脑磺酸 ,99%C137946 氯{[(1R,2R)-(-)-2 - 氨基- 1,2 -二苯基乙基](五氟苯磺酰)氨基}(对伞花烃)钌(II) ,≥90.0%C282705 氯{N-[(1R,2R)-2-[(S)-[2-[[1,2,3,4,5,6-η)-4-甲基苯基]甲氧基]乙基]氨基]-1 ,2-二苯基乙基甲磺酰胺合}钌(II) ,Ru 15%D121000 (R)-5,5-联苯-2-甲基-3,4-丙醇-1,3,2-噁唑硼烷 ,97%D121025 (R)-(+)-2-二苯膦-2'-甲氧基-1,1'-联萘 ,98%D282689 二乙酸根[(S)-(-)-5,5′-双[二(3,5-二甲苯基)膦基]-4,4′-二-1,3-苯并二噁茂]钌(II) ,97%R138419 氯[[(1R,2R)-(-)-2-氨基-1,2-二苯基乙基](对甲苯磺酰基)氨基)](对伞花烃)钌(II) ,98%B129153 二氯双[二叔丁基-(4-二甲基氨基苯基)膦]钯(II) ,95%H121103 (S)-(+)-2-[羟基(二苯基)甲基]-1-甲基吡咯烷 ,97%H121106 (R)-(-)-2-[羟基(二苯基)甲基]-1-甲基吡咯烷 ,99%H283157 氢化(二甲基次膦酸-kP)[氢双(二甲基次膦酸-kP)]铂(II) [Ghaffar-Parkins catalyst] ,95%L117136 L-亮氨醇 ,96%M121139 (R)-2-(甲氧甲基)吡咯烷 ,99%M124083 (S)-(-)-2-甲基-CBS-噁唑硼烷 ,95%N281657 N-[3,5-双(三氟甲基)苯基]-N'-[(9R)-6'-甲氧基辛可宁-9-基]脲 ,95%,99% eeO282015 11bS)-8,9,10,11,12,13,14,15-八氢-4-羟基-2,6-二苯基-4-氧化物-二萘并[2,1-d:1',2'-f] [1,3,2]二氧杂磷醚 ,98%R138379 RuCl[(R,R)-TsDPEN](均三甲苯) ,≥90.0% (HPLC)R138672 二乙酸根[(R)-(+)-2,2'-二(二苯基膦基)-1,1'-联萘基]钌(II) ,97%
应用实例
2023.07.10
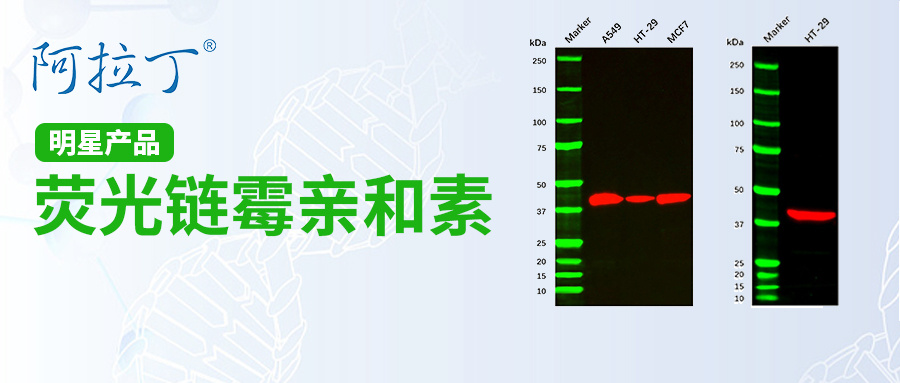
阿拉丁荧光链霉亲和素:解锁细胞秘密的光之钥匙!
新品
2023.07.07

Baeyer-Villiger氧化----阿拉丁试剂
简介Baeyer-Villiger氧化反应是以过氧化氢、间氯过氧苯甲酸(m-CPBA)、过氧乙酸或过氧三氟乙酸作为氧化剂使羰基的C-C键断裂从而引入氧原子,将醛、酮氧化为酯的反应。[1] 此反应最早由Adolf von Baeyer和Victor Villiger在1899年研究环酮的裂解时发现。Baeyer-Villiger氧化中迁移基团的立体化学保持不变,并且反应具有一定的区域选择性是其重要特征。区域特异性是该反应的本质,且给电子能力强(取代较多的碳)的基团优先迁移。一般的迁移顺序是:叔碳 > 环己基 > 仲碳 > 苄基 > 苯基 > 伯碳 > 甲基。因此它在有机合成中对官能团转化和环扩张有重要意义。图1. Baeyer-Villiger氧化反应图2. 环酮通过Baeyer-Villiger氧化合成内酯(环酯)机理在第一步中,羰基被质子化以增加其亲电性,然后过氧酸加入到该阳离子物质中,形成所谓的 Criegee 中间体(加合物)。图3. Baeyer-Villiger氧化反应机理1当羧酸离开该中间体时,形成缺电子氧取代基,其立即发生烷基迁移。这种烷基迁移和羧酸的损失都是在协同的过程中发生的。并假设迁移基团必须位于与过氧化物的断裂的氧-氧单键相反的位置。图4. Baeyer-Villiger氧化反应机理2应用Baeyer-Villiger氧化反应可应用于以下研究:1. 内消旋环己酮可通过酶催化Baeyer-Villiger氧化合成具有对映选择性的内酯。[2]2. 采用Baeyer–Villiger氧化可提高吲哚-2-羧酸盐转化为3-羟基吲哚-2-羧酸酯的产率,且该方法对各种吲哚取代基的底物通用。[3] 3. 从未活化的[18F]氟苯甲醛经Baeyer-Villiger氧化转化为[18F]-氟苯酚。[4]4. Baeyer-Villiger氧化可用于通过二苯并-18-冠-6的乙酰化和Baeyer-Villiger氧化制备二苯并18-冠-6的二羟基衍生物。使用三氟乙酸可以显著促进Baeyer-Villiger氧化。[5]5. 采用一锅化学酶法,通过Baeyer - Villiger反应从相应的酮开始合成γ-丁内酯。[6]6. 乙酸乙烯酯的无金属Baeyer-Villiger氧化合成。氧化剂Oxone通过Baeyer–Villiger氧化将的α,β-不饱和酮转化为相应的乙酸乙烯酯。[7]研究进展1.在过渡金属氧化物上使用过氧化氢水溶液作为氧化剂将环酮通过Baeyer–Villiger氧化合成相应的内酯。[8]2.在Mukaiyama条件下环己酮的Baeyer-Villiger氧化中,通过浸渍方法制备的具有不同钴负载量的二氧化硅负载的三钴四氧化物(Co3O4/SiO2)催化剂。Co3O4/SiO2催化剂具有成本低、效率高、稳定性好等优点,在环己酮氧化中的应用前景广阔。[9]3.以氯化锡为锡源,原硅酸四乙酯为硅源,在氢氧化钠介质中于室温下通过稀溶液制备的尺寸为几百纳米的亚微米级含锡MCM-41颗粒(Sn-MCM-41/SMP)。该材料被证明对H2O2水溶液Baeyer-Villiger氧化金刚烷酮具有活性和选择性。研究表明,在亚微米尺度上减小Sn-MCM-41的粒径是获得具有改善催化性能的Baeyer-Villiger氧化催化剂的有效途径。[10]4.一种化学酶催化环酮Baeyer-Villiger氧化为内酯的新方法。使用悬浮在离子液体中的南极假丝酵母脂肪酶B或Novozyme-435所催化的氧化环己酮和环丁酮,反应速率和产率都得到了提高。[11]5.通过高度区域和对映选择性的Baeyer-Villiger氧化,成功地开发了外消旋2-取代环戊酮的动力学拆分。[12]参考文献1.Yachnin BJ, Sprules T, McEvoy MB, Lau PCK, Berghuis AM. 2012. The Substrate-Bound Crystal Structure of a Baeyer?Villiger Monooxygenase Exhibits a Criegee-like Conformation. J. Am. Chem. Soc.. 134(18):7788-7795. https://doi.org/10.1021/ja211876p2.Taschner MJ, Black DJ. 1988. The enzymatic Baeyer-Villiger oxidation: enantioselective synthesis of lactones from mesomeric cyclohexanones. J. Am. Chem. Soc.. 110(20):6892-6893. https://doi.org/10.1021/ja00228a0533.Hickman ZL, Sturino CF, Lachance N. 2000. A concise synthesis of 3-hydroxyindole-2-carboxylates by a modified Baeyer?Villiger oxidation. Tetrahedron Letters. 41(43):8217-8220. https://doi.org/10.1016/s0040-4039(00)01456-84.Castillo Meleán J, Ermert J, Coenen H. 2014. [18F]Fluorophenols from non-activated [18F]fluorobenzaldehydes by Baeyer-Villiger oxidation. J Nucl Med. 55(1):155.5.Utekar DR, Saman SD. 2014. Application of Bayer-Villiger Reaction to the Synthesis of Dibenzo-18-crown-6, Dibenzo-21-crown-7 and Dihydroxydibenzo-18-crown-6. Journal of the Korean Chemical Society. 58(2):193-197. https://doi.org/10.5012/jkcs.2014.58.2.1936.González-Martínez D, Rodríguez-Mata M, Méndez-Sánchez D, Gotor V, Gotor-Fernández V. 2015. Lactonization reactions through hydrolase-catalyzed peracid formation.Use of lipases for chemoenzymatic Baeyer?Villiger oxidations of cyclobutanones. Journal of Molecular Catalysis B: Enzymatic. 11431-36. https://doi.org/10.1016/j.molcatb.2014.09.0027.Poladura B, Martínez-Castañeda &, Rodríguez-Solla H, Llavona R, Concellón C, del Amo V. 2013. General Metal-Free Baeyer?Villiger-Type Synthesis of Vinyl Acetates. Org.Lett.. 15(11):2810-2813. https://doi.org/10.1021/ol401143q8.Ma Q, Xing W, Xu J, Peng X. 2014. Baeyer?Villiger oxidation of cyclic ketones with aqueous hydrogen peroxide catalyzed by transition metal oxides. Catalysis Communications. 535-8. https://doi.org/10.1016/j.catcom.2014.04.0179.Zang J, Ding Y, Pei Y, Liu J, Lin R, Yan L, Liu T, Lu Y. 2014. Efficient Co3O4/SiO2 catalyst for the Baeyer?Villiger oxidation of cyclohexanone. Reac Kinet Mech Cat. 112(1):159-171. https://doi.org/10.1007/s11144-014-0687-110.Chen N, Jiang Y, Cheng W, Lin K, Xu X. 2015. Synthesis of submicrometer-sized Sn-MCM-41 particles and their catalytic performance in Baeyer-Villiger oxidation. Chem. Res. Chin.Univ.. 31(1):138-143. https://doi.org/10.1007/s40242-014-4204-x11.Drod A, Erfurt K, Bielas R, Chrobok A. Chemo-enzymatic Baeyer-Villiger oxidation in the presence of Candida antarctica lipase B and ionic liquids. New J. Chem.. 39(2):1315-1321. https://doi.org/10.1039/c4nj01976h12.Zhou L, Liu X, Ji J, Zhang Y, Wu W, Liu Y, Lin L, Feng X. 2014. Regio- and Enantioselective Baeyer?Villiger Oxidation: Kinetic Resolution of Racemic 2-Substituted Cyclopentanones. Org.Lett.. 16(15):3938-3941. https://doi.org/10.1021/ol501737a阿拉丁相关产品列表产品货号产品名称R115618(R)-(+)-2,2'-联[二-(4-甲基苯基)膦基]-1,1'-联萘 ,98%C425871(±)-樟脑(合成) ,10mM in DMSOM134785薄荷酮 ,异构体混合物, 98%C295202环己酮 ,99.8%B1291631,2-双(二苯基膦)苯 ,98%B3017611,2-双(二苯基膦)苯 ,95%C1064923-氯过氧苯甲酸 ,85%C108567环戊酮 ,>99.0%(GC)C108568环戊酮 ,CP,97%C110690(±)-樟脑(合成) ,96%C116449环己酮 ,AR,99.5%C116451环己酮 ,>99.5%(GC)C116453环己酮 ,ACS, ≥99.0%
应用实例
2023.07.06

关于催化剂的基本概念----阿拉丁试剂
过渡态理论[1]过渡态理论,也称活化复合体理论或绝对反应速率理论,是对化学反应和其他过程的研究,认为它们是通过组成原子和分子的相对位置和势能的连续变化来进行的。在原子或分子的初始排列和最终排列之间的反应路径上,存在一种中间态,处于这个中间态时,势能达到最大值。这个最大值对应的构型被称为活化络合物,它的状态被称为过渡状态。过渡态能量与初始态能量的差值与反应的实验活化能密切相关;它表示反应或流动系统为进行转换必须获得的最小能量。在过渡态理论中,活化络合物被认为是在与处于初始状态的原子或分子的平衡状态下形成的,因此可以确定其统计和热力学性质。达到终态的速率由形成的激活复合物的数量和它们进入终态的频率决定。对于简单的系统,这些量可以用统计力学原理计算出来。用这种方法,化学或物理过程的速率常数可以用原子和分子的尺寸、原子质量和原子间或分子间的力来表示。过渡状态理论也可以用热力学术语来表述。图1:势能曲线活化能是指在化学反应中将反应物转化为产物所需的最小能量。活化能的值等于处于中间构型(称为过渡态或活化络合物)的粒子与处于初始态的反应物粒子之间的位能差。因此,活化能可以看作是反应物在生成生成物之前必须克服的障碍。反应热[2]反应热,也叫反应焓,在化学反应过程中,为了使所有物质在相同的温度下存在,必须增加或除去的热量。如果反应系统容器中的压力保持恒定值,则测量的反应热也表示随反应过程而发生的热力学量的变化,称为焓或热含量。即反应结束时物质的焓与反应开始时物质的焓之差。因此,在恒压下确定的反应热也被指定为反应焓,用符号ΔH表示。若反应热为正,则称该反应为吸热反应;如果是负的,则是放热的。伴随化学变化的热效应的预测和测量对于理解和运用化学反应是很重要的。如果包含反应系统的容器隔热程度非常高,没有热量流入或流出系统(绝热状态),则伴随转化的热效应可能表现为温度的升高或降低(视具体情况而定)。确切的反应热值对于正确设计化学过程中使用的设备是必要的。因为对每一个反应进行热测量是不现实的,而且对某些反应这样的测量甚至可能是不可行的,所以习惯上是从已编制的标准热数据的适当组合来估计反应的热。这些数据通常采用标准生成热和燃烧热的形式。标准生成热的定义是:当1摩尔化合物由其组成元素形成时,每种物质在其正常物理状态(气体、液体或固体),在25°C(77°F)和1大气压下吸收或演化的热量。元素的生成热可以随意地赋值为零。标准燃烧热的定义与此类似,即1摩尔物质在过量的氧气中燃烧时,在25°C和一大气压下所产生的热量。从测量的生成热和燃烧热计算反应热的方法是基于热总和的原理称为赫斯定律。图2:催化和热反应(非催化)反应的能量分布催化剂[3]催化剂是在化学中经常使用到的一种物质,能在不消耗自身的情况下加快反应进度。酶是自然发生的催化剂,负责许多基本的生化反应的进程加速。大多数固体催化剂是金属或金属元素以及半金属元素硼、铝和硅的氧化物、硫化物和卤化物。气态和液态催化剂通常以其纯物质形式或与适当的载体或溶剂结合使用;固体催化剂通常分散在其他被称为催化剂载体的物质中。一般来说,催化作用是催化剂和反应物之间的化学反应,形成化学中间体,这些中间体能够更容易地相互反应或与另一种反应物反应,以形成所需的最终产物。在化学中间体和反应物之间的反应中,催化剂被再生。催化剂和反应物之间的反应模式变化很大,在固体催化剂中往往是复杂的。典型的这些反应有酸碱反应、氧化还原反应、配位络合物的形成和自由基的形成。对于固体催化剂,反应机理受到表面性质和电子或晶体结构的强烈影响。某些固体催化剂,称为多功能型催化剂,能够与反应物发生多种形式的相互作用;双功能催化剂则被广泛应用于石油工业的重整反应。催化反应是许多工业化学过程的基础,且催化剂制造本身就是一个快速发展的工业过程。催化过程及其催化剂催化过程对应所使用的催化剂氨的合成铁硫酸的生产氮(II)氧化物、铂石油裂解沸石不饱和烃的加氢反应镍、铂或钯汽车尾气中碳氢化合物的氧化氧化铜、氧化钒、铂、钯正丁烷向异丁烷的异构化氯化铝、氯化氢图3:乙烯聚合工业中用到的齐格勒-纳塔催化剂乙烯气体在压力下被泵入反应容器,在该反应容器中,它在齐格勒-纳塔催化剂的影响下,在溶剂存在下进行聚合。一种由聚乙烯、未反应的乙烯单体、催化剂和溶剂组成的浆液从反应器中流出。未反应的乙烯被分离并返回反应器,而催化剂被酒精洗涤中和并过滤。多余的溶剂从热水浴中回收并回收,干燥器将湿聚乙烯脱水为其最终的粉末形式。 化学中间体[4]化学中间体,指在某些反应物转化为产物过程中产生的任意化学物质。大多数合成过程都涉及通过一系列步骤将一些现成的、通常价格较为低廉的物质转化为所需的产品。一个步骤产生并用于后续步骤的所有物质都被认为是中间体。除了在中间不稳定分子生成时停止反应的物质可以作为产物回收的物质外,有些化学物质即使还没有被分离出来,也已知或假设为中间体。在一般不稳定的中间体的类别中,被研究最为充分的是自由基、碳烯和碳离子。这些中间体属于是分子的高度活性碎片,通常只在很短的时间内保持不结合。参考文献1. Laidler, Keith J.. "transition-state theory". Encyclopedia Britannica, 23 Sep. 2019, https://www.britannica.com/science/transition-state-theory.2. Britannica, The Editors of Encyclopaedia. "heat of reaction". Encyclopedia Britannica, 25 Aug. 2022, https://www.britannica.com/science/heat-of-reaction.3. Britannica, The Editors of Encyclopaedia. "catalyst". Encyclopedia Britannica, 30 Oct. 2022, https://www.britannica.com/science/catalyst.4. Britannica, The Editors of Encyclopaedia. "chemical intermediate". Encyclopedia Britannica, 20 Jul. 1998, https://www.britannica.com/science/chemical-intermediate.阿拉丁相关产品列表产品货号产品名称C105368 氯化铯 ,AR,≥99%H135562 氯铂酸 水合物 ,99.995% trace metals basisC111683 氯化铜,二水 ,99.99% metals basisD105295 十二烷基三甲基氯化铵(DTAC) ,99%G109501 硝酸镓(III) 水合物 ,99.9% metals basisH157071 六氨基氯化钴 ,>99.0%(T)H105309 十六烷基三甲基氯化铵(CTAC) ,97%I106504 氯化亚铁,无水 ,99.5%,粉末L107012 硼氢化锂四氢呋喃溶液(易制爆) ,2.0M in THFL107011 硼氢化锂(易制爆) ,95%L108933 溴化锂溶液 ,55 wt. % in H2OL101684 碳酸锂 ,99.99% metals basisL298760 氯化锂 ,无水级,98%L130113 氯化锂 ,无水级,99.9% metals basisL104230 氟化锂 ,99.9% metals basisN108314 氧化镍 ,AR,99.0 %P116795 钯碳 ,10%Pd,contains 40-60% H2OP116273 溴化钾 ,SP,99%P116293 氟化钾,无水 ,AR,powder,99.0%P105228 氯铂酸钾 ,98%P123846 氯铂酸钾 ,99.9% metals basisP112221 磷酸氢二钾,无水 ,色谱级,≥99.0%(T)R109233 三氯化铑(III) 水合物 ,Rh 38.5-42.5%S111733 无水碳酸钠 ,GR,≥99.8%S111736 无水碳酸钠 ,99.99% metals basisS110860 氢化钠 ,60% dispersion in mineral oilS105269 高氯酸钠,无水(易制爆) ,99.99% metals basisS105271 高氯酸钠,无水(易制爆) ,色谱级,99.0%S109393 高氯酸钠 一水合物(易制爆) ,AR,99%T105045 四丁基氢氧化铵溶液 ,10% in H2OT100882 四甲基氢氧化铵 ,AR,25%水溶液T105417 二氧化钛 ,99.99% metals basisT103857 三氧化钨 ,99.8% metals basisZ112532 氯化锌 ,99.95% metals basisZ100682 三氟甲烷磺酸锌 ,98%Z106340 正丙醇锆 ,70 wt. % 正丙醇溶液M164461 [2-(甲基丙烯酰基氧基)乙基]二甲基-(3-磺酸丙基)氢氧化铵 ,≥97%C105060 碳酸铯 ,99.99% metals basisC105063 碳酸铯 ,99.9% metals basisC111535 氯化钠 ,GR,99.8%M118110 二氧化锰 ,99% metals basisP116279 碘化钾 ,AR,≥99.0%
应用实例
2023.07.04

羟醛缩合反应----阿拉丁试剂
什么是羟醛缩合反应?醛醇缩合反应是由Charles Wurtz提出的一种有机反应,他于1872年首次从乙醛中制备出β-羟基醛[1]。在醛醇缩合反应中,烯酸盐离子与羰基化合物在酸/碱催化剂的存在下反应生成β-羟基醛或β-羟基酮,随后脱水得到共轭的烯酮。是一种有效的碳-碳成键反应。醛醇缩合反应的基本步骤是:1. 醛醇(醛+醇)反应-醛(或酮)烯酸酯与醛(或酮)的另一分子在NaOH或KOH存在下反应,形成β-羟基醛(或酮)。2. 脱水/消除反应——从β-羟基醛(或酮)中除去水分子,形成α,β-不饱和醛或α,β-不饱和酮。注意事项请参阅安全数据表,以了解有关危害和安全处理措施的信息。应用场景醛醇缩合反应可用于以下合成:· 脂肪酸的酶促合成[2]· 埃博霉素B的高效全合成[3]· (E)-6-(2,2,3-三甲基-环戊-3-烯基)-己-4-烯-3-酮的制备[4]:将2-甲基乙酰乙酸乙酯(2)(727 mg, 4.53 mmol)添加至NaH (184 mg, 4.60 mmol)存于二氧六环(25 mL)中之搅拌溶液中。然后添加莰烯醛(1)(707 mg, 3.72 mmol)并将混合物回流15 h。然后添加2N HCl (15 mL)并用Et2O (3×15 mL)萃取混合物。用2N HCl (2×15 mL)、饱和Na2CO3 (2×15 mL)和盐水(3×15 mL)洗涤合并的有机层。经无水Na2SO4干燥有机相并且在减压下蒸发溶剂,得到残余物(900 mg),在减压下通过蒸馏纯化残余物,得到标题化合物3 (505 mg, 2.63 mmol, 58%)。· 聚戊二醛高聚物的合成[5]· (±)-麻黄碱的立体选择性合成[6]· 大环内酯类和离子载体抗生素(天然产物)的合成[7]· 两种结构独特的四环喹诺酮类化合物——双螺旋胺A和B的全合成[8]该路线分为三步,通过5-溴-4-甲氧基羰基甲氧基喹啉与乙烯基硼酸酯的Suzuki交叉偶联,然后进行α-酮羟基化和分子内醛醇缩合和内酯化的双环化,从而获得吡喹喹啉丁烯内酯环。随后通过对侧链的操作,引入胍片段,完成了双螺旋胺B的合成,而双螺旋胺A的同系物是由后期中间体脱羧作用得到的。最新研究及未来发展趋势· 采用水溶性杯芳烃作为反相转移催化剂,对活化的甲基和亚甲基化合物进行了醛缩合和迈克尔加成反应[9]· 采用含铯离子催化剂,在SBA-15介孔分子载体上进行乙酸甲酯与甲醛的缩合反应[10]该催化剂在缩合反应中表现出较高的催化活性。XRD表征表明,低于5 wt%负载的硝酸铯在SBA-15支架上高度分散。FT-IR和XPS结果证实催化剂表面形成Si-O-Cs。NH3-TPD和CO2-TPD结果表明,弱Lewis酸碱对负载在表面,这些弱酸碱活性位点可能有利于醛醇缩合反应。5Cs/SBA-15催化剂表现出最高的乙酸甲酯转化率(48.4%)和95.0%的丙烯酸甲酯选择性。失活催化剂经煅烧完全再生。催化剂再生9次,总操作时间超过60 h,乙酸甲酯的初始转化率和对丙烯酸甲酯的选择性均未发生变化。催化剂具有较高的催化活性,主要是由于载体表面的Si-O-Cs基团具有较强的弱酸碱性质。· 环己酮与对硝基苯甲醛在水中的有机催化不对称醛醇反应[11]· 一锅法铜催化醚化/醛醇缩合级联反应制备二苯并西平内酰胺[12]· 通过烷基化、区域选择性碘化、醛醇缩合、Suzuki偶联和[1,3]-sigma重排等方法合成一系列二戊烯化和二癸基化查尔酮类似物[13]· 在KOH存在的甲醇溶剂中,苊醌与苯乙酮进行迈克尔加成和醛醇缩合,可形成多米诺骨牌序列,从而进一步形成了不同的2:2加合物[14]· 缩醛反应通常会利用酸性或碱性催化剂进行,因此,MCM-41的改性是通过两个酸性基团(3-丙基磺基、3-丙基羧基)和一个碱性基团(3-(1,2-二乙基氨基)丙基)后接枝法实现的,采用的是经硫酸和硝酸处理的MCM-41[15]。用BET、元素分析和UV-Vis光谱对所制备的功能化材料进行了表征。结果表明,最佳反应条件为反应混合物温度100℃,反应物4-异丙基苯甲醛:丙醛的摩尔比为1:2,催化剂的用量和种类为50 wt%的MCM SO3H(与4-异丙基苯甲醛的用量相比),丙醛的加入时间为90分钟,反应的适宜溶剂为甲苯。在最佳反应条件下,甲醛的收率为45%。将所得结果与H2SO4均相催化的效果进行了比较,其收率为15%。参考文献1. Nielsen AT, Houlihan WJ. The Aldol Condensation. 1-438. https://doi.org/10.1002/0471264180.or016.012. Brady RO. 1958. The Enzymatic Synthesis of Fatty Acids by Aldol Condensation. Proceedings of the National Academy of Sciences. 44(10):993-998. https://doi.org/10.1073/pnas.44.10.9933. Balog A, Haris C, Savin K, Zhang X, Chou T, Danishefsky S. 1998. Angew. Chem. Int. Ed.. 372675.4. Badía C, Castro J, Linares Palomino P, Salido S, Altarejos J, Nogueras M, Sánchez A. (E)-6-(2,2,3-Trimethyl-cyclopent-3-enyl)-hex-4-en-3 one. Molbank. 2004(1): M388. https://doi.org/10.3390/m3885. Tashima T, Imai M, Kuroda Y, Yagi S, Nakagawa T. 1991. Structure of a new oligomer of glutaraldehyde produced by aldol condensation reaction. J. Org. Chem.. 56(2):694-697. https://doi.org/10.1021/jo00002a0386. Heathcock CH, Buse CT, Kleschick WA, Pirrung MC, Sohn JE, Lampe J. 1980. Acyclic stereoselection. 7. Stereoselective synthesis of 2-alkyl-3-hydroxy carbonyl compounds by aldol condensation. J. Org. Chem.. 45(6):1066-1081. https://doi.org/10.1021/jo01294a0307. Masamune S, Choy W, Kerdesky Francis A. J., Imperiali B. 1981. Stereoselective aldol condensation. Use of chiral boron enolates. J. Am. Chem. Soc.. 103(6):1566-1568. https://doi.org/10.1021/ja00396a0508. Jolibois AER, Lewis W, Moody CJ. 2014. Total Synthesis of (±)-Distomadines A and B. Org. Lett.. 16(4):1064-1067. https://doi.org/10.1021/ol403598k9. Shimizu S, Shirakawa S, Suzuki T, Sasaki Y. 2001. Water-soluble calixarenes as new inverse phase-transfer catalysts. Their application to aldol-type condensation and Michael addition reactions in water. Tetrahedron. 57(29):6169-6173. https://doi.org/10.1016/s0040-4020(01)00572-510. Yan J, Zhang C, Ning C, Tang Y, Zhang Y, Chen L, Gao S, Wang Z, Zhang W. 2015. Vapor phase condensation of methyl acetate with formaldehyde to preparing methyl acrylate over cesium supported SBA-15 catalyst. Journal of Industrial and Engineering Chemistry. 25344-351. https://doi.org/10.1016/j.jiec.2014.11.01411. Mase N, Nakai Y, Ohara N, Yoda H, Takabe K, Tanaka F, Barbas CF. 2006. Organocatalytic Direct Asymmetric Aldol Reactions in Water. J. Am. Chem. Soc.. 128(3):734-735. https://doi.org/10.1021/ja057331212. Lim HS, Choi YL, Heo J. 2013. Synthesis of Dibenzoxepine Lactams via a Cu-Catalyzed One-Pot Etherification/Aldol Condensation Cascade Reaction: Application toward the Total Synthesis of Aristoyagonine. Org. Lett.. 15(18):4718-4721. https://doi.org/10.1021/ol402036t13. Wang H, Zhang L, Liu J, Yang Z, Zhao H, Yang Y, Shen D, Lu K, Fan Z, Yao Q, et al. 2015. Synthesis and anti-cancer activity evaluation of novel prenylated and geranylated chalcone natural products and their analogs. European Journal of Medicinal Chemistry. 92439-448. https://doi.org/10.1016/j.ejmech.2015.01.00714. Domino reaction sequences leading to the formation of 2:2 adducts between acenaphthenequinone and acetophenone. 2014(6):127. https://doi.org/10.3998/ark.5550190.p008.83415. Vrbková E, Vyskocilová E, Cerveny L. 2015. Functionalized MCM-41 as a catalyst for the aldol condensation of 4-isopropylbenzaldehyde and propanal. Reac Kinet Mech Cat. 114(2):675-684. https://doi.org/10.1007/s11144-014-0811-2阿拉丁提供相关产品:产品货号产品名称C106972 1,3-环己二酮 ,97%B104602 2,3-丁二酮 ,standard for GC,≥99.0%(GC)B1046012,3-丁二酮 ,98%B104603 2,3-丁二酮 ,分析标准品,≥99.0%(GC)H1066962,3-己二酮 ,90%P106293 2,3-戊二酮 ,98%C139450 2-环戊烯-1-酮 ,≥96.0%(GC)C352403 2-环戊烯-1-酮缩乙醛 ,97%C107526 2-环戊基环戊酮 ,97%H103803 2-庚酮 ,98%H1038272-庚酮 ,分析标准品,≥99.8%(GC)H105744 2-己酮 ,98%H105743 2-己酮 ,standard for GC,>99.5%(GC)H117489 2-己基环戊酮 ,≥96%(GC)P103783 2-戊酮 ,色谱级,99.0%P103782 2-戊酮 ,分析标准品,用于环境分析,≥99.8%(GC)H117488 2-庚基环戊酮 ,>99.0%(GC)A106311 乙醛 ,99.5%B110460 苯甲醛 ,AR,≥98%(GC)B103861 二苯甲酮 ,99%B103860 二苯甲酮 ,CPC105368 氯化铯 ,AR,≥99%C108569 环戊酮 ,分析标准品D105295十二烷基三甲基氯化铵(DTAC) ,99%H157071 六氨基氯化钴 ,>99.0%(T)H105309 十六烷基三甲基氯化铵(CTAC) ,97%L108933 溴化锂溶液 ,55 wt. % in H2OL101684 碳酸锂 ,99.99% metals basisL298760 氯化锂 ,无水级,98%N108314氧化镍 ,AR,99.0 %P165286 丙酮酸 ,≥70%R109233 三氯化铑(III) 水合物 ,Rh 38.5-42.5%S111733 无水碳酸钠 ,GR,≥99.8%S110860 氢化钠 ,60% dispersion in mineral oilS140966 高氯酸钠,无水(易制爆) ,99%S105271 高氯酸钠,无水(易制爆) ,色谱级,99.0%T103857 三氧化钨 ,99.8% metals basisZ112532 氯化锌 ,99.95% metals basisH399657 盐酸(易制毒) ,37%B131582 2-溴苯乙酮 ,用于GC衍生化, ≥99.0%
参数原理
2023.06.29

免疫沉淀技术----阿拉丁试剂
免疫沉淀简介免疫沉淀(IP)是应用最广泛的免疫化学技术之一。免疫沉淀,然后是SDS-PAGE和免疫印迹,通常用于各种应用,例如:确定蛋白质抗原的分子量,研究蛋白质间相互作用,确定特异性酶活性,监测蛋白质翻译后修饰,并确定蛋白质的存在和数量。IP技术还能够检测稀有蛋白质,否则很难检测,因为它们可以通过免疫沉淀浓缩到1×104倍。在IP方法中,来自细胞或组织匀浆的蛋白质通过免疫复合物在适当的裂解缓冲液中沉淀,所述免疫复合物包括抗原(蛋白质)、第一抗体和蛋白质A-、G-或L-蔗糖缀合物或第二抗体琼脂糖缀合物。琼脂糖缀合物的选择取决于初级抗体的物种来源和同种型。所描述的方法是可比较的,并且方法的选择取决于特异性抗原抗体系统。试剂和设备1.琼脂糖缀合物:固定在Sepharose®CL-4B(产品编号P405880)或G蛋白琼脂糖4B(产品编号R394624)或L蛋白琼脂糖(产品编号P394623)上的A蛋白,或抗体-琼脂糖缀合体(根据第一抗体的物种来源和同种型的第二抗体缀合物)2.细胞或组织裂解物制备3.免疫沉淀一级抗体和非相关抗体(阴性对照)4.SDS-PAGE和免疫印迹试剂及设备5.HNTG缓冲液:20mM HEPES缓冲液,pH 7.5(产品编号H109406),含有150mM NaCl(产品编号S301560)、0.1%(w/v)Triton X-100(产品编号T434565)和10%(w/v)甘油(产品编号G424796)6.洗涤缓冲液(冰冷):HNTG缓冲液,或PBS pH 7.4(产品编号R355028),或根据所需洗涤严格程度的其他缓冲液(RIPA缓冲液 产品编号 R488105)7.具有或不具有2-巯基乙醇(2-ME(产品编号M301574))的莱姆利样品缓冲液(例如1x、2x或3x)8.微型离心管9.微型离心机和振动筛免疫步骤特异性抗原的免疫沉淀注意:除非另有说明,否则在冰上使用微型离心管执行所有IP步骤。用洗涤缓冲液洗涤琼脂糖缀合物两次,在室温下以12000xg离心10秒,丢弃上清液。注意:如果琼脂糖缀合物是粉末,则用去离子H2O将其重建,并使其膨胀5分钟。在洗涤缓冲液(50%悬浮液)中重新悬浮琼脂糖缀合物。根据需要,继续使用方法A或方法B。方法A1.在微量离心管中将琼脂糖缀合物分成50-100µL(约25-50µL琼脂糖/床体积)的等分试样。2.在每根试管中加入10µL适当稀释的初级抗体。3.在室温下培养15-60分钟,在合适的摇壶上轻轻混合样品。4.在4°C下以3000xg离心2分钟,丢弃上清液。5.用1mL洗涤缓冲液洗涤每个样品,在4°C下以3000xg离心2分钟,重复此步骤至少两次。6.向每根试管中加入0.1-1.0 mL的细胞裂解物。7.在4°C下培养90分钟至过夜,在合适的摇壶上轻轻混合样品。8.通过在4°C下以3000xg离心2分钟来收集免疫沉淀的复合物,丢弃上清液。9.用1 mL洗涤缓冲液洗涤颗粒,在4°C下以3000xg离心2分钟。重复此步骤至少3次。方法B1.向细胞裂解物样品(0.1-1.0 mL)中加入10µL适当稀释的抗体(参考产品规范)。2.在4°C下培养90分钟至过夜,在合适的摇壶上轻轻混合样品。3.加入50-100µL琼脂糖缀合物悬浮液(约25-50µL琼脂糖/床体积)。4.在4°C下培养15-60分钟,用摇壶轻轻混合样品。5.通过在4°C下以3000xg离心2分钟来收集免疫沉淀的复合物,丢弃上清液。6.通过重悬浮和在4°C下以3000xg离心2分钟,用1ml洗涤缓冲液洗涤颗粒。重复此步骤至少3次。SDS-PAGE的制备1.将每个颗粒重新悬浮在25-100µL莱姆利样品缓冲液中,最终浓度为1x样品缓冲液,将样品在95°C下加热5分钟。2.在室温下以12000xg离心30秒。收集上清液(IP样品)。如果需要,(在蛋白质稳定性允许的情况下)IP样品可以储存在-70°C的样品缓冲液中。3.在SDS-PAGE上运行已知浓度的样品和MW标准品(聚丙烯酰胺凝胶的适当百分比取决于蛋白质的分子大小)。4.转移到硝化纤维上,进行免疫印迹。免疫沉淀(IP)的常见问题及解答1. 用于一般免染的抗体是否都可以用作免疫沉淀实验? 答:抗体的性质对免疫沉淀实验影响很大。抗体不同,和抗原以及Protein G或Protein A的结合能力也就不同。所以一般免染能结合的抗体未必能用于IP反应。一般来说,多抗是沉淀反应最好的选择。另外,纯化的单抗、腹水和杂交瘤上清液也可用于免疫沉淀。2. 为什么溶解抗原的缓冲液中要加入一定量的蛋白酶抑制剂? 答:从活性细胞裂解出来的样品中,往往含有一定量的蛋白酶。为防止预检测蛋白的分解,修饰,溶解抗原的缓冲液必须加蛋白每抑制剂,并且在低温下进行实验。3. 溶解抗原的缓冲液的配制需要注意什么? 答:多数的抗原是细胞构成的蛋白,特别是骨架蛋白,缓冲液必须要使其溶解。为此,必须使用含有强表面活性剂的缓冲液,尽管它有可能影响一部分抗原抗体的结合。另一面,如用弱表面活性剂溶解细胞,就不能充分溶解细胞蛋白。即便溶解也产生与其它的蛋白结合的结果,抗原决定族被封闭,影响与抗体的结合。4. 免疫沉淀实验应如何把握抗体和缓冲液的比例? 答:每次沉淀实验之前,考虑抗体/缓冲液的比例十分重要。抗体过少就不能检出抗原,过多则就不能沉降在beads上,残存在上清;缓冲剂太少则不能溶解抗原,过多则抗原被稀释。具体比例视具体情况而定。5. 免疫沉淀应该如何配制细胞裂解液? 答:适当的细胞裂解液的选择取决于所要研究蛋白的特性。TritonX-100,NP40是非离子界面活性剂,作用温和;DOC(sodium de-oxycholate),SDS是离子性的强活性剂。所以裂解液的配制时所用去污剂的种类和浓度以及盐浓度等条件需实验者进行优化。注意如果忘记加TritonX-100,只加DOC或SDS,可能使抗体完全失去活性。参考文献[1] Harlow E, Lane D. 1988. Antibodies: A Laboratory Manual. Laboratory Press New York: Cold Spring Harbor.阿拉丁提供相关产品:产品货号产品名称G424796甘油 ,10mM in DMSOH109406N-2-羟乙基哌嗪-N'-2-乙磺酸 ,99%P394623蛋白质L琼脂糖R488105RIPA 缓冲液 ,R355028ReadytouseTM PBS速溶颗粒(pH 7.4) ,R394624重组蛋白G琼脂糖 ,S301560氯化钠溶液 ,5mol/LT434565Triton™ X-102 ,M301574β-巯基乙醇 ,分子生物学,用于电泳,细胞培养,99%
应用实例
2023.06.29

傅-克酰基化反应----阿拉丁试剂
反应简介傅-克酰基化反应是指一类在路易斯酸作为催化剂的条件下,芳烃与酰氯或酸酐进行酰化的反应。该反应由于羰基吸电子效应的影响,一般不会像烷基化反应生成多重酰基化产物,仅通过亲电芳香取代形成单酰化产物。[1,2]图1. 傅-克酰基化反应反应机理Ⅰ. 路易斯酸催化剂(AlCl3)和酰基的氯原子形成络合物,氯的解离形成酰基碳正离子。Ⅱ. 酰基离子(RCO+)继续对芳环进行继续对芳烃进行亲电攻击。随着络合物的形成,其芳香性暂时消失。Ⅲ. 中间态去质子化,恢复芳环的芳香性。电荷转移至氯离子形成HCl,AlCl3催化剂重新形成。[1]图2. AlCl3傅-克酰基化反应机理应用傅-克酰基化反应可应用于以下化合物的合成:1. 芳基乙酸衍生物[4]2. 聚醚醚酮(PEEK)或mPEK[5]3. 1,5-双(4-氟苯甲酰基)-2,6-二甲基萘[6]4. 芳香酮[7]5. 不对称芳香胺[8]6. 环酮,例如1-四烯酮和1-茚满酮[9]7. 2-乙酰基-6-甲氧基萘,用于合成非甾体抗炎镇痛新药萘普生及萘普酮的关键中间体[10]研究进展1.PVP-TfOH已经作为傅-克酰基化反应中高效且易于后处理的固体超强酸催化剂体系得以应用。在温和的反应条件下,芳烃和乙醛酸通过傅-克酰基化反应,实现了无溶剂一锅法合成二芳基乙酸衍生物。[4] 图3. 芳烃和乙醛酸通过傅-克酰基化反应,无溶剂一锅法合成二芳基乙酸衍生物2. 一种基于咪唑的离子液体可作为催化剂,催化芳烃与乙酰氯进行傅-克酰基化反应。[11]3. 据报道,三氟甲磺酸铒是一种含有给电子基团且可用于微波辅助下芳烃的傅-克酰基化的高效催化剂。[12]4. 虎皮楠生物碱是一类结构高度复杂多样的三萜类生物碱,可通过分子内的傅-克酰基化反应直接且快速的构建其ACDE环系统。[13]5. 酸催化的的多米诺傅-克酰基化反应,可用于高效构建台湾杉醌的6,5,6-ABC三环骨架结构。还可用于二萜(±)-甲萘醌B和(±)-二氯酮的合成。[14]6. 通过亲电傅克酰基化缩聚反应可合成两种含1,4-萘单元的单体,新型聚(芳基酮)和聚(芳基醚酮砜)。[15]7. 据研究,SBA-15中三苯基锡的甲苯和乙酸酐可发生傅-克酰基化反应。[16]8. 离子液体1-异丁基-3-甲基咪唑二氢磷酸([i-BMIM]H2PO4)中的三氟甲磺酸铟在芳香族化合物与酸酐的傅-克酰基化反应中显示出强力的催化活性。[17]9. 25,27-二烷氧基杯芳烃的傅-克酰化反应。使用酰氯和AlCl3在1,2-二氯乙烷中直接酰化脱叔丁基杯芳烃,以高产率区域选择性地提供相应的二酰基衍生物。[18]参考文献:1.Fox MA, Whitesell JK. 1994. Organic Chemistry. Boston: Jones and Bartlett.2.Li JJ. 2009. Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications. Springer.3.Sartori G, Maggi R. Advances in Friedel-Crafts Acylation Reactions. https://doi.org/10.1201/97814200679344.Prakash G, Paknia F, Kulkarni A, Narayanan A, Wang F, Rasul G, Mathew T, Olah GA. 2015. Taming of superacids: PVP-triflic acid as an effective solid triflic acid equivalent for Friedel?Crafts hydroxyalkylation and acylation. Journal of Fluorine Chemistry. 171102-112. https://doi.org/10.1016/j.jfluchem.2014.08.0205.Baek J, Lyons CB, Tan L. 2004. Grafting of Vapor-Grown Carbon Nanofibers via in-Situ Polycondensation of 3-Phenoxybenzoic Acid in Poly(phosphoric acid). Macromolecules. 37(22):8278-8285. https://doi.org/10.1021/ma048964o6.Ohno M, Takata T, Endo T. 1995. Synthesis of a novel naphthalene-based poly(arylene ether-ketone) by polycondensation of 1,5-bis(4-fluorobenzoyl)-2,6-dimethylnaphthalene with bisphenol a. J. Polym.Sci. A Polym.Chem.. 33(15):2647-2655. https://doi.org/10.1002/pola.1995.0803315117.de Noronha RG, Fernandes AC, Romão CC. 2009. MoO2Cl2 as a novel catalyst for Friedel?Crafts acylation and sulfonylation. Tetrahedron Letters. 50(13):1407-1410. https://doi.org/10.1016/j.tetlet.2009.01.0398.Nordlander JE, Payne MJ, Njoroge FG, Balk MA, Laikos GD, Vishwanath VM. 1984.Friedel-Crafts acylation with N-(trifluoroacetyl)-.alpha.-amino acid chlorides.Application to the preparation of .beta.-arylalkylamines and 3-substituted 1,2,3,4-tetrahydroisoquinolines. J.Org.Chem.. 49(22):4107-4111. https://doi.org/10.1021/jo00196a0019.Tran PH, Huynh VH, Hansen PE, Chau DN, Le TN. 2015. An Efficient and Green Synthesis of 1-Indanone and 1-Tetralone via Intramolecular Friedel-Crafts Acylation Reaction. Asian Journal of Organic Chemistry. 4(5):482-486. https://doi.org/10.1002/ajoc.20140227410.Kobayashi S, Komoto I. 2000. Remarkable Effect of Lithium Salts in Friedel?Crafts Acylation of 2-Methoxynaphthalene Catalyzed by Metal Triflates. Tetrahedron. 56(35):6463-6465. https://doi.org/10.1016/s0040-4020(00)00610-411.Cai M, Wang X. 2014. Activity of Imidazolium-Based Ionic Liquids as Catalysts for Friedel-Crafts Acylation of Aromatic Compounds. Asian J. Chem.. 26(18):5981-5984. https://doi.org/10.14233/ajchem.2014.1635412.Tran PH, Hansen PE, Nguyen HT, Le TN. 2015. Erbium trifluoromethanesulfonate catalyzed Friedel?Crafts acylation using aromatic carboxylic acids as acylating agents under monomode-microwave irradiation. Tetrahedron Letters. 56(4):612-618. https://doi.org/10.1016/j.tetlet.2014.12.03813.Wang W, Li G, Wang S, Shi Z, Cao X. 2015. Direct and Short Construction of the ACDE Ring System of Daphenylline. Chem. Asian J.. 10(2):377-382. https://doi.org/10.1002/asia.20140315214.Tang S, Xu Y, He J, He Y, Zheng J, Pan X, She X. 2008. Application of a Domino Friedel?Crafts Acylation/Alkylation Reaction to the Formal Syntheses of (±)-Taiwaniaquinol B and (±)-Dichroanone. Org.Lett.. 10(9):1855-1858. https://doi.org/10.1021/ol800513v15.Wen H, Wang P, Cheng S, Yan T, Cai M. 2015. Synthesis and characterization of novel organosoluble poly(aryl ether ketone)s and poly(aryl ether ketone sulfone)s containing 1,4-naphthylene units. High Performance Polymers. 27(6):705-713. https://doi.org/10.1177/095400831455770716.Deng Q, Qin Z, Yang Y, Song W. 2015. Synthesis, characterization of triphenyltin grafted on SBA-15 mesoporous silica and its catalytic performance for the synthesis of 4-methylacetophenone. Chinese Journal of Chemical Engineering. 23(2):384-388. https://doi.org/10.1016/j.cjche.2013.12.00117.Tran PH, Hansen PE, Hoang HM, Chau DN, Le TN. 2015. Indium triflate in 1-isobutyl-3-methylimidazolium dihydrogen phosphate: an efficient and green catalytic system for Friedel?Crafts acylation. Tetrahedron Letters. 56(17):2187-2192. https://doi.org/10.1016/j.tetlet.2015.03.05118.Skácel J, Budka J, Eigner V, Lhoták P. 2015. Regioselective Friedel?Crafts acylation of calix[4]arenes. Tetrahedron. 71(13):1959-1965. https://doi.org/10.1016/j.tet.2015.02.021阿拉丁相关产品列表产品货号产品名称A108664乙酰氯 ,99%,无色A140638乙酰氯 ,1M in DichloromethaneE1321645-甲基吲哚-2-甲基酸酯 ,99%S106648三氟甲烷磺酸钪 ,98%T398955三氟甲烷磺酸 ,≥99.5%Y113955三氟甲烷磺酸镱水合物 ,99.9% metals basisT104298三氟甲烷磺酸 ,98%A108662乙酰氯 ,AR,98%P1356203-苯氧基苯甲酸 ,≥98.0%I119317吲哚-4-甲醛 ,98%
企业动态
2023.06.26

【阿拉丁】大包装试剂全面升级——高品质、保供应!
企业动态
2023.06.26
白蛋白的应用----阿拉丁试剂
白蛋白简介白蛋白是人体血浆中最主要的蛋白质,对维持血容量和体液平衡有重要作用。它是一种溶于水且遇热凝固的球形单纯蛋白,分子结构于1975年阐明,其为含585个氨基酸残基的单链多肽,分子形状呈椭圆形。在体液pH 7.4的环境中,它表现为负离子,每个分子带有200个以上的负电荷,对缓冲酸碱平衡紊乱有一定作用。白蛋白是血浆中最主要的载体,许多水溶性差的物质可以通过与白蛋白的结合而被运输,包括胆红素、长链脂肪酸(每个白蛋白分子可结合4-6个脂肪酸分子)、胆汁酸盐、前列腺素、类固醇激素、金属离子(如Cu2+、Ni+、Ca2+)、药物(如阿司匹林、青霉素)等。白蛋白的生理作用1.维持血浆胶体渗透压 血浆胶体渗透压是使静脉端组织间液返回血管内的主要动力,而胶体渗透压主要由白蛋白产生。当血浆白蛋白浓度下降时,血浆胶体渗透压下降,可导致血液水分过多的进入组织间液而出现水肿。2.运输功能血浆白蛋白能与体内许多难溶性的小分子有机物和无机离子可逆地结合形成易溶性的复合物,并成为这些物质在血液循环中的运输形式。由此可见白蛋白属于非专一性的运输蛋白,具有重要生理意义,与人体健康密切相关。3.对球蛋白的稳定作用血浆白蛋白含量远比球蛋白多,亲水作用又比球蛋白大,对球蛋白起到一种胶体保护的稳定作用。白蛋白浓度明显下降可使血浆球蛋白失去胶体保护作用,而稳定性下降。血浆球蛋白稳定性下降将引起红细胞沉降率( ESR )增快,容易导致微循环障碍和血栓。4.营养作用白蛋白是人体的重要营养物质。白蛋白在血浆中不断代谢,分解产生氨基酸,可用于合成组织蛋白;也可氧化分解供应能量或转变为其他含氮物质。5.酸碱缓冲作用在体液pH 7.4的环境中,白蛋白为负离子,每个分子带有200个以上负电荷,对酸碱平衡紊乱有一定缓冲作用,其中缓冲酸中毒的作用较强。6.调节血浆中物质的活性 具有活性的激素或药物与白蛋白结合后,可以不表现其活性,而为储存形式。由于这种结合的可逆性和处于动态平衡,白蛋白在调节这些激素和药物的代谢上具有重要意义。7.保护作用白蛋白是具有黏性、胶质性的物质,在人体内遇到重金属离子时,会自动与其结合,由排泄系统排出体外,起到解毒作用。因此,食用含白蛋白丰富的食物,可避免重金属离子吸收。白蛋白对胃壁也有保护作用。白蛋白的检查血浆白蛋白常规在肝功能检查中完成。不同年龄段的白蛋白正常值有所不同,新生儿正常值范围为28-44g/L,14岁后为38-54g/L,成人为35-50g/L,60岁后34-48 g/L,常规选择35-45g/L。1.白蛋白浓度升高 主要见于血液浓缩,如严重脱水和休克、严重烧伤、急性出血、慢性肾上腺皮质功能低下;腹泻呕吐、高热时,急剧失水也可导致血白蛋白浓度升高。2.白蛋白浓度降低见于合成不足、分解代谢增强加或渗出增加,常见于慢性消耗性疾病;毛细血管通透性增高性疾病,如急性肺损伤创伤、手术、危重病;肝脏疾病,如肝硬化、腹水,急性肝坏死,中毒性肝炎;糖尿病;严重肾脏疾病,如肾病综合征、重症肾炎、肾功能不全等。当白蛋白降低至25g/L以下易产生水肿。消化道疾病,不能很好地消化、吸收,缺乏制造蛋白的原料,如胃癌、肠癌、肝癌。人血白蛋白使用的五个误区提高机体免疫力:有人认为注射人血白蛋白可以增强体质、提高免疫力、延缓衰老等,因此主动要求注射白蛋白的健康人群越来越多。然而,参与人体免疫机制形成的是球蛋白, 而不是白蛋白。大剂量输注白蛋白,不仅不能提高免疫力,反而可能引起机体免疫功能下降。这是因为白蛋白制剂中含有某些生物活性物质,如微量内毒素、血管舒缓素、微量α1-酸性糖蛋白等这些物质可能对人体的免疫功能产生“干扰”作用。作为健康人群的营养补充剂:事实上,白蛋白作为氮源合成蛋白质的速度较慢,且白蛋白的半衰期长达16~21天,其输入人体后需要经过一段时间分解为游离的氨基酸后,才能合成机体自身需要的蛋白质,因此,当天输入的白蛋白并不能很快发挥营养作用。而且研究发现,白蛋白所含的必需氨基酸比例十分不均衡,尤其是用于合成其他蛋白质的重要氨基酸之一的色氨酸缺乏,其营养价值低,在费用效益比方面更远低于平衡氨基酸制剂,因此白蛋白并不适宜作为营养补充剂。使用前未进行血清蛋白浓度的检测:临床上使用人血白蛋白的重要参考指标是血清白蛋白浓度,其正常浓度范围是35 ~ 50g/L。在实际的临床实践中,对于血清白蛋白浓度在正常范围的患者,或者没有对血清白蛋白进行浓度检测的患者,却给予输注人血白蛋白,这两类患者均属于无指征用药。通常来说,血清白蛋白浓度低于25g/L才有指征应用人血白蛋白。白蛋白可能会传播乙肝等传染病:虽然从理论上来说不能完全否认这种可能性。但是目前使用的人血白蛋白是在有稳定剂时,经60℃加温灭活病10小时。在这种条件下,HBV、HCV、HIV 等病毒均已丧失传染性,且白蛋白无抗原性,可反复输注。所以,输注白蛋白其实比输注血浆或全血安全很多。当然,导致感染的可能性也不能完全排除。白蛋白安全无副作用,任何人都可以用:药品是一把双刃剑,既有治疗的作用,也有可能导致副作用的发生。白蛋白也一样,有的患者对白蛋白产生过敏反应,因此,对于对白蛋白有过敏反应史者,应禁用白蛋白。另外,对于严重贫血和患有心力衰竭的病人也禁止使用。输注白蛋白还偶尔会发生恶心、呕吐、发烧、头痛、寒颤、荨麻疹、心动过速等不良反应。对于过敏性休克则可能危及生命,应及时救治。阿拉丁提供相关产品列表产品货号产品名称D35569011β-13,14-二氢-15-酮前列腺素F2α ,A275085肾上腺皮质激素(甲吗啡) ,≥95%A107821白蛋白 来源于鸡蛋白 ,冻干粉, ≥90% (agarose gel electrophoresis)A107820白蛋白 来源于鸡蛋白 ,冻干粉, ≥98% (agarose gel electrophoresis)A351789阿司匹林-精氨酸 ,B425226胆红素 ,2mM in DMSOB104211胆红素 ,98%L391392亮氨酰-苯丙氨酰基-异亮氨酰-谷氨酰胺-色氨酸-亮氨酰-赖氨酸三氟乙酸酯 ,P34561311β-前列腺素F2α-d4 ,A solution in ethanolP35572111β-前列腺素E2 ,0.5 wt. % in methyl acetateP35590011β-前列腺素E1 ,P35720411β-前列腺素F1β ,A104912牛血清白蛋白,组分 V ,分子生物学级B265991牛血清白蛋白(BSA) ,新西兰货源, 标准级 pH 7.0, ≥ 96.0%B265993牛血清白蛋白(BSA) ,新西兰源,精制级, ≥ 98.0%B265994牛血清白蛋白(BSA) ,新西兰源, 巯基封闭型, ≥ 98.0%R283924重组人血清白蛋白OsrHSA ,冻干粉,99%R283928重组人血清白蛋白OsrHSA ,细胞培养级,99%R283936重组人血清白蛋白OsrHSA ,细胞培养级(高辛酸钠),99%H304436白蛋白 人 ,96%A116563牛血清白蛋白 ,生物技术级,96%
应用实例
2023.06.21


代谢信号通路----阿拉丁试剂
细胞内稳态是由几个关键代谢途径的协调活动调节。这些过程包括碳水化合物代谢、脂质代谢、谷氨酰胺代谢和核苷酸代谢,它们维持细胞的能量状态,并提供必要的成分以确保细胞正常功能。反过来,这些代谢途径中的许多是由细胞外信号转导调节的。例如,胰岛素通过其同源受体作为主要激素,控制关键的能量功能,如葡萄糖和脂质代谢。碳水化合物代谢碳水化合物代谢包括所有负责碳水化合物(由碳、氢和氧组成的分子(CHO))形成、分解和相互转化的生化过程,以确保向活细胞持续供应能量。 l葡萄糖是代谢的主要底物,它被小肠吸收到血液中,并循环到身体的所有组织中,在这些组织中,葡萄糖的吸收受到胰岛素信号的调节,因此提供了个人日常所需的大部分能量。 l葡萄糖通过糖酵解进一步分解为丙酮酸,生成净三磷酸腺苷(ATP),这是活细胞的重要能量来源。 l它主要通过肝脏和骨骼肌中的糖原生成转化为多糖糖原,在那里它被用作紧急燃料储备,随后通过糖原分解释放为游离葡萄糖。图1 α-淀粉酶免疫组化染色胰岛素信号传导胰岛素是控制关键能量功能的主要激素,包括葡萄糖和脂质代谢。 l胰岛素结合并激活胰岛素受体酪氨酸激酶,主要通过PI3K/AKT和ERK1/2途径启动下游信号转导,随后磷酸化和底物连接的招募,包括IRS蛋白家族。 l通过刺激脂肪和肌肉细胞摄取葡萄糖和减少肝脏中的葡萄糖合成来维持葡萄糖稳态。 l胰腺中的胰岛素分泌受血糖水平的调节。图2脂质代谢脂质代谢脂质代谢包括合成或降解脂质的生物过程,脂质是一类有机化合物,包括脂肪酸或其衍生物,不溶于水,但可溶于有机溶剂。l 脂质是细胞膜等关键细胞结构的基石,在许多细胞信号网络中发挥作用,是用于支持细胞功能的能量丰富的燃料来源。l 被称为甘油三酯的复杂脂类在口腔和小肠消化过程中被称为脂肪酶的酶分解,并通过脂蛋白在血液中运输。l 脂肪酸既是细胞的能量来源,也是能量储存单位。脂肪酸在脂肪酸合成酶的催化下,由乙酰辅酶A和NADPH在细胞质中合成,并代谢成磷脂。磷脂是细胞膜的主要成分,在细胞信号转导通路中发挥作用。图3磷酸乙酰辅酶a羧化酶IHC 图4脂肪酸合酶(绿色)和肌动蛋白(红色)IF谷氨酸代谢氨基酸谷氨酰胺是细胞快速增殖的重要代谢燃料。l谷氨酰胺是血液循环和细胞内库中含量最多的游离氨基酸。l作为底物,以满足不断增长的需求ATP,生物合成前体和还原剂在分裂细胞。l特定的氨基酸转运体允许谷氨酰胺进入细胞,然后在线粒体中转化为谷氨酸,这是TCA循环中间产物α酮戊二酸的前体。l癌细胞通常依赖谷氨酰胺代谢来满足其增加的能量需求。图5核苷酸代谢核苷酸代谢核苷酸代谢是合成和降解核酸、DNA和RNA的基本构件所必需的一系列生化反应。l嘌呤(腺嘌呤和鸟嘌呤)和嘧啶(胞苷、尿苷和胸苷)是核苷酸的两大类。所有核苷酸都由戊糖和磷酸基组成,但嘌呤和嘧啶的氮基大小不同。l三磷酸核苷(ATP, GTP, CTP和UTP)形式的核苷酸作为许多细胞功能所需的化学能的存储,包括氨基酸和蛋白质合成,细胞迁移和分裂,这些能量通过去除磷酸盐而释放。l环核苷酸cGMP和cAMP在许多细胞信号级联中起关键的第二信使作用,这些信号级联被一类称为环核苷酸磷酸二酯酶的酶修饰。图6 CNPase(绿色)和α/β鞘绿色蛋白(Syn205)(红色)IF阿拉丁相关产品列表产品货号产品名称T113170胰蛋白酶抑制剂 ,试剂级A1072023-氨基-1,2,4-三唑 ,96%A111014氯化乙酰胆碱 ,99%A128786苯磺酸氨氯地平 ,98%A349467甲基胺草磷 ,≥98%C129997环孢菌素 ,≥99%C110501β-胡萝卜素 ,≥96%(HPLC)D140542敌敌畏标准溶液 ,analytical standard,1.00mg/ml in methanolE101312反油酸 ,98%E1101287-乙基-10羟基喜树碱 ,98%E110145雌二醇 ,99%,用于细胞培养E126032伊利司莫(STA-4783) ,≥98%F110742反丁烯二酸 ,99.5%F118532呋喃唑酮 ,98%G115746光甘草定 ,分析标准品,≥99%H107675羟基酪醇 ,98%I193491Obeticholic acid ,98%K107145山奈酚 ,97%L107329木犀草素 ,≥98%(HPLC)L168455Luminol 钠盐 ,98%(HPLC)M107060(-)-薄荷醇 ,99%N164488柚皮素 ,97%P106426黄体酮 ,98%T106804可可碱 ,99%C111282(S)-(+)-喜树碱 ,97%T101160他克莫司 ,≥98%(HPLC)F1823652,5-呋喃二甲醇 ,98%I129291布洛芬 ,≥98%(GC)L130208羊毛甾醇 ,>95%N119039N-苯基-N-(4-哌啶基)哌啶酰胺 ,98%R132741DL-去甲变肾上腺素盐酸盐 ,≥95%R134561(+/-)-变肾上腺素盐酸盐 ,≥98% (HPLC)G127944吉西他滨 ,≥99%
应用实例
2023.06.16

阿兹海默症信号通路----阿拉丁试剂
阿兹海默症信号通路图1.阿兹海默症分子与细胞生物学原理阿尔茨海默病是世界上最常见的神经退行性疾病之一。临床表现为细胞外淀粉样斑块和细胞内神经纤维缠结,导致神经元功能障碍和细胞死亡。这种疾病的核心是淀粉样前体蛋白(APP)的差异加工。APP是一种完整的膜蛋白,经过蛋白水解处理。APP最初被α-分泌酶裂解,生成sAPPα和C83羧基端片段。sAPPα的存在与正常的突触信号传递有关,并调节神经元存活和突触可塑性等过程,这些过程有助于学习和记忆等更高阶的大脑功能,以及其他行为。APP也可以被β-分泌酶和γ-分泌酶依次裂解,释放出不同大小的胞外单体,其中最重要的是Aβ40/42。在疾病状态下,APP处理途径之间的不平衡导致神经毒性单体聚集增加,产生Aβ寡聚和斑块形成。致病性Aβ聚集导致离子通道阻塞、钙稳态破坏、线粒体氧化应激、能量代谢受损和葡萄糖调节异常、突触功能改变,最终导致神经元细胞死亡。一些胶质细胞类型,包括星形胶质细胞和小胶质细胞,在淀粉样单体、寡聚物和斑块积聚的背景下,被认为是神经保护和致病的。阿尔茨海默病的另一个特点是存在神经原纤维缠结,它由微管相关蛋白Tau的过度磷酸化形式组成。GSK-3α/β和CDK5是主要负责Tau磷酸化的激酶,尽管其他激酶如PKC, PKA和Erk2也参与其中。Tau蛋白的过度磷酸化导致Tau蛋白从微管分离,随后是微管失稳和Tau蛋白的寡聚,最终导致细胞内的神经纤维缠结。这些缠结的逐渐积累导致神经元凋亡。虽然阿兹海默症给人类的神经系统健康带来巨大的威胁,但目前也只有少数药物得到批准可用于该疾病的治疗。并且这类药物的作用通常只是控制症状,很难改变疾病的进程。通过对大量的病 例及临床结果的研究与总结,研究人员也提出了各种不同的理论来阐述阿兹海默病的发病过程,其中广为接受并开展药物设计与研究的主要有以下几种:β-淀粉样蛋白假说 (Amyloid cascade hypothesis) 、Tau蛋白假说 (Tau hypothesis) 和胆碱能假说 (Cholinergic hypothesis) 等。阿拉丁可为您提供近百种用于阿兹海默症研究治疗的活性化合物。参考文献1. Bossy-Wetzel E, Schwarzenbacher R, Lipton SA (2004) Molecular pathways to neurodegeneration. Nat. Med. 10 Suppl, S2–9.2. Chen JX, Yan SS (2010) Role of mitochondrial amyloid-beta in Alzheimer's disease. J. Alzheimers Dis. 20 Suppl 2, S569–78.3. Claeysen S, Cochet M, Donneger R, Dumuis A, Bockaert J, Giannoni P (2012) Alzheimer culprits: cellular crossroads and interplay. Cell. Signal. 24(9), 1831–40.4. Marcus JN, Schachter J (2011) Targeting post-translational modifications on tau as a therapeutic strategy for Alzheimer's disease. J. Neurogenet. 25(4), 127–33.5. Müller WE, Eckert A, Kurz C, Eckert GP, Leuner K (2010) Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer's disease--therapeutic aspects. Mol. Neurobiol. 41(2-3), 159–71.6. Nizzari M, Thellung S, Corsaro A, Villa V, Pagano A, Porcile C, Russo C, Florio T (2012) Neurodegeneration in Alzheimer disease: role of amyloid precursor protein and presenilin 1 intracellular signaling. J Toxicol 2012, 187297.7. Thinakaran G, Koo EH (2008) Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 283(44), 29615–9.8. Guo, T., Zhang, D., Zeng, Y., Huang, T. Y., Xu, H., & Zhao, Y. (2020). Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Molecular Neurodegeneration 15.9. Lane, C. A., Hardy, J., & Schott, J. M. (2018). Alzheimer’s disease. European Journal of Neurology 25(1), 59–70.阿拉丁相关产品列表 货号 产品名称H352298羟基酪醇3-硫酸钠盐P386209苯基[(噻吩-2-基磺酰基)氨基]乙酸Z302592羟基锡酸锌 ,Sn≥42%A339694皂甙元C303410(氯甲基)二甲基硅基丁烷 ,97%C353869(±)顺-2,5-双(3,4,5-三甲氧基苯基)-1,3-二氧戊环 ,98%D412148DWK-1339 ,H107336石杉碱甲 ,98%I340588茚唑啉L118998白细胞激肽I ,≥98% (HPLC)
企业动态
2023.06.15


Fries重排----阿拉丁试剂
简介Fries重排反应是指在路易斯酸或布朗斯特酸( 如HF、AlCl3, BF3, TiCl4, 或 SnCl4)催化下,以酚酯作为原料重排合成邻位或对位酰基酚的反应[1]。Fries重排反应由德国化学家 Karl Theophil Fries 首先报道,以此以其名字命名,也是重要的人名反应之一。图 1.Fries重排反应Fries重排也可以在没有催化剂的情况下进行,但是需要有紫外光的存在。产物仍然是邻位或者对位羟基芳酮。这种类型的Fries重排称为“光Fries重排”。光Fries重排产率很低,很少用于合成。不够苯环上连有间位定位基时仍然可以进行光Fries重排[1]。硫杂-Fries重排是指在二氯甲烷体系中,氯化铝的催化下,芳基三氟甲磺酸酯重排生成为三氟甲磺酸亚砜基酚的过程[2]。阴离子磷酸Fries重排会生成含有邻位碳-磷键的酚,可将芳基磷酸酯 [ArOP(=O)(OR)2] 重排为邻羟基芳基膦酸酯[o-HO-Ar-P(=O)(OR)2][3]。应用Fries重排目前应用于以下领域:n应用于以离子熔体[1-丁基-3-甲基咪唑鎓氯铝酸盐([BMIm]Cl·xAlCl3)]作为溶剂,以路易斯酸作为催化剂的合成条件下,苯甲酸苯酯重排反应生成邻位-羟基二苯甲酮和对位-羟基二苯甲酮[4]。图2.对羟基二苯甲酮的合成n医药中间体邻羟基苯乙酮和对羟基苯乙酮的合成。邻羟基苯乙酮和对羟基苯乙酮是重要的有机合成原料之一,是医药领域中用途广泛的中间体。而邻羟基苯乙酮和对羟基苯乙酮主要由乙酸苯酯的Fries重排反应合成[5]。n维生素E(α-生育酚)的合成[6]。n在TiCl4的催化下利用Fries重排并发生直接区域选择性酰化反应,合成邻位酰基羟基[2.2]对环戊烷[7]。n农用化学中间体o-羟基苯丁酮和p-羟基苯丁酮的合成。o/p-羟基苯丁酮,为o-羟基苯丁酮和p-羟基苯丁酮的混合物,是一种新型农用杀菌剂。目前常以丁酸苯酯为原料,经过Friesc重排制备o/p-羟基苯丁酮[8]。n在三氟甲磺酸钪的催化下,酰基萘经Fries重排,合成成羟基萘酮[9]。n以3-甲基-2-丁烯酸芳基酯为原料,通过光-Fries重排和碱催化的分子内oxa-Michael加成反应,使用光化学一锅法合成5-、6-和7-取代的苯并二氢吡喃-4-酮[10]。上述综合方案:图3.Oxa Michael加成反应研究进展n关于硫代芳烃在无溶剂、微波介质加热条件下硫杂-Fries重排的相关研究[8]。n光反应液晶聚合物薄膜通过具有轴选择的光-Fries重排,且在暴露于线性偏振紫外光下(LPUV)时表现出光诱导的光学各向异性[1]。n反式和顺式solamin生物合成中的不饱和推定前体muricadienin的合成中,Fries重排是至关重要的步骤[10]。n手性二茂铁基磷酸酯通过阴离子磷酸-Fries重排合成含有非对映异构体的1,2-P,O-磷酸酯,而后其将再进一步转化为对映体纯的磷烷[13]。n关于负载在二氧化硅或其盐Cs2.5H0.5PW12O40 (CsPW)上的杂多酸H3PW12O40(PW)催化芳基酯的液相-Fries重排反应的研究[14]。n阴离子磷酸-Fries已在二茂铁化学的相关研究中的成功应用[15]。nFries重排以2,6-二甲氧基醌为原料,已用于合成抗病毒类黄酮先导化合物[16]。图4. 2,6-二甲氧基醌n杂多酸H3PW12O40作为一种性能高效且环境友好的催化剂,已经应用于乙酸苯酯的Fries重排。图5.乙酸苯酯参考文献:1.Bansal R K. 1996. Synthetic Approaches in Organic Chemistry. Jones & Bartlett Learning.2.Chen X, Tordeux M, Desmurs J, Wakselman C. 2003. Thia-Fries rearrangement of aryl triflinates to trifluoromethanesulfinylphenols. Journal of Fluorine Chemistry. 123(1):51-56. https://doi.org/10.1016/s0022-1139(03)00106-43.Taylor C, Watson A. 2004. The Anionic Phospho-Fries Rearrangement. COC. 8(7):623-636. https://doi.org/10.2174/13852720433707174.Harjani JR, Nara SJ, Salunkhe MM. 2001. Fries rearrangement in ionic melts. Tetrahedron Letters. 42(10):1979-1981. https://doi.org/10.1016/s0040-4039(01)00029-65.Jayat F, Picot MJS, Guisnet M. 1996. Solvent effects in liquid phase Fries rearrangement of phenyl acetate over a HBEA zeolite. Catal Lett. 41(3-4):181-187. https://doi.org/10.1007/bf008114886.Termath AO, Velder J, Stemmler RT, Netscher T, Bonrath W, Schmalz H. 2014. Total Synthesis of (2RS)-?-Tocopherol through Ni-Catalyzed 1,4-Addition to a Chromenone Intermediate. Eur. J. Org.Chem.. 2014(16):3337-3340. https://doi.org/10.1002/ejoc.2014022407.Rozenberg V, Danilova T, Sergeeva E, Vorontsov E, Starikova Z, Lysenko K, Belokon .Y.Eur J. 2000. Org.Chem. 193295.8.Moghaddam FM, Dakamin MG. 2000. Thia-Fries rearrangement of aryl sulfonates in dry media under microwave activation. Tetrahedron Letters. 41(18):3479-3481. https://doi.org/10.1016/s0040-4039(00)00402-09.Kobayashi S, Moriwaki M, Hachiya I. 1995. The catalytic Fries rearrangement of acyloxy naphthalenes using scandium trifluoromethanesulfonate as a catalyst. J. Chem. Soc., Chem. Commun..(15):1527. https://doi.org/10.1039/c3995000152710.Iguchi D, Erra-Balsells R, Bonesi SM. 2014. Expeditious photochemical reaction toward the preparation of substituted chroman-4-ones. Tetrahedron Letters. 55(33):4653-4656. https://doi.org/10.1016/j.tetlet.2014.06.08111.Uraoka H, Kondo M, Kawatsuki N. 2014. Influence of End Groups in Photoinduced Reorientation of Liquid Crystalline Polymer Films Based on Axis-Selective Photo-Fries Rearrangement. Molecular Crystals and Liquid Crystals. 601(1):79-87. https://doi.org/10.1080/15421406.2014.94050812.Adrian J, Stark CBW. 2014. Total Synthesis of Muricadienin, the Putative Key Precursor in the Solamin Biosynthesis. Org.Lett.. 16(22):5886-5889. https://doi.org/10.1021/ol502849y13.Korb M, Lang H. 2014. Planar Chirality from the Chiral Pool: Diastereoselective Anionic Phospho-Fries Rearrangements at Ferrocene. Organometallics. 33(22):6643-6659. https://doi.org/10.1021/om500953c14.Kozhevnikova E. 2004. Fries rearrangement of aryl esters catalysed by heteropoly acid: catalyst regeneration and reuse. Applied Catalysis A: General. 260(1):25-34. https://doi.org/10.1016/j.apcata.2003.10.00815.Korb M, Schaarschmidt D, Lang H. 2014. Anionic Phospho-Fries Rearrangement at Ferrocene: One-Pot Approach to P,O-Substituted Ferrocenes. Organometallics. 33(8):2099-2108. https://doi.org/10.1021/om500282716.Martin-Benlloch X, Elhabiri M, Lanfranchi DA, Davioud-Charvet E. 2014. A Practical and Economical High-Yielding, Six-Step Sequence Synthesis of a Flavone: Application to the Multigram-Scale Synthesis of Ladanein. Org.Process Res. Dev.. 18(5):613-617. https://doi.org/10.1021/op400364217.Kozhevnikova EF, Derouane EG, Kozhevnikov IV. 2002. Heteropoly acid as a novel efficient catalyst for Fries rearrangement. Chem. Commun..(11):1178-1179. https://doi.org/10.1039/b202148j
企业动态
2023.06.13

贴壁细胞培养指南:接种、增殖和收获----阿拉丁试剂
细胞培养是一项艰难的工作。因为这是一个高度技术性的过程,当你从一个来源提取细胞并在另一个条件下去对他们进行操作时,可能会出现很多问题,比如受到外界的污染,或者生长速度非常缓慢。但其中也有许多是我们可以去控制的。在过去的100多年里,贴壁细胞培养实验为现代医学的一些最具革 命性的进步做出了贡献,促进了我们对癌症等疾病的理解,并支撑了安全有效的治疗方法的发展。幸运的是,现在人们对细胞培养的了解比20世纪初体外培养先驱罗斯·哈里森(Ross Harrison)在玻片上分析青蛙组织时多得多。在本指南中,我们分享了一个多世纪来培育活细胞的基本知识,包括如何接种、增殖和收获。低温状态下的细胞接种在购买细胞培养品系时,产品是冷冻保存的,这对长期保持细胞活力是必要的。在这种状态下,你需要解冻并进行细胞接种,这个过程应当是非常谨慎小心的,即便它发生得很快。然而,这一重要的步骤的经常匆忙进行,有时很多细节甚至被忽视,这就存在了细胞被污染的风险,也没有给细胞提供它们所需要的良好的、无菌的开始。以下是进行细胞接种的正确方法:• 准备好所需的所有的器具。你需要一个冷冻的小瓶子、一个烧杯或水浴槽,装满预热的水,和一个装有预平衡介质的烧瓶。如果你打算降低细胞的活跃度并去除DMSO或其他低温保护剂,你还需要一个锥形瓶用于盛装这些介质。• 加热小瓶子。在装有预热过的水的烧杯中轻轻搅动冷冻的小瓶子,直到里面的组分几乎完全融化。然后,用酒精湿巾清洁小瓶,以减少被污染的风险。• 接种细胞。使用移液管,从冷冻液中取出细胞并转移到烧瓶中。然后用移液器反复吸取混匀溶液,以便接种。这样,你的细胞就会被解冻、转移、并准备好进入培养箱(或用于去除DMSO的离心机)。细胞数量随着细胞增殖而扩大当细胞生长到一定密度后,需要对细胞进行分离传代。你需要掌握正确的细胞分离方法。细胞增殖是细胞培养的必要部分,依赖于效率和一致性。如果使用的工作流程效率低下,可能会提高供应和人工成本;如果遵循了前后不一致的工作流程,则可能会给您的细胞带来过度压力,并杀死它们。达到目的的方式取决于你的对于实验容器的选择。市面上有非常多的选择,比如使用i-Quip®容器,可以帮助生命科学家在更小的空间中建立友好的环境,以实现更快的生长,也更多的减少劳动力和污染风险。因为不是所有的容器都具有相同的生长面积,研究人员通常会考虑细胞/cm2的产量来决定他们的容器选择和相关试剂的用量。然后,他们将利用这一技术持续地进行细胞接种实验,使其达到研究人员所需的细胞数量。为了获得最佳生长效果,通过以下操作保持细胞/cm2和mL/cm2的比例稳定来选择容器和试剂:• 使用这个公式来计算您的细胞数量,以容器为单位:[(所需细胞/cm2)×(容器的面积cm2)];• 应用这个公式来计算每个容器所需的试剂量:[(期望mL数/ cm2)×(容器的面积cm2)]。为了达到最佳的细胞生长和气体扩散,大多数情况下每cm2的细胞生长面积将需要0.2 mL到0.5 mL的培养基。收获你的贴壁细胞当细胞在显微镜下呈现为单层时,你就知道它们已经准备好被收获了。实验员们一般会通过化学或物理手段分离细胞。一种方法是通过解离试剂进行化学分离,需要针对细胞类型和应用进行优化,以确保细胞不受试剂的负面影响。相比之下,对于可能不耐受解离试剂的强黏附、敏感细胞,通过细胞刮除器进行物理清除可能是更好的方法。同时还需要考虑下游应用场景,以及如何或是否解离试剂可能影响您的研究。如果您选择使用解离试剂,请使用移液管无菌地遵循以下操作步骤:• 从培养瓶中取出培养基;• 向培养瓶中加入PBS等缓冲溶液,以去除可能干扰解离剂的任何微量血清。轻轻摇动容器,使里面的溶液均匀。浸泡10到15分钟以分离哪些难以分离的细胞系。移去缓冲液;• 以0.02到0.03 mL/cm2的比例添加解离试剂。根据细胞系或解离试剂的特性,在37℃孵育可以促进解离;• 当细胞状态呈现圆形,但还没有开始聚集时,它们就已经准备好了——就好像你在看一个充满了成千上万颗星星的夜空。溶液开始变得浑浊是一个好迹象,表明它们已经准备好被分离了;• 用缓冲液或含血清的培养基稀释解离试剂,通常以1:1的比例稀释。在去除整个溶液并将其转移到试管内之前,上下吸取几次以混匀;• 如果细胞对试剂特别敏感,则建议通过离心法实现细胞的分离。现在你的细胞可以计数了,同时也可以对其细胞密度和活力进行测量了。通过细胞培养达成你的研究贴壁细胞培养可以为你的实验室打开一个实验和下游应用的全新世界——它同样也可以很有趣。但当你操纵活体细胞时,可能会有很多风险,所以正确的操作非常重要。通过遵循这些基本的指示,并努力达成更完善的操作流程,你可以逐步成长为一个细胞培养专家。
应用实例
2023.06.01

DNA损伤和修复----阿拉丁试剂
DNA损伤和修复的机制细胞DNA的损伤与突变和癌症的发生有关。人类细胞中的DNA每天要经历数千到数百万次的破坏事件,这些事件是由外部(外源性)和内部代谢(内源性)过程产生的。细胞基因组的改变会在DNA转录中产生错误,并随后翻译成信号传递和细胞功能所必需的蛋白质。如果突变在有丝分裂之前没有修复,基因组突变也可以传递到子代细胞。一旦细胞失去有效修复受损DNA的能力,就有三种可能的反应(图1)。1. 细胞可能会衰老,即不可逆地休眠。2005年,多个实验室报告说,在体内和体外,癌细胞都可能发生衰老,阻止有丝分裂,并阻止细胞进一步进化。2. 细胞可能发生凋亡。足够的DNA损伤可能触发凋亡信号级联,迫使细胞进入程序性细胞死亡。3. 细胞可能变成恶性的,也就是说,发展出不朽的特征并开始不受控制的分裂。图1所示。导致衰老、细胞凋亡或癌症的细胞DNA损伤和修复的途径为了补偿DNA损伤的程度和类型,细胞已经发展出多种修复过程,包括错配、碱基切除和核苷酸切除修复机制,几乎没有过程冗余。如果发生了压倒性的损伤,而不是消耗能量来有效地修复损伤,细胞可能已经进化到凋亡或衰老。细胞修复的速度取决于细胞类型和细胞年龄等因素。DNA损伤的来源多年来,外源性损伤被认为是DNA突变导致癌症的主要原因。然而,Jackson和Loeb提出,DNA损伤的内源性来源也对导致恶性肿瘤的突变起着重要作用环境和细胞来源都可能导致类似类型的DNA损伤。DNA可以被物理和化学诱变剂攻击。物理诱变剂主要是辐射源,包括来自太阳的紫外线(200-300纳米波长)辐射。紫外线辐射产生共价键,交联相邻的嘧啶(胞嘧啶和胸腺嘧啶)碱基在DNA链。电离辐射(x射线)通过在细胞内产生自由基引发DNA突变,自由基产生活性氧(ROS),并导致双螺旋中的单链和双链断裂。化学诱变剂可以将烷基共价连接到DNA碱基上;能使DNA碱基甲基化或乙基化的氮芥化合物是DNA烷基化剂的例子。致癌物原是化学惰性前体,经代谢转化为高活性致癌物。这些致癌物可以与DNA反应形成DNA加合物,即附着在DNA上的化学实体。苯并[a]芘是一种多芳杂环,本身并不致癌。它在细胞色素P450酶的介导下经历了两次连续的氧化反应,产生苯并[a]芘二醇环氧化物(BPDE),这种致癌代谢物能够形成共价DNA加合物(图2)。图2。苯并[a]芘被P450酶氧化生成具有高度致癌性的苯并[a]芘二烯烃。DNA损伤也可由内源性代谢和生化反应引起,其中一些尚不清楚水解反应可以部分或完全将核苷酸碱基从DNA链上裂解。连接嘌呤碱基(腺嘌呤或鸟嘌呤)和脱氧核糖磷酸链的化学键可以在被称为脱嘌呤的过程中自发断裂。据估计,哺乳动物细胞中每天发生10,000次脱嘌呤事件脱嘧啶(从胸腺嘧啶或胞嘧啶中失去嘧啶基)也会发生,但其速率比脱嘌呤低20至100倍。脱氨发生在细胞内,腺嘌呤、鸟嘌呤和胞嘧啶环的胺基丢失,分别产生次黄嘌呤、黄嘌呤和尿嘧啶。DNA修复酶能够识别和纠正这些不自然的碱基。然而,在随后的DNA复制过程中,未经纠正的尿嘧啶碱基可能被误读为胸腺嘧啶,并产生C→T点突变。DNA甲基化,一种特殊形式的烷基化,发生在细胞内,由于与s -腺苷蛋氨酸(SAM)的反应。SAM是一种含有高活性甲基的细胞内代谢中间体。在哺乳动物细胞中,甲基化发生在距离鸟苷基(G) 5′的胞嘧啶碱基(C)胞嘧啶环的5位,即序列CpG。突变误差的一个重要来源是甲基化的5-甲基胞嘧啶产物的自发脱氨。胺基的损失导致胸腺嘧啶碱基,它不被DNA修复酶检测到作为一个非自然的碱基。由此产生的取代保留在DNA复制中,产生C→T点突变(图3)。图3。胞嘧啶的2期突变导致胸腺嘧啶,产生C→T点突变。正常的代谢过程产生活性氧(ROS),通过氧化修饰碱基。嘌呤和嘧啶碱都容易氧化。最常见的突变是鸟嘌呤氧化为8-oxo-7,8-二氢鸟嘌呤,导致核苷酸8-oxo-脱氧鸟嘌呤sin (8-oxo-dG)。8-oxo-dG能够与脱氧腺苷碱基配对,而不是像预期的那样与脱氧胞苷碱基配对。如果这个错误没有被错配修复酶检测到并纠正,随后复制的DNA将包含C→a点突变。ROS还可能导致脱氧核糖核酸的脱嘌呤、脱嘧啶和单链或双链断裂。在细胞周期S期的DNA复制过程中可能引入其他基因组突变。复制模板DNA的聚合酶有一个小但显著的错误率,并且可能包含基于沃森-克里克配对的与模板DNA不正确的核苷酸。化学改变的核苷酸前体可以被聚合酶合并到生成的DNA中,而不是正常的碱基。此外,当复制含有大量重复核苷酸或重复序列的DNA片段(微卫星区)时,聚合酶容易“口吃”。这种酶的“口吃”是由于链滑移,当模板和复制的DNA链脱离正确的排列时。结果,聚合酶无法插入模板DNA所指示的正确数量的核苷酸,导致子链中的核苷酸过少或过多。DNA可能发生单链和双链裂解。单链断裂可能是由于脱氧核糖基磷酸链的脱氧核糖部分受损造成的。断裂也是ap -核酸内切酶去除磷酸脱氧核糖后碱基切除修复途径的中间步骤。当单链断裂发生时,核苷酸碱基和脱氧核糖主干都从DNA结构中丢失。双链裂解最常发生在细胞经过s期时,因为DNA在展开作为复制模板时更容易断裂。 DNA修复机制虽然细胞能够进化到凋亡或衰老状态,但这些行动是最后的手段。对于每一种类型的DNA损伤,细胞已经进化出一种修复损伤或消除破坏性化合物的特定方法。o6 -甲基鸟嘌呤DNA甲基转移酶(MGMT;DNA烷基转移酶)根据DNA结构从鸟嘌呤中分离甲基和乙基加合物。该反应不是催化(酶促)反应,而是化学计量(化学)反应,每去除一个加合物就消耗一个MGMT分子。经过过表达MGMT的细胞对癌症的抵抗力更强,可能是因为它们能够抵消大量的烷基化损伤。Niture等人最近的一项研究报告称,使用半胱氨酸/谷胱甘肽增强药物和天然抗氧化剂可增加MGMT的表达。DNA聚合酶,如聚合酶-δ,含有校对活性,主要参与复制错误修复。当检测到错误时,这些聚合酶就会停止DNA复制的过程,然后反向从子DNA链中移除核苷酸,直到不正确的核苷酸明显消失,然后重新启动正向复制过程。在Pold1基因的两个拷贝中都有点突变的小鼠表现出DNA聚合酶-δ的校对活性的丧失,并且与具有野生型基因或单拷贝突变的小鼠相比,发生上皮性癌症的几率显著更高。一组被称为错配切除修复(MMR)酶的蛋白质能够纠正DNA聚合酶校对活动未检测到的复制错误。MMR酶从子DNA中去除一个不正确的核苷酸,并使用W-C配对和父DNA链作为正确的模板来修复链这对于微卫星区域复制过程中产生的错误尤其重要,因为DNA聚合酶的校对活动不会检测到这些错误。在较小程度上,MMR酶还纠正了由DNA氧化或烷基化引起的各种碱基对异常。这些突变包括含有o6 -甲基鸟嘌呤和8-氧鸟嘌呤的修饰碱基对,以及致癌物和顺铂加合物。12,13人错配切除修复基因MSH2和MLH1突变与遗传性非息肉病性结直肠癌(HNPCC)综合征相关。碱基切除修复和核苷酸切除修复碱基切除修复(BER)涉及多个酶切除和替换单个受损的核苷酸碱基。主要由BER酶修复的碱基修饰是内源性氧化和水解破坏的碱基修饰。DNA糖基化酶切断核苷酸碱基和核糖之间的键,使DNA的核糖磷酸链完整,但产生无嘌呤或无嘧啶(AP)位点。8-Oxoguanine DNA糖基化酶I (Ogg1)去除7,8-二氢- 8-Oxoguanine (8-oxoG),这是活性氧产生的碱基突变之一。人类OGG1基因的多态性与肺癌和前列腺癌等各种癌症的风险相关。尿嘧啶DNA糖基化酶,另一种BER酶,切除胞嘧啶脱氨的产物尿嘧啶,从而防止随后的C→T点突变n -甲基嘌呤DNA糖基化酶(MPG)能够去除多种修饰的嘌呤碱基。DNA中由BER酶作用产生的AP位点,以及由脱嘧啶和脱嘌呤作用产生的AP位点,可通过AP-核酸内切酶1 (APE1)的作用进行修复。APE1将磷酸二酯链5′裂解到AP位点。DNA链含有一个3 ' -羟基和一个5 ' -碱性脱氧核糖磷酸。DNA聚合酶β (Polβ)根据相应的W-C配对插入正确的核苷酸,并通过其相关的ap裂解酶活性去除脱氧核糖磷酸。x射线修复交叉互补基团1 (XRCC1)的存在是与DNA连接酶III (LIG3)形成异二聚体所必需的。XRCC1作为一个支架蛋白,为Polβ提供一个非活性的结合位点,并在修复位点将Polβ和LIG3酶结合在一起Poly(adp -核糖)聚合酶(PARP-1)与XRCC1和Polβ相互作用,是BER途径的必要组成部分。18,19修复的最后一步由LIG3完成,它将替换核苷酸的脱氧核糖连接到脱氧核糖基磷酸主干。这一途径被命名为“短补丁误码率”。另一种被称为“长补丁误码率”的途径用最少2个核苷酸的长度替换一条核苷酸链。据报道,修复长度为10到12个核苷酸。21,22 Longpatch BER需要增殖细胞核抗原(PCNA)的存在,它作为重组酶的支架蛋白其他DNA聚合酶,可能是Polδ和Polε, 24用于产生寡核苷酸瓣。现有的核苷酸序列被皮瓣内切酶-1 (FEN1)去除。然后寡核苷酸被DNA连接酶I (LIG1)连接到DNA上,封闭断裂并完成修复用于确定短补丁与长补丁误码率路径选择的过程仍在研究中(图4)。图4。短补丁和长补丁误码率路径示意图。虽然误码率可能通过长补丁途径取代多个核苷酸,但短补丁和长补丁误码率的起始事件都是对单个核苷酸的损伤,从而对DNA双螺旋结构的影响最小。核苷酸切除修复(NER)修复至少含有2个碱基的核苷酸链的损伤,并造成DNA的结构扭曲。NER的作用是修复单链断裂以及外源的一系列损伤,如笨重的DNA加合物和紫外线辐射。同样的途径也可以用来修复氧化应激造成的损伤在哺乳动物细胞中,有超过20种蛋白质参与NER通路,包括XPA、XPC-hHR23B、复制蛋白A (RPA)、转录因子TFIIH、XPB和XPD DNA解旋酶、ERCC1- xpf和XPG、Polδ、Polε、PCNA和复制因子c。切除修复交叉互补(ERCC1)基因的过表达与非小细胞肺癌细胞对顺铂的耐药有关29,并对应于增强的DNA修复能力30全球基因组NER (GGR)修复整个基因组的损伤,而一种称为转录偶联修复(TCR)的特定NER通路在活性RNA聚合酶转录期间修复基因。双股断裂的修复DNA中的双链断裂会导致基因组序列的丢失和重排。这些断裂可以通过非同源端连接(NHEJ)或同源重组(HR)进行修复,也称为重组修复或模板辅助修复。当细胞处于S/G2晚期且模板最近被复制时,HR通路被激活。这种机制需要存在一个相同或几乎相同的序列,通过着丝粒连接到受损的DNA区域,作为修复模板。由该机制修复的双链断裂通常是由复制机制试图通过单链断裂或未修复的病变进行合成引起的,导致复制叉的崩溃。当姐妹染色单体不能用作HR模板时,非同源末端连接(NHEJ)用于细胞周期的其他点。当这些断裂发生时,细胞还没有复制包含断裂的DNA区域,因此与HR途径不同,没有相应的模板链可用。在NHEJ中,Ku异二聚体蛋白在没有可用模板的情况下定位断裂DNA链的两端进行修复,在此过程中丢失序列信息。多个酶参与了再连接过程,包括DNA连接酶IV, XRCC4和DNA依赖蛋白激酶(DNA- pk)。32,33 NHEJ具有固有的诱变性,因为它依赖于两个待连接DNA片段的单链尾部之间的偶然配对,称为微同源性(图5)。在高等真核生物中,NHEJ修复需要DNA- pk,无论是通过主要机制还是通过替代备份机制(D-NHEJ)。图5。NHEJ修复DNA双链断裂的一般机制。未来的应用虽然DNA损伤是癌细胞发育和进化的关键因素,但持续的损伤被用作癌症临床治疗的一部分,迫使恶性细胞凋亡或衰老。许多化疗药物,如博莱霉素、丝裂霉素和顺铂,都是有效的,因为它们会进一步破坏癌细胞的DNA,而癌细胞的复制速度比周围组织更快。细胞DNA修复机制是一把双刃剑;通过减少可能导致癌症的突变,这些过程努力保持基因组的完整性,但在恶性细胞中,同样的机制允许这些细胞在额外的DNA损伤中幸存下来,并继续不受控制地生长。为了阻断癌细胞内的这种生存机制,目前正在进行临床试验,使用抑制剂抑制特定的DNA修复酶,包括MGMT、PARP和DNA- pk。35-38
应用实例
2023.05.30
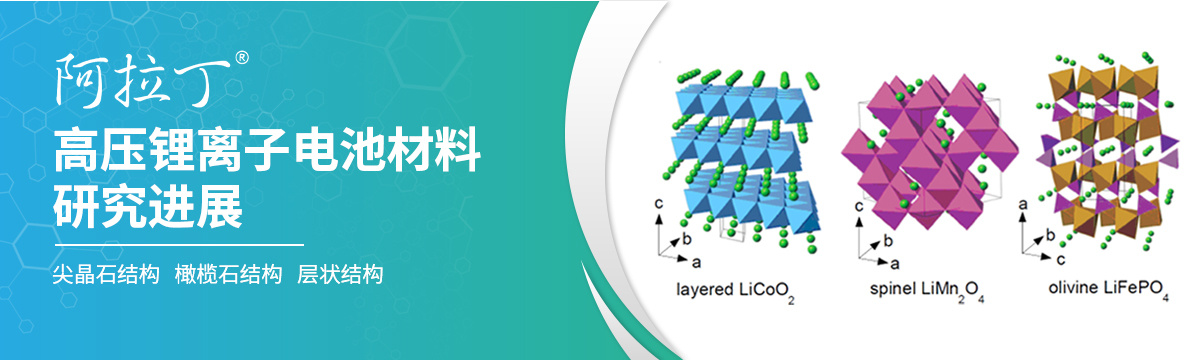
高压锂离子电池材料研究进展
引言锂离子电池(LIBs)由于其高能量密度、高库仑效率、低自放电特性以及不同电极设计可获得的一系列化学势而被广泛应用于广泛的应用领域。随着人们经济,生活方式的飞速发展,锂离子电池的应用领域被进一步拓展,这也对其性能提出了新要求,锂离子电池中关键的参数有很多,电池的能量密度和电池容量就是其中一部分。通过提高工作电压或电荷存储容量都可以提高锂离子电池的能量密度。但由于目前最常见的石墨负极的工作电压已经接近Li/Li+电位。因此提高电池电压的唯一选择是提高正极的工作电压。目前,有三种正极结构(层状、尖晶石和橄榄石)提供的工作电压 (~ 4.3 V vs Li/Li+)较高[1,2],正在受到广泛关注。本文介绍了层状、尖晶石和橄榄石结构三种正极材料的合成以及研究进展,为相关研究提供思路。图1 三种不同类型正极材料的晶体结构表1:主流商用正极材料的电化学特性对比[3]尖晶石结构尖晶石正极材料LiMn1.5Ni0.5O4被认为是一种先进的锂离子电池材料,因其高工作电压(约4.8 V)而受到主要研究关注(图1)。由于LiyNi1−yO(杂质)作为第二相存在,影响了电化学行为,因此很难合成具有化学计量性的纯Li1−x[Ni0.5Mn1.5]O4尖晶石[4,5]。LiMn1.5Ni0.5O4具有两种不同的晶体结构:(i)可化学计量的有序结构,仅由Mn4+组成,和 (ii)无法化学计量的无序结构,由Mn3+和Mn4+组成,面心立方结构(Fd3m)。有序相(LiMn1.5Ni0.5O4)为原始简单立方结构(P4332),其中Ni、Mn和Li原子分别占据4a、12d和8c位,而O原子位于8c和24e位。非化学结构(LiNi0.5Mn1.5O4-δ)为面心立方结构(Fd3m),其中Ni和Mn原子随机分布在16d位,而Li和O原子分别占据8a和32e位。在这两种晶体结构制备过程中,退火温度都很关键[6]。尽管LiMn1.5Ni0.5O4具有许多优点,但在高速率应用中仍然遇到了许多障碍。Jafta等人研究了微波辐射对LiMn1.5Ni0.5O4尖晶石中Mn3+浓度和电化学性能的影响[6]。结果表明,微波能够调节尖晶石中的Mn3+含量,以增强电化学性能(高容量,高容量保持,优异的速率能力和快速的Li+插入/提取动力学)。这一发现有望彻底改变微波辐照提高LiMn1.5Ni0.5O4尖晶石性能的应用,特别是在高速率应用中。图2 微波处理前后LiMn1.5Ni0.5O4形貌及放电性能对比图橄榄石结构橄榄石结构正极材料因其具有优越的安全性、大可逆性、高优化潜力、宽能量密度和高功率应用能力受到了研究者们的广泛关注。在橄榄石结构中,一个MO6八面体与两个LiO6八面体,一个PO4四面体边相连[7]。晶体内离子扩散发生在1D通路(010) [8],该方向由于MO6八面体和LiO6四面体聚阴离子分离,阻碍了离子的快速插入和提取。LiMPO4的有序橄榄石结构是由铁(Fe)、锰(Mn)、钴(Co)、镍(Ni)等过渡d金属获得的[8,9]。橄榄石结构目前存在的技术问题主要集中在循环稳定性和倍率性能两个方面[7,10,11]。这些缺点可以通过减小粒径[7,9,10,12,13]、掺杂其他d元素[7,8]和碳涂层[13-19]来改善。除此之外,表面改性技术还可以避免正极与有机电解质的直接接触,从而防止正极的降解,因为正极可以与电解质分解的产物HF发生反应[20,21]。层状结构由于能量密度高,成本低,环境友好,富镍层状过渡金属氧化物被认为是构建下一代锂离子电池以满足电动汽车需求的极有前途的正极候选材料。但在速率性能、结构稳定性和安全性等方面仍存在一些问题,阻碍了该技术的实际应用。直到现在,它仍然在锂离子电池市场上扮演着重要的角色。由于这些优点,进一步提高LiCoO2的充电截止电压以保证更高的能量密度是不可阻挡的发展趋势。然而,使用过高的充电截止电压可能会引起很多负面影响,特别是循环容量的快速衰减。这主要是由于循环过程中在高压下晶体结构的快速破坏和界面副反应的加剧造成的。因此,如何保持LiCoO2稳定的晶体结构,同时保证在高压下具有优异的长周期性能,是LiCoO2应用中的一个热点研究问题。Guo等人开发了一种一步综合共改性方法,以实现LiCoO2在3.0-4.5 V范围内的长循环寿命。在这项研究中,磷酸表面层抑制了电解液副反应和Co溶解,Mn的掺杂提高了LiCoO2的结构稳定性。结果表明,当截止电压为4.5 V时,经过700次循环后,改性LiCoO2的容量保持率高达83.7%[22]。图3:LiCoO2材料改性后的电化学性能图噻吩衍生物(THs)作为一种新型功能添加剂被用于提高高压LiCoO2的循环性能。Liu等人的研究表明,2,20-bithiophene (2TH)和2,20:50,20-terthiophene (3TH)可以在电解质溶剂分解之前进行电化学聚合,形成导电聚合物保护层在正极表面镀上一层薄膜,阻止电解液在高压下的严重分解,从而提高了高LiCoO2阴极的循环稳定性[23]。图4 不含添加剂的液态电解质中高电位阴极表面的示意图模型结果显示,在4.4 V的高截止电压下循环100次后,基底电解液中放电容量保持率为50%,而在含有0.1 wt% 3TH的电解液中循环的LiCoO2阴极在0.25 C的速率下放电容量保持率为84.8%。这项工作也展现了噻吩衍生物作为功能性添加剂在高压锂离子电池中的应用具有相当大的潜力。图5 Li/LiCoO2电池在1 molL-1 LiPF6/EC+DMC (3/7,v/v)无添加剂电解质溶液中的循环性能图(流密度为0.25 C,截止电压3.0-4.4 V)参考文献[1] Julien C M, Mauger A, Zaghib K, et al. Comparative issues of cathode materials for Li-ion batteries[J]. Inorganics, 2014, 2(1): 132-154. https://doi.org/10.3390/inorganics2010132[2] Bruce P G. Solid-state chemistry of lithium power sources[J]. Chemical Communications, 1997 (19): 1817-1824.https://pubs.rsc.org/en/content/articlehtml/1997/cc/a608551b[3] Tolganbek N, Yerkinbekova Y, Kalybekkyzy S, et al. Current state of high voltage olivine structured LiMPO4 cathode materials for energy storage applications: A review[J]. Journal of Alloys and Compounds, 2021, 882: 160774. https://doi.org/10.1016/j.jallcom.2021.160774[4] Liu J, Manthiram A. Understanding the improved electrochemical performances of Fe-substituted 5 V spinel cathode LiMn1.5Ni0.5O4[J]. The Journal of Physical Chemistry C, 2009, 113(33): 15073-15079. https://doi.org/10.1021/jp904276t[5] Liu D, Han J, Goodenough J B. Structure, morphology, and cathode performance of Li1−x [Ni0.5Mn1.5] O4 prepared by coprecipitation with oxalic acid[J]. Journal of Power Sources, 2010, 195(9): 2918-2923. https://doi.org/10.1016/j.jpowsour.2009.11.024[6] Jafta C J, Mathe M K, Manyala N, et al. Microwave-assisted synthesis of high-voltage nanostructured LiMn1.5Ni0.5O4 spinel: tuning the Mn3+ content and electrochemical performance[J]. ACS applied materials & interfaces, 2013, 5(15): 7592-7598. https://doi.org/10.1021/am401894t[7] Yamada A, Hosoya M, Chung S C, et al. Olivine-type cathodes: Achievements and problems[J]. Journal of Power Sources, 2003, 119: 232-238.https://doi.org/10.1016/S0378-7753(03)00239-8[8] Sgroi M F, Lazzaroni R, Beljonne D, et al. Doping LiMnPO4 with cobalt and nickel: a first principle study[J]. Batteries, 2017, 3(2): 11.https://doi.org/10.3390/batteries3020011[9] Zaghib K, Guerfi A, Hovington P, et al. Review and analysis of nanostructured olivine-based lithium recheargeable batteries: Status and trends[J]. Journal of Power Sources, 2013, 232: 357-369.https://doi.org/10.1016/j.jpowsour.2012.12.095[10] J.B. Goodenough, Cathode materials: a personal perspective, J. Power Sources 174 (2007) 996–1000, https://doi.org/10.1016/j.jpowsour.2007.06.217[11] M.M. Doeff, J.D. Wilcox, R. Kostecki, G. Lau, Optimization of carbon coatings on LiFePO4, J. Power Sources 163 (2006) 180–184, https://doi.org/10.1016/j. jpowsour.2005.11.075[12] Zhang H, Gong Y, Li J, et al. Selecting substituent elements for LiMnPO4 cathode materials combined with density functional theory (DFT) calculations and experiments[J]. Journal of Alloys and Compounds, 2019, 793: 360-368. https://doi.org/10.1016/j.jallcom.2019.04.191[13] J. Li, W. Yao, S. Martin, D. Vaknin, Lithium-ion conductivity in single crystal LiFePO4, Solid State Ion. 179 (2008) 2016–2019, https://doi.org/10.1016/j.ssi. 2008.06.028[14] H.-C. Dinh, I.-H. Yeo, W. Il Cho, S. Mho, Characteristics of conducting polymer- coated nanosized LiFePO4 cathode in the Li+ batteries, ECS Trans. 28 (2019) 167–175, https://doi.org/10.1149/1.3490696[15] S.L. Bewlay, K. Konstantinov, G.X. Wang, S.X. Dou, H.K. Liu, Conductivity improvements to spray-produced LiFePO4 by addition of a carbon source, Mater. Lett. 58 (2004) 1788–1791, https://doi.org/10.1016/j.matlet.2003.11.008[16] M. Safari, C. Delacourt, Modeling of a commercial graphite/LiFePO4 cell, J. Electrochem. Soc. 158 (2011) A562, https://doi.org/10.1149/1.3567007 [17] Y. Hu, M.M. Doeff, R. Kostecki, R. Fiñones, Electrochemical performance of sol- gel synthesized LiFePO4 in lithium batteries, J. Electrochem. Soc. 151 (2004) A1279, https://doi.org/10.1149/1.1768546 [18] V. Srinivasan, J. Newman, Existence of path-dependence in the LiFePO4 electrode, Electrochem. Solid-State Lett. 9 (2006) A110, https://doi.org/10.1149/1.2159299 [19] C. Gong, Z. Xue, S. Wen, Y. Ye, X. Xie, Advanced carbon materials/olivine LiFePO4 composites cathode for lithium-ion batteries, J. Power Sources 318 (2016) 93–112, https://doi.org/10.1016/j.jpowsour.2016.04.008[20] N. Laszczynski, A. Birrozzi, K. Maranski, M. Copley, M.E. Schuster, S. Passerini, Effect of coatings on the green electrode processing and cycling behaviour of LiCoPO4, J. Mater. Chem. A 4 (2016) 17121–17128, https://doi.org/10.1039/ c6ta05262b[21] S.M. Oh, S.T. Myung, Y.K. Sun, Olivine LiCoPO4-carbon composite showing high rechargeable capacity, J. Mater. Chem. 22 (2012) 14932–14937, https://doi.org/ 10.1039/c2jm31933k[22] Gu R, Qian R, Lyu Y, et al. One-step integrated comodification to improve the electrochemical performances of high-voltage LiCoO2 for lithium-ion batteries[J]. ACS Sustainable Chemistry & Engineering, 2020, 8(25): 9346-9355. https://doi.org/10.1021/acssuschemeng.0c01491[23] Xia L, Xia Y, Liu Z. Thiophene derivatives as novel functional additives for high-voltage LiCoO2 operations in lithium-ion batteries[J]. Electrochimica Acta, 2015, 151: 429-436. https://doi.org/10.1016/j.electacta.2014.11.062
应用实例
2023.05.29

【阿拉丁】荣获2022年“上海市专利工作示范企业”称号!
作为一家专注于高端化学、生命科学、分析科学和材料科学领域的上市企业,阿拉丁一直高度重视知识产权保护和管理。公司在试剂研发过程中坚持技术创新,积极运用专利保护手段,全面提升自身的市场竞争力。近日,阿拉丁荣获“上海市专利工作示范企业”的称号,这既是对公司多年来专利保护和管理工作的肯定,也是对公司未来发展的鞭策。众所皆知,知识产权和专利对于企业来说具有至关重要的意义。在当今激烈竞争的市场环境下,要想提高核心竞争力,企业必须依靠自身的技术创新和专利保护。为此,阿拉丁专门设立了专利管理办公室,由专业人员负责管理。该办公室制定了一系列专利管理规章制度,以保障专利等技术信息的机密性,并加强信息的收集、整理、保存和应用。我们的专利管理办公室与科研团队密切合作,及时收集科研成果,并对相关专利进行申请和保护。在实践中,阿拉丁积极利用专利技术进行产品研发和生产,不断提高产品质量和技术含量,通过将自身的专利优势转化为市场竞争力,促进企业的可持续发展。我们还会不断完善专利布局,把握市场动态,积极进行技术创新和研发投入,以满足客户需求和市场变化,保持企业在行业中的领先地位。阿拉丁自2009年成立以来,一直致力于科研试剂的开发和生产。公司凭借着其国内科研试剂品种最为齐全供应商之一的美誉,成为了业内的佼佼者。目前,公司拥有超过5.7万种常备库存产品、36万个SKU数量和280万瓶库存商品数。近三年来,公司新增入库的科研试剂品种超过1.1万种,展示了阿拉丁在科研试剂领域的雄厚实力。阿拉丁在技术与人才优势方面表现出色。我们拥有一支高素质的研发团队,其中包括多名博士、硕士等高级别人才,专业覆盖化学、生物、医学、材料等多个领域。阿拉丁掌握了113项专利,其中包括11项发明专利。此外,公司还掌握了超过3,200种分析方法,主导了46项行业标准的起草,参与了15项行业标准的验证,并完成了超过4.3万项企业产品标准的制订。同时,阿拉丁积极引进国内外高端科研设备和先进技术,加强自主创新能力。这些技术和人才的优势为公司在市场竞争中提供了坚实的支撑。我们将继续坚持技术创新和知识产权保护,不断提升核心竞争力,为广大客户提供更加优质的科研试剂产品和服务。此次荣誉的颁发是对阿拉丁多年来在知识产权保护方面不懈努力的认可,我们将以此为动力,不断提高技术水平和专利创新能力,为行业和社会做出更多贡献。未来,阿拉丁将继续深入推进知识产权保护和管理工作,加强专利申请和布局,拓展国内外专利布局,助力企业持续发展。同时,公司也将加强与国内外知名高校和科研机构的合作,共同开展前沿科技研究,推动科技成果转化,为社会经济发展做出更大贡献。
企业动态
2023.05.29

如何提高锂离子电池中电解液的安全性?----阿拉丁试剂
如何提高锂离子电池中电解液的安全性?引言锂离子电池作为一种流行的储能设备,正被广泛应用于便携式电子设备、电动汽车、大型储能电站等动力领域[1-3]。随着能源结构的转变和大型电气设备的更新换代,锂离子电池无疑给人们的生活带来了巨大的变化和便利,但与此同时,其安全性问题始终存在。电解液作为锂离子电池中最易燃的成分,一直被认为与其安全性密切相关。电解液中主要易燃的成分是有机碳酸盐溶剂,仅靠提高锂盐的热稳定性是不够的。最直接的方法是在溶剂中加入阻燃剂或彻底放弃易燃溶剂。考虑到整个电解液体系的复杂性,较大的变化可能导致完整的电化学性能失效,初步研究主要集中在使用少量的阻燃添加剂。它们通常可以增加电解质的闪点,使其不那么易燃。本文讨论了电解液中各类阻燃剂的特点,作用机理以及研究进展,旨在为相关研究者提供新型电解液的设计思路。磷阻燃添加剂有机磷阻燃添加剂因其种类丰富、毒性低、物理相容性好、成本低等特点,在一开始就得到了广泛的研究[4]。用于锂离子电池电解质的常见磷阻燃剂包括磷酸三甲酯(TMP) [5]、磷酸三乙基(TEP) [6]、磷酸三丁酯(TBP) [7]、甲基膦二甲基酯(DMMP) [8, 9]、二乙基磷酸二乙酯(DEEP) [10]、磷酸三苯酯(TPP)[11]和4-异丙基磷酸苯基二苯酯(IPPP)[12](图1)。图1:常见的几种磷阻燃添加剂的结构总的来说,这些初步研究的磷阻燃剂的阻燃性能是值得肯定的,其较低的价格也是实际应用中一个很大的优势。但在有限用量的前提下,还是较难对整体电解质造成影响。它们的高粘度有导致电化学性能衰减的风险。磷阻燃添加剂的另一个常见问题是其电化学窗口不够宽,阳极表面经常会发生还原性分解反应,造成阻抗增加和容量退化[13]。有必要对这些磷酸盐进行进一步改性,以进一步提高阻燃性能,减少用量。氟阻燃添加剂由于具备高闪点和良好的热稳定性,卤素阻燃添加剂也常用于锂离子电池电解质。锂离子电池中使用的含卤素阻燃剂主要是有机氟化物(图2)。图2:常见的几种氟阻燃添加剂的结构与有机磷添加剂不同,有机氟添加剂的粘度较低,溶解度高,低温性能表现好[14]。因为C-F键的键能(105.4 kcal mol−1)比C-H键的键能(98.8 kcal mol−1)大,这就意味着破坏C-F键需要的能量越大,即有机氟添加剂具备更好的热稳定性(图3)。此外,通过氟原子取代氢原子,可以减少H·自由基,使得材料的易燃性大大降低[15]。因此,有机氟化物被认为是构建不可燃溶剂的最佳选择之一。Arai等以三氟碳酸丙烯(TFPC)为助溶剂合成了碳酸氯乙烯(ClEC) [16]。与PC/TFPC和EC/TFPC电解质相比,含有1 mol L-1 LiPF6的ClEC/TFPC二元电解质在石墨负极和Li1+xMn2O4负极材料上均表现出良好的循环性能。图3:从键能和LUMO两方面阐释氟阻燃剂的不易燃成膜性能对于高压锂离子电池,Xia等[17]报道了一种使用1,1,1,3,3,3-六氟异丙基甲基醚的不易燃电解质(HFPM)作为共溶剂,在4.9 V的高截止电压下,循环200次后仍能保持82%的容量。因此,与磷酸盐阻燃剂相比,氟化阻燃添加剂在维持电化学性能方面具有优势。这是因为这些有机氟添加剂的最低未占据分子轨道(LUMO)比电解质溶剂低[18]。在特有的氟原子电子效应影响下,有机氟添加剂可以提高电解液溶剂的还原电位,在阳极上形成更稳定的SEI膜(图3)。氟阻燃剂通常表现出良好的电化学性能和阻燃效果,其低粘度有助于降低溶剂分子之间的吸引力,提高导电性。此外,氟元素可以改善SEI膜的组成和形貌,可以缓解使用高浓度电解质添加剂引起的容量衰减。但在大剂量的情况下,LiPF6与氟化阻燃剂的相容性普遍较差。因此,有必要探索合适的锂盐或对氟化阻燃剂进行改性,以提高其与LiPF6基电解质的兼容性。此外,目前氟化阻燃剂的成本仍然很高,需要特殊的设备和严格的制备工艺。离子液体阻燃添加剂离子液体作为一种不挥发、不易燃、无污染的液体,具有较宽的电化学窗口,近年来被合成并报道为电解质添加剂[19]。部分研究的锂离子电池电解质离子液体的化学结构如图4所示。图4:常见的几种离子液体阻燃添加剂的结构离子液体通常是指室温下完全由阴离子和阳离子组成的液态盐。因此,离子液体电解质有望取代传统的有机电解质,提高锂离子电池的安全性。良好的电化学性能的基础是保证合适的电导率和合适的电化学窗口。但是,离子液体的使用可能会受到限制,因为普通离子液体会在石墨阳极中分解,从而影响SEI膜的热稳定性。以具有良好的电化学性能和热稳定性的三氟甲基磺酰亚胺(TFSI)为基础,对其作为阴离子进行了深入研究。Guerfi等使用由1-乙基-3-甲基咪唑(EMI)和TFSI组成的离子液体与包括EC和DEC在内的商业电解质混合。得到的混合电解质具有与普通液体电解质相当的电化学性能[20]。当离子液体添加量为40%时,混合电解质不易燃(图5)。图5:不同ECDEC-VC混合物与EMI-TFSI电解质的可燃性试验Ishikawa报道了一种含有双氟磺酰亚胺(FSI)阴离子和EMI阳离子以及Nmethyl-的纯离子液体对n-丙基吡咯吡啶(P13)与石墨的相容性进行了详细的分析,对于半电池的测试结果,石墨阳极的可逆容量可以达到360 mAh g-1 [21]。综上所述,离子液体作为一种新型的安全电解质,具备较高的热稳定性和低挥发性,具有良好的发展前景。但离子液体普遍存在的问题是高粘度导致室温下电导率低,且对纯度要求高,限制了合成工艺。另一个非常重要的方面是离子液体与传统电解质相比成本较高,这在很大程度上限制了目前的实际应用。因此,绝大多数的相关研究仍然是离子液体与传统有机电解质的结合。复合阻燃添加剂多年来,关于电解液阻燃添加剂的研究从未停止过,锂离子电池的阻燃添加剂种类繁多,但很少有添加剂能在少量添加的情况下显著提高阻燃效率的同时保证电化学性能。单一的阻燃剂往往需要较大的添加量,添加剂的溶解度和电解液的兼容性都非常有限。因此,有必要通过综合各种类型的阻燃剂的优点和缺点来配置更适合锂离子电池的添加剂[22]。复合添加剂的引入有利于减少添加剂的用量,提高阻燃效率。并且几种阻燃元素可以起到协同作用[23],甚至在提高热稳定性的同时提高循环性能。目前研究的复合阻燃电解质添加剂主要有磷-氮化合物[24]和磷-氟化合物[23]。氟化磷酸盐是典型的F-P复合添加剂。烷基磷酸盐的氟化是降低粘度和氟化程度最有效的方法之一,氟化基团的位置和类型对阻燃剂和电化学性能有显著不同的影响。Shiga等发现,通过烷基氟化,TFEP及其与带电电极的混合物的热稳定性可以比TMP等未氟化的磷酸盐有所提高[25]。Zhu等使用二乙基膦酸酯、二甲基甲酰胺、三氟乙醇和三甲胺进行合成TFEP化合物作为电解质阻燃添加剂。普通碳酸盐电解质在TFEP用量为20%时可完全不燃[26]。参考文献[1] Armand M, Tarascon J M. Building better batteries[J]. nature, 2008, 451(7179): 652-657.https://www.nature.com/articles/451652a[2] Goodenough J B, Kim Y. Challenges for rechargeable Li batteries[J]. Chemistry of materials, 2010, 22(3): 587-603.https://doi.org/10.1021/cm901452z[3] Nitta N, Wu F, Lee J T, et al. Li-ion battery materials: present and future[J]. Materials today, 2015, 18(5): 252-264.https://doi.org/10.1016/j.mattod.2014.10.040[4] Green J. A review of phosphorus-containing flame retardants[J]. Journal of fire sciences, 1992, 10(6): 470-487.https://doi.org/10.1177/073490419201000602[5] Yao X L, Xie S, Chen C H, et al. Comparative study of trimethyl phosphite and trimethyl phosphate as electrolyte additives in lithium-ion batteries[J]. Journal of power sources, 2005, 144(1): 170-175.https://doi.org/10.1016/j.jpowsour.2004.11.042[6] Wang X, Yasukawa E, Kasuya S. Nonflammable trimethyl phosphate solvent-containing electrolytes for lithium-ion batteries: I. Fundamental properties[J]. Journal of The Electrochemical Society, 2001, 148(10): A1058.https://iopscience.iop.org/article/10.1149/1.1397773/meta[7] Hyung Y E, Vissers D R, Amine K. Flame-retardant additives for lithium-ion batteries[J]. Journal of power sources, 2003, 119: 383-387. https://doi.org/10.1016/S0378-7753(03)00225-8[8] Xiang H F, Xu H Y, Wang Z Z, et al. Dimethyl methylphosphonate (DMMP) as an efficient flame retardant additive for the lithium-ion battery electrolytes[J]. Journal of Power Sources, 2007, 173(1): 562-564.https://doi.org/10.1016/j.jpowsour.2007.05.001[9] Zeng Z, Wu B, Xiao L, et al. Safer lithium-ion batteries based on nonflammable electrolyte[J]. Journal of Power Sources, 2015, 279: 6-12.https://doi.org/10.1016/j.jpowsour.2014.12.150[10] Feng J, Ma P, Yang H, et al. Understanding the interactions of phosphonate-based flame-retarding additives with graphitic anode for lithium-ion batteries[J]. Electrochimica Acta, 2013, 114: 688-692.https://doi.org/10.1016/j.electacta.2013.10.104[11] Högström K C, Lundgren H, Wilken S, et al. Impact of the flame retardant additive triphenyl phosphate (TPP) on the performance of graphite/LiFePO4 cells in high power applications[J]. Journal of Power Sources, 2014, 256: 430-439.https://doi.org/10.1016/j.jpowsour.2014.01.022[12] Wang Q, Sun J, Yao X, et al. 4-Isopropyl phenyl diphenyl phosphate as flame-retardant additive for lithium-ion battery electrolyte[J]. Electrochemical and Solid-State Letters, 2005, 8(9): A467.https://iopscience.iop.org/article/10.1149/1.1993389/meta[13] Xu K, Ding M S, Zhang S, et al. An attempt to formulate nonflammable lithium-ion electrolytes with alkyl phosphates and phosphazenes[J]. Journal of the Electrochemical Society, 2002, 149(5): A622.https://iopscience.iop.org/article/10.1149/1.1467946/meta[14] Chandrasekaran R, Koh M, Ozhawa Y, et al. Electrochemical cell studies on fluorinated natural graphite in propylene carbonate electrolyte with difluoromethyl acetate (MFA) additive for low temperature lithium battery application[J]. Journal of chemical sciences, 2009, 121: 339-346.https://link.springer.com/article/10.1007/s12039-009-0039-2[15] Nagasubramanian G, Fenton K. Reducing Li-ion safety hazards through use of non-flammable solvents and recent work at Sandia National Laboratories[J]. Electrochimica Acta, 2013, 101: 3-10.https://doi.org/10.1016/j.electacta.2012.09.065[16] Arai J, Katayama H, Akahoshi H. Binary mixed solvent electrolytes containing trifluoropropylene carbonate for lithium secondary batteries[J]. Journal of the Electrochemical Society, 2002, 149(2): A217.https://iopscience.iop.org/article/10.1149/1.1433749/meta[17] Xia L, Xia Y, Wang C, et al. 5 V‐class electrolytes based on fluorinated solvents for Li‐ion batteries with excellent cyclability[J]. ChemElectroChem, 2015, 2(11): 1707-1712.https://doi.org/10.1002/celc.201500286[18] Zhang Z, Hu L, Wu H, et al. Fluorinated electrolytes for 5 V lithium-ion battery chemistry[J]. Energy & Environmental Science, 2013, 6(6): 1806-1810.https://doi.org/10.1039/C3EE24414H[19] Lewandowski A, Świderska-Mocek A. Ionic liquids as electrolytes for Li-ion batteries—An overview of electrochemical studies[J]. Journal of Power sources, 2009, 194(2): 601-609.https://doi.org/10.1016/j.jpowsour.2009.06.089[20] Guerfi A, Dontigny M, Charest P, et al. Improved electrolytes for Li-ion batteries: Mixtures of ionic liquid and organic electrolyte with enhanced safety and electrochemical performance[J]. Journal of Power Sources, 2010, 195(3): 845-852.https://doi.org/10.1016/j.jpowsour.2009.08.056[21] Ishikawa M, Sugimoto T, Kikuta M, et al. Pure ionic liquid electrolytes compatible with a graphitized carbon negative electrode in rechargeable lithium-ion batteries[J]. Journal of power sources, 2006, 162(1): 658-662.https://doi.org/10.1016/j.jpowsour.2006.02.077[22] Wilhelm H A, Marino C, Darwiche A, et al. Significant electrochemical performance improvement of TiSnSb as anode material for Li-ion batteries with composite electrode formulation and the use of VC and FEC electrolyte additives[J]. Electrochemistry communications, 2012, 24: 89-92.https://doi.org/10.1016/j.elecom.2012.08.023[23] Zeng Z, Jiang X, Wu B, et al. Bis (2, 2, 2-trifluoroethyl) methylphosphonate: an novel flame-retardant additive for safe lithium-ion battery[J]. Electrochimica Acta, 2014, 129: 300-304.https://doi.org/10.1016/j.electacta.2014.02.062[24] Izquierdo-Gonzales S, Li W, Lucht B L. Hexamethyl phosphoramide as a flame retarding additive for lithium-ion battery electrolytes[J]. Journal of power sources, 2004, 135(1-2): 291-296.https://doi.org/10.1016/j.jpowsour.2004.04.011[25] Shiga T, Kato Y, Kondo H, et al. Self-extinguishing electrolytes using fluorinated alkyl phosphates for lithium batteries[J]. Journal of Materials Chemistry A, 2017, 5(10): 5156-5162.https://doi.org/10.1039/C6TA09915G[26] Zhu X, Jiang X, Ai X, et al. Bis (2, 2, 2-Trifluoroethyl) ethylphosphonate as novel high-efficient flame retardant additive for safer lithium-ion battery[J]. Electrochimica Acta, 2015, 165: 67-71.https://doi.org/10.1016/j.electacta.2015.02.247
应用实例
2023.05.25

稳定性同位素在药物开发和定制化医学中的应用----阿拉丁试剂
稳定性同位素在药物开发和定制化医学中的应用: 揭示生物标记与动力学生化网络的奥秘在新兴的定制化医疗时代,稳定同位素标记与强大的质谱分析技术的结合正在为药物开发和临床诊断提供越来越重要的诊断工具。问题:当代药物开发中的高流失率虽然人们普遍认为我们生活在一个新药研发生产突破的黄金时代,但事实恰恰相反。近年来,尽管制药行业投资大幅增加,但新药批准率仍达到了一直以来的最低水平[1]。这些令人失望的情况适用于所有类别的疾病,但尤其令人担忧的是日益流行的慢性疾病,如阿尔茨海默病、糖尿病、骨关节炎和肥胖相关疾病。这个问题的出现不在于缺乏分子靶点或候选药物。基于分子靶点的药物发现方法在过去20年的药物研究中占据主导地位,它产生了大量的基因、蛋白质和潜在的药物疗法。问题是药物线索的减损率变得更糟,而不是更好,现在98%的线索由于疗效或安全性原因而失效,包括人体试验中90%的失败率[2,3]。这种减损在很大程度上导致了最终批准的每一种成功药物的高成本。药物复杂网络中的难以预测性药物线索的减损,其主要原因是由生命系统组成的复杂网络的不可预测性组成的,而这些网络是对特定节点的针对性干预措施做出反应的[2],在这些系统中,有针对性的干预措施会带来意想不到的功能后果(可能是有害或者是有益的),这是非常常见的情况而非特例(图1)。每种疾病中个体之间的致病异质性放大了这一问题,因此需要针对不同患者亚群采取不同的干预策略。后一个问题体现在私人定制化医疗的理念上。图1:药物复杂网络中的难以预测性。缺失的一环:探索疾病复杂生物学的指标在了解疾病的复杂生物学过程中,关键缺失的因素是指导药物研发人员朝着安全有效结局目标前进的客观指标[4]。这些指标被称为生物标记物,必须能够预测临床结局,并且可以从临床前模型转化到人类。实现这些目标的最可靠方法是捕获驱动每种疾病的基础生物学过程(即疾病修饰通路或基础发病机制)。这类指标可用于指导合理的药物发现和开发,并用于监测临床应答情况。“定制化医学”领域对功能信息生物标记物的需求最大,即正确的患者、正确的药物、正确的时间和正确的剂量。伴随诊断检查是这一趋势的价值极高的例子。稳定性同位素对于一类新型生物标记物至关重要:通过揭示基础疾病过程的功能可解读信息来预测临床检测的结果我们需要能够预测临床结果的一类新型生物标记物[4,5]。构成慢性疾病基础的生物学通路(负责疾病启动、进展、严重程度和治疗逆转的因果过程)通常涉及分子流动,而这一通路本身复杂,且受到许多因素的影响(图2)[5-8]。稳定同位素技术已使所有这些因果通路在高等生物中可测量。图2:通路通量作为分子靶点和临床结果之间的联系。稳定同位素为诊断生物标记物带来了什么稳定同位素允许测量通过代谢途径的通量和全球生化网络的动态,且无毒性,通常无创,原因有二:第一,稳定同位素的实验性施用在时间维度上引入了“不对称”(本身没有标记,随后加入标记),这使得动态过程耗费的时间可以被测量;其次,过去一个世纪的生化研究已经建立了连接细胞和生物体分子的途径,使得标记底物在体内的运动行为得以被追踪。重要的是,稳定同位素已经在人类和实验动物中使用了70多年,几乎没有已知的毒性。FDA对稳定同位素标记产品的政策是明确的,并且几十年来一直是一致的:除了管理其天然丰度同系物(无菌性、无热原性等)所需的审批之外,管理稳定同位素标记的化合物不需要任何监管批准。需要注意的是,稳定同位素质谱生物标记物不是放射成像技术,但需要来自身体的样本(血液、尿液、脑脊液、组织、唾液)。两大类基于稳定同位素的可用动力学生物标记物有两大类基于稳定同位素的生物标记物,它们在药物开发和诊断中是最有用的:(1)靶向因果途径的动力学,以及(2)网络动力学的研究,以无偏倚地发现动力学特征和其无法被预测的因果途径。这两种类型在药物发现和开发中都是有效的[5-15]。作为药物发现和开发生物标记物的靶向因果通路动力学表1列出了疾病致病途径的一些常见例子。其中包括:纤维化疾病中胶原和细胞外基质的合成;多发性硬化的髓鞘合成和代谢;阿尔茨海默病中淀粉样斑块的周转和淀粉样β1-42的合成;肌少症患者肌肉肌球蛋白合成及线粒体生物发生的研究肿瘤血管生成与肿瘤细胞增殖和死亡;在神经退行性疾病中通过轴突运送分子;亨廷顿病、帕金森病和其他以蛋白质聚集为特征的疾病中的自噬流;血栓栓塞性疾病中的凝块形成和溶解;胰岛素抵抗状态下胰岛素介导的葡萄糖摄取与胰岛β细胞增殖肥胖时脂肪组织脂质动力学及重塑;动脉粥样硬化中的胆固醇逆向转运;炎症状态下补体级联的激活;艾滋病患者的HIV复制和CD4+ T细胞的转换等其他案例。测量被认为在疾病中起因果作用的任何功能相关过程的活动的能力,对这些领域的药物发现和开发具有潜在的变革作用(如帕金森病)[10,11]。A) 神经生物学通过轴突进行的递送淀粉样β蛋白合成和斑块周转神经发生髓鞘形成/髓鞘再生神经递质释放和周转神经元线粒体生物发生神经炎症,小胶质细胞的活化细胞因子释放亨廷顿蛋白周转朊病毒的周转突触可塑性B) 肥胖/II型糖尿病胰腺β细胞增殖胰岛素介导的葡萄糖摄取肝葡萄糖的生成脂肪生成和TG沉积脂肪组织重塑脂肪组织脂肪酸氧化/棕色脂肪转化动脉粥样硬化、胆固醇清除和沉积脂肪组织巨噬细胞增殖和活化肌肉线粒体β-氧化和生物发生肝脏TG的合成与释放C) 癌症/肿瘤形成肿瘤细胞增殖和死亡率血管新生淋巴管生成/转移扩散DNA甲基化/脱甲基核糖核苷酸还原酶活性组蛋白去乙酰化癌前进化到侵袭性表型肿瘤特异性T细胞增殖细胞外基质转换表1:因果途径的例子网络动力学探究也许近年来稳定同位素生物标记最令人兴奋的进展是“网络动力学”的出现:对构成生命系统的复杂生化网络的动态行为进行全面的探究。这已经成功地应用于临床前模型和人类的全球蛋白质组动力学或动态蛋白质组学[12,13]。这提供了一种新型的系统生物学,作为生物标记物发现的无偏倚筛选工具具有巨大的潜力。动态蛋白质组学代表了“组学”技术中功能上最可解释的技术,不仅提供热图或信息学,而且提供功能上可解释的系统生物学信息。图3显示了测量蛋白质组动态的操作流程图。该方法已成功应用于以下场景,如热量限制对细胞蛋白稳态的影响,包括线粒体生物发生和线粒体自噬;慢性淋巴细胞白血病肿瘤细胞不良预后的蛋白质组动力学特征区分成功补偿肥胖动物胰岛素抵抗的胰岛和衰竭并变得“疲惫”的胰岛;运动对肌肉蛋白质组转换的影响神经炎症对脑脊液蛋白质组周转的影响;血脂异常状态下高密度脂蛋白(HDL)蛋白质组的动态变化;以及生理学和病理生理学中的其他问题。图3:动态蛋白质组学:在体内通过稳定同位素标记测量蛋白质组动力学和浓度。用于细胞内通路非侵入性生物标记物的“虚拟活检”方法组织中蛋白质组动力学的无偏筛选也可以导致发现可在体液中采样的靶向蛋白质生物标记物。被称为“虚拟活检”技术(图4),这是一种强大的方法,用于测量难以接近的原始组织(如骨骼肌、心脏、大脑、肾脏、肝脏或癌症组织)中蛋白质合成或蛋白质分解的速率,通过测量可接近的体液(如血液、脑脊液、唾液或尿液)。该方法包括给药稳定同位素示踪剂(例如,氧化氘(D, 70%);L-亮氨酸(13C6, 99%);甘氨酸(15N, 98%);螺旋藻全细胞(冻干粉)(U-15N, 98%),代谢并入新合成的蛋白质。然后这些蛋白质逃逸到可接触的体液中,从中分离并分析同位素含量或模式。所测量的逃逸蛋白的替换率反映了蛋白质在原始组织中的合成或分解率。因此,可以称作是对原始组织进行了“虚拟活检”。虚拟活检方法可用于发现和验证生物标记物,用于药物发现和开发、识别个性化医疗中的疾病子集,以及患者的临床诊断和管理。这种方法已被开发并应用于基于血液的组织纤维化和骨骼肌蛋白合成测量,以及基于CSF的轴突运输和神经炎症测量[10]。一个例子是血浆肌酸激酶MM(来源于骨骼肌),用于从血液测试中测量骨骼肌蛋白合成代谢。还有许多其他的应用可以设想。图4:动态生物标记物的“虚拟活检”技术。从血浆肌酸激酶M型(CK-M)合成骨骼肌蛋白的案例原位动力学组织化学:结合组织病理学与稳定同位素和质谱现在也有可能在组织病理学标本中,在空间上可视化感兴趣的目标分子的动力学[14]。将空间组织学信息与分子通量率联系起来,为病理诊断和疾病监测提供了一个显著的新维度,这可以通过激光显微解剖或物理显微切片来实现(图5)。在前列腺癌中引入稳定同位素后组织显微解剖的例子已经发表。例如,前列腺细胞的增殖梯度已被证明与前列腺癌患者活检标本的组织学分级密切相关,并通过从精液中分离的前列腺上皮细胞的增殖率反映出来,作为一种潜在的非侵入性生物标记物[14]。图5:用于质谱动力学分析的正常和肿瘤组织的显微解剖。组织样本的动力学成像通过将稳定同位素标记与组织质谱成像相结合,通过基于NIMS或MALDI的组织切片空间可视化,现在可以实现动力学或代谢通量成像。肿瘤模型中空间定义的动力学脂质组学揭示了小鼠乳腺癌模型中与体内侵袭性相关的肿瘤行为的解剖学差异[15]。稳定同位素生物标记物在药物开发中的实际应用基于同位素的稳定生物标记物在药物发现和开发中有许多用途(表2)。将临床前研究结果迅速转化进行人体实验;“快速杀伤”的代理或类对目标路径的活性差;确定适当的患者亚群进行治疗;确定最佳剂量、状态、测量终点和受试者间反应变异性;医疗个性化(伴随诊断);通过调整剂量来预测毒性或避免毒性。可预测疾病结果的转译标记物还允许选择最能反映人类疾病的动物模型,或者从药物开发过程中减少甚至逐渐消除动物模型。减少推测过程:1. 选择目标;2. 选择化学类别及类别中最佳化合物;3. 确定合适的受试者(排除有毒性风险的无反应亚群);4. 为临床试验寻找最佳剂量和方案;5. 在患者中选择终点进行测量和预期的变异性;6. 给药以最大限度地减少毒性;7. 测试个性化调整是否能改善反应;8. 决定是否提前退出(快速杀伤)。表2:因果路径指标的应用基于稳定同位素的动态生物标记物与静态生物标记物相比具有优势,同时具有补充作用传统的静态生物标记提供了有关生命系统中分子的浓度、存在或结构的信息。相比之下,动力学生物标记物揭示了通向和离开这些分子的途径的动态行为。例如,组织中胶原蛋白的数量并不能显示在疾病环境中或开始治疗干预后胶原蛋白合成(成纤维)的速度。线粒体蛋白质的含量也没有告诉我们线粒体生物发生或线粒体自噬是由干预引起的程度。同样,脑脊液中蛋白质的浓度也能告诉我们大脑神经元将这种分子通过轴突运送到神经末梢的效率。后一种过程的核心都是分子通过复杂的途径和网络流动。这些致病过程或疾病途径的活性原则上是与疾病的发生、严重程度、进展和治疗逆转最密切相关的指标。如上所述,测量分子通量速率的唯一方法是引入同位素标签。虽然静态参数可以提供关键的补充信息,如池大小和分子组分的净增益或损失,但只能通过动力学测量来揭示潜在致病过程的功能活性。与静态“组学”生物标记物相比,同样的考虑适用于“网络动力学”,如动态蛋白质组学,但有一个额外的点值得注意。蛋白质的合成和分解速率通常代表细胞或有机体的主动决定,在健康或疾病的背景下,这种决定在功能上是可解释的。以蛋白质为例,基于泛素蛋白组的去除,转录因子刺激的合成,细胞器生物发生过程中的组装,囊泡中的包装和分泌,通过未折叠蛋白反应的调节,作为细胞外基质的沉积,作为蛋白质信号级联的一部分的诱导等等,这些都可以被生理学家、毒理学家和临床医生从功能角度考虑。对于蛋白质的简单存在或浓度并不总是如此。由于内在功能意义和广泛、无假设筛选之间的结合,动态蛋白质组学是一种特别强大的生物标记物和靶标发现技术。小结总之,将基于稳定同位素的生物标记物添加到诊断库中,为药物开发带来了一个新的和迅速扩大的维度。这些生物标记物提供了关于疾病基础生物学的功能可解释的决策相关信息,捕捉了驱动疾病和治疗的因果途径的活动。因此,动力学生物标记物可预测临床应答及其与靶点接触或临床治疗方案效果的关系。稳定的基于同位素的动力学生物标记物在个性化医学的新兴时代是特别强大的补充。参考文献1. Swann, J.P. 2011. Summary of NDA Approvals & Receipts, 1938 to the present, FDA History Office, http://www.fda.gov/AboutFDA/WhatWeDo/History.2. Duyk, G. 2003. Attrition and translation. Science, 302(5645), 603-605. https://www.science.org/doi/10.1126/science.10905213. Biotechnology Industry Organization (BIO) analysis, 2012.4. FDA, Innovation or stagnation: challenge and opportunity on the critical path to new medical technologies. March 2004.5. Hellerstein, M.K. 2008. A critique of the molecular target-based drug discovery paradigm based on principles of metabolic control: advantages of pathway-based discovery. Metab Eng, 10(1), 1-9. https://doi.org/10.1016/j.ymben.2007.09.0036. Hellerstein, M.K. 2003. In vivo measurement of fluxes through metabolic pathways: the missing link in functional genomics and pharmaceutical research. Annu Rev Nutr, 23, 379-402. https://doi.org/10.1146/annurev.nutr.23.011702.0730457. Turner, S.M.; Hellerstein, M.K. 2005. Emerging applications of kinetic biomarkers in preclinical and clinical drug development. Curr Opin Drug Discov Devel, 8(1), 115-126. https://www.x-mol.com/paper/1212977911043006473?adv8. Hellerstein, M.K. 2008. Exploiting complexity and the robustness of network architecture for drug discovery. J Pharmacol Exp Ther, 325(1), 1-9; https://doi.org/10.1124/jpet.107.1312769. Shankaran, M.; King, C.; Lee, J.; et al. 2006. Discovery of novel hippocampal neurogenic agents by using an in vivo stable isotope labeling technique. J Pharmacol Exp Ther, 319(3), 1172-1181. https://doi.org/10.1124/jpet.106.11051010. Fanara, P.; Wong, P.Y.; Husted, K.H.; et al. 2012. Cerebrospinal fluid-based kinetic biomarkers of axonal transport in monitoring neurodegeneration, J Clin Invest, 122(9), 3159-3169. https://www.jci.org/articles/view/6457511. Potter, W.Z. 2012. Mining the secrets of the CSF: developing biomarkers of neurodegeneration. J Clin Invest, 122(9), 3051-3053. https://www.jci.org/articles/view/6530912. Price, J.C.; Khambatta, C.F.; Li, K.W.; et al. 2012. The effect of long term calorie restriction on in vivo hepatic proteostatis: a novel combination of dynamic and quantitative proteomics. Mol Cell Proteomics, 11(12), 1801-1814. https://doi.org/10.1074/mcp.M112.021204
应用实例
2023.05.22

重磅发布!阿拉丁2022年年度报告
阿拉丁2022年年度报告 公司是国内科研试剂品种最齐全的生产商之一,自成立以来一直以进口替代为己任,坚持打造“阿拉丁”自主试剂品牌,通过自主研发扩充产品线,缓解了国内企业进口试剂价格昂贵、发货周期长等局面,在业内享有较高声誉。 报告期内,公司主营业务收入逐年提升,产品市场占有率逐步提高。公司部分产品达到了国际同等技术水平并实现了部分进口替代,逐步打破了外企绝对垄断的态势。 业务增长 财务数据 研发成果 营收分析 口碑荣誉 阿拉丁是中国化学试剂工业协会团体标准委员会副主任委员单位和团体会员单位,也是中国分析测试协会会员以及上海化学试剂产业技术创新战略联盟成员。公司作为牵头单位共主导 46 项行业标准的起草,参与 15 项行业标准的验证,完成接近 8 万项企业产品标准的制订,连续 11 年被评为“最受用户欢迎试剂品牌”,连续 6 年被评为“中国化学试剂行业十强企业”。在全国化学试剂信息站进行的国内试剂品牌综合评价调查中,“阿拉丁”连续 4 年在品种选择方面位列榜首,获得了客户的高度信赖。 作为一家技术密集型企业,公司一直以科技领先性和技术创新性为核心优势,深受广大客户的认可。报告期内,公司荣获上海市第 28 批市级企业技术中心称号,并被上海市知识产权局评为“上海市专利工作示范企业”。此外,公司开展的研发项目“一种离子对色谱级试剂十二烷基三甲基氯化铵的制备工艺”在第三十四届上海市优秀发明选拔赛中荣获优秀创新“金奖”。
企业动态
2023.05.12

蛋白样品制备流程----阿拉丁试剂
一、蛋白提取蛋白分析的第一步是蛋白提取,蛋白提取方法包括化学抽提法、物理抽提法和生物学抽提法。蛋白提取无论通过机械处理还是基于去垢剂的提取方法,都会不可避免地破坏细胞稳态,导致蛋白降解或不稳定。因此,提取过程中蛋白的完整性直接决定了下游蛋白样品分析所得数据的质量。样品类型关于内源性蛋白和表达系统蛋白的研究来源主要包括哺乳动物细胞,哺乳动物组织和原代细胞。当提取哺乳动物组织中的蛋白质时,需要一些温和的酶或机械破碎手段来帮助从较复杂组织基质中分离细胞。对于仅通过质膜将细胞内容物与环境隔离开的培养哺乳动物细胞和原代细胞来说,试剂中的去垢剂可打破蛋白-脂质膜双层结构,使总蛋白提取更为容易。通常所研究的或用于重组蛋白表达系统的其他生物体包括细菌、酵母和植物。这些细胞类型含有细胞壁,需要额外使用酶解或机械破碎的方法才能有效释放蛋白。然而,目前已开发出基于去垢剂的解决方案,可有效提取和溶解这些细胞中的蛋白,而无需机械外力破坏。该方法对于处理更多样品数量或使用自动提取和纯化方案来说尤其重要。亚细胞分级分离对于大多数研究来说,只需直接制备全细胞裂解物以获取可溶蛋白样品,用于下游直接检测或进一步纯化与分离。但是,如果在蛋白提取之前将细胞分为不同的区室或细胞器,可以显著提高特定蛋白的产量或富集率。机械裂解通常会破坏所有细胞区室,使分离特定细胞组分变得更加困难。但是,通过谨慎优化试剂,已开发出基于逐级差异去垢剂的方法来分离核蛋白、胞质蛋白和膜蛋白组分。该方法可以将疏水性膜蛋白溶解,使其与亲水蛋白分离,并且可以分离完整细胞核、线粒体和其他细胞器,用于直接研究或分步提取蛋白。二、蛋白质降解细胞裂解时会破坏细胞膜和细胞器,导致蛋白水解活性不受调控,从而降低蛋白产量并影响蛋白功能。为了防止提取的蛋白降解并保持其活性,通常需要在细胞裂解试剂中添加蛋白酶和磷酸酶抑制剂。 蛋白酶抑制剂通过与蛋白酶活性位点结合而起作用。由于蛋白水解机制的差异,单一化合物无法有效抑制所有蛋白酶,因此,需要使用几种不同抑制剂的混合物来确保蛋白提取物在下游分析之前不被降解。典型的抑制剂混合物包括丝氨酸、半胱氨酸和天冬氨酸蛋白酶等小分子抑制剂,以及氨基肽酶和金属蛋白酶。尽管有些抑制剂是不可逆的,但很多抑制剂是可逆的,它们需要长时间存在于粗制样品中,直到后续进行了纯化,免除了蛋白水解活性的风险为止。同样,磷酸酶也各有不同,因此建议使用有效的磷酸酶混合物(含丝氨酸、苏氨酸、酪氨酸、酸性和碱性磷酸酶的抑制剂)来保持脆弱的磷酸化翻译后修饰。通过使用正确的保存方法来防止靶蛋白降解:快速操作并保持样品低温(可使用液氮冷冻样品);抑制或灭活内源性蛋白酶和磷酸酶;添加保护或稳定化合物,例如还原剂和酶抑制剂;通过沉淀稳定或灭活蛋白。三、蛋白样品除杂蛋白提取后,蛋白样品通常含有影响蛋白稳定性或与下游应用不兼容的杂质。透析、脱盐和浓缩是去除蛋白样品中常见小分子污染物(例如盐和去垢剂)的三种常用方法。根据下游应用要求,选择方法时的考虑因素可能包括样品起始量、蛋白功能和处理时间。透析、脱盐和浓缩的有多种规格和包装形式。透析透析是一种经典分离技术,它通过半透膜选择性扩散,去除蛋白溶液中的小分子和不需要的化合物。将样品和缓冲液置于膜的相对侧。大于膜孔的蛋白保留在膜的样品侧,较小的分子(污染物)通过膜自由扩散,直至达到平衡浓度。这种技术可以使样品中小分子污染物的浓度降低至可接受水平。脱盐常用的脱盐方法有透析法、电透析法和凝胶过滤法。透析法及电透析法耗时长,样品稀释度大,不易放大进行大规模生产,所以工业生产中应用较少。凝胶过滤层析脱盐过程中盐分子和蛋白质分子大小差异巨大,蛋白质溶液中小分子的盐分子随着层析流动相进入孔径较小的固定相,使其在层析中的迁移速率小,而蛋白质因分子尺寸较大,不能随流动相进入固定相中,因此在层析柱中的迁移速率大,首先从层析术中流出,实现脱盐。浓缩蛋白浓缩与透析法类似,其使用半透膜将蛋白与小分子量化合物分离。与透析法的被动扩散原理不同,浓缩是通过离心使溶液穿过膜而实现的。在离心过程中,缓冲液和小分子量溶质均被动穿过膜,并在另一侧收集(滤液)。大分子(蛋白)保留在膜的样品侧,其随着试剂被动穿过膜到达另一侧而浓缩为小体积液体(截留液)。四、蛋白定量紫外分光光度法蛋白质分子中存在含有共轭双键的酪氨酸和色氨酸,使蛋白质对280nm的光波具有最大吸收值,在一定的范围内,蛋白质溶液的吸光值与其浓度成正比,可作定量测定。该法操作简单、快捷,并且测定的样品可以回收,低浓度盐类不干扰测定结果,故在蛋白质和酶的生化制备中广泛被采用。但此方法存在以下缺点:1.当待测的蛋白质中酪氨酸和色氨酸残基含量差别较大会产生一定的误差,故该法适用于测定与标准蛋白质氨基酸组成相似的样品。 2.若样品中含有其他在280nm吸收的物质如核酸等化合物,就会出现较大的干扰。但核酸的吸收高峰在260nm,因此分别测定280nm和260nm两处的光吸收值,通过计算可以适当的消除核酸对于测定蛋白质浓度的干扰作用。但因为不同的蛋白质和核酸的紫外吸收是不同的,虽经校正,测定结果还存在着一定的误差。荧光法蛋白定量基于荧光进行蛋白定量是除比色法以外的另一选择。荧光检测法具有出色的灵敏度,所需蛋白样品量少,可节省更多样品用于其他实验。此外,读取时间并非关键因素,因此这种检测方法可轻松应用于自动化高通量分析。可使用荧光计或酶标仪检测荧光信号。Bradford法1976年Bradford建立了用考马斯亮蓝G-250与蛋白质结合的原理,是一种能够迅速并且准确的定量蛋白质的方法。染料与蛋白质结合后引起染料最大吸收光的改变,从465nm变为595nm。蛋白质-染料复合物具有高的消光系数,因此大大提高了蛋白质测定的灵敏度(最低检出量为1μg)。染料与蛋白质的结合是很迅速的过程,大约只需2min,结合物的颜色在1h内是稳定的。一些阳离子如K+,Na+,Mg2+、(NH4)2SO4、乙醇等物质不干扰测定,而大量的去污剂如TritonX-100,SDS等严重干扰测定,少量的去污剂可通过用适当的对照而消除。由于染色法简单迅速,干扰物质少,灵敏度高,现已广泛用于蛋白质含量的测定。总氮量的测定——微量凯氏定氮法:当被测定的含氮有机物与浓硫酸共热消化时,分解出氮、二氧化碳和水,其中氮与硫酸化合成硫酸铵。由于分解反应进行得很慢,故可加入硫酸铜和硫酸钾或硫酸钠,其中硫酸铜为催化剂,硫酸钾或硫酸钠可提高消化液的沸点,氧化剂(过氧化氢)也能加速反应。消化终止后,在凯氏定氮仪中加入强碱碱化消化液,使硫酸氨分解放出氨;用水蒸气蒸馏,将氨蒸入过量的标准无机酸溶液中,全部蒸完之后,用标准的盐酸溶液滴定收集的氨量,准确测定氨量,从而折算出蛋白质含量。双缩脲法具有两个或两个以上肽键的化合物皆有双缩脲反应,蛋白质在碱性溶液中能与Cu2+络合呈紫红色,颜色深浅与蛋白质浓度成正比,故可用比色法进行测定,根据标准曲线进行计算可以确定蛋白质浓度。Folin-酚试剂法测定蛋白质含量的经典方法,它是在双缩脲法的基础上发展而来的。它操作简单、迅速、灵敏度高,较双缩脲法灵敏100倍。Folin-酚法所用的试剂由两部分组成,试剂A相当于双缩脲试剂。蛋白质中的肽键与试剂A中的碱性硫酸铜反应形成铜-蛋白质复合物。这个复合物可与试剂B中磷钼酸-磷钨酸发生氧化还原反应。由于磷钼酸与磷钨酸易被酚类化合物还原而呈蓝色反应。而蛋白质中的酪氨酸和色氨酸均可发生此呈色反应。颜色的深浅与蛋白质的浓度成正比。故可用比色法测定蛋白质的含量。此法易受蛋白质样品中酚类化合物及柠檬酸的干扰。另外,试剂B中的磷钼酸-磷钨酸仅在酸性pH值时稳定,故在将试剂B加入到碱性的铜-蛋白质溶液时,必须立即混合均匀。以确保还原反应能正常发生。此法也适用与酪氨酸和色氨酸的定量测定。五、蛋白检测根据实验需要,可以使用多种方法来检测和分析靶蛋白。以下是用于检测和分析复杂混合物(例如裂解物和血清)中蛋白质的常见技术,以及各种技术的特点和常见要求。ELISA蛋白质免疫印迹质谱分析优势可进行 96-孔或 384-孔板的高通量分析可定量靶蛋白分子量测定和蛋白鉴定可以对靶蛋白进行分离和区分在同一份样品中定性和定量多个靶标检测蛋白翻译后修饰或不同亚型灵敏度从低飞克到高阿克摩尔范围(1018)裂解液兼容性对于非活性检测 ELISA:基于离子型去垢剂的裂解液对于活性检测 ELISA:基于非离子型去垢剂的裂解液(例如 NP-40、Triton X-100)对于 SDS-PAGE(变性):RIPA 或含其他含离子型去垢剂的裂解液对于非变性聚丙烯酰胺凝胶电泳(Native-PAGE):基于非离子型去垢剂的裂解液(例如 NP-40、Triton X-100)质谱分析前必须去除样品中的去垢剂和高盐物质常规所需蛋白量0.1–1 µg/mL1–50 µg所需设备酶标仪X 光胶片或 CCD 成像系统质谱仪相关产品列表D304861 无花果蛋白酶,来源于无花果树乳胶 ,≥ 100u/mgSDS| 价格A274819 钙蛋白酶抑制剂I ,≥95%SDS| 价格A118288 活性依赖性神经保护蛋白片段 74-81,小鼠,大鼠 ,≥97.0% (HPLC)SDS| 价格A116563 牛血清白蛋白 ,生物技术级,96%SDS| 价格A104912 牛血清白蛋白,组分 V ,分子生物学级SDS| 价格A107820 白蛋白 来源于鸡蛋白 ,冻干粉, ≥98% (agarose gel electrophoresis)SDS| 价格A107821 白蛋白 来源于鸡蛋白 ,冻干粉, ≥90% (agarose gel electrophoresis)SDS| 价格A302887 β-淀粉样蛋白(40-1) 三氟乙酸盐 ,95%SDS| 价格A107853 β-淀粉样蛋白片段 25-35 ,97%SDS| 价格A103033 亲合素 来源于鸡蛋白 ,冻干粉, 10-15 units/mg protein (E1%/280), ≥98%SDS| 价格A302087 偶氮酪蛋白 ,protease substrateSDS| 价格C304284 酸水解酪蛋白 ,BRSDS| 价格C194606 酪蛋白磷酸肽 ,Protein ≥80%SDS| 价格S304898 酪蛋白酸钠 ,BR,来源于牛奶SDS| 价格C275652 组织蛋白酶G抑制剂 ,95%SDS| 价格C275083 组织蛋白酶G底物 ,≥95%SDS| 价格C129408 组织蛋白酶抑制剂1 ,≥99%SDS| 价格C113165 胰凝乳蛋白酶抑制剂 ,≥95%SDS| 价格C390608 人IV型胶原蛋白 ,0.5-5 mg/ml in 0.01mol/L PBS,>95%SDS| 价格C275407 钙调蛋白抑制剂 ,SDS| 价格C388022 交联别藻蓝蛋白 ,≥10 mg/mL in Ammonium sulfateSDS| 价格D195752 中性蛋白酶 ,BR,50u/mgSDS| 价格E128657 弹性蛋白酶 来源于猪胰腺(冻干) ,≥3 units/mg proteinSDS| 价格E128664 弹性蛋白酶 来源于猪胰腺(悬浮液) ,≥3 units/mg proteinSDS| 价格E107150 弹性蛋白酶,来源猪胰腺 ,30 units/mgSDS| 价格P128655 蛋白内切酶 Glu-C 来源于金黄色葡萄球菌 V8 ,≥500 units/mg dry weightSDS| 价格E382042 蛋白内切酶Lys-C ,≥3.0AU/mgSDS| 价格F118924 纤维蛋白肽 B 人类 ,≥97% (HPLC)SDS| 价格G304672 谷蛋白 (来自小麦) ,SDS| 价格G304749 牛乳清糖蛋白 ,80%SDS| 价格H304436 白蛋白 人 ,96%SDS| 价格H195714 血红蛋白 ,来源于牛血液SDS| 价格H276277 组蛋白乙酰转移酶抑制剂II ,≥98%SDS| 价格L344777 乳清蛋白 ,SDS| 价格L116859 水解乳蛋白 ,BRSDS| 价格L302916 乳铁蛋白(来自牛乳) ,95%SDS| 价格L389977 脂蛋白,低密度,来源于人血浆 ,Solution in 150 mM NaCl and 0.01% EDTASDS| 价格L128640 溶菌酶 来源于鸡蛋白 ,≥5,000 units/mg dry weightSDS| 价格L128641 溶菌酶 来源于鸡蛋白(纯化,无盐) ,≥8,000 units/mg dry weightSDS| 价格L105521 溶菌酶 来源于鸡蛋白 ,≥20,000U/mgSDS| 价格M489394 甲基丙烯酰化丝素蛋白 ,分子量 6-10KDaSDS| 价格M304751 粘蛋白来源牛颌下腺 ,BSSDS| 价格M302050 肌红蛋白 来源于马骨骼肌 ,90%SDS| 价格M119690 马心肌红蛋白相对分子质量标准物质 ,马心肌红蛋白:16951SDS| 价格N128694 中性蛋白酶 来源于多粘芽孢杆菌(部分纯化) ,≥0.1 units/mg dry weightSDS| 价格N128693 中性蛋白酶 来源于多粘芽孢杆菌(纯化) ,≥4 units/mg dry weightSDS| 价格O302792 卵清蛋白三氟乙酸盐(OVA Peptide 257-264) ,98%SDS| 价格P123425 木瓜蛋白酶 ,lyophilized powder, ≥10 units/mg,with BAEE as substrateSDS| 价格P164463 木瓜蛋白酶 ,≥2000units/mg,以酪蛋白为底物SDS| 价格P110928 胃蛋白酶(猪源) ,1:15000SDS| 价格P110927 胃蛋白酶(猪源) ,1:3000SDS| 价格P128678 胃蛋白酶 来源于猪胃 ,≥2,500 units/mg dry weightSDS| 价格P113168 胃蛋白酶抑制剂 ,≥75% (HPLC)SDS| 价格P422930 胃蛋白酶抑制剂 ,10mM in DMSOSDS| 价格P139522 酵母蛋白胨 ,用于微生物SDS| 价格P304957 大豆蛋白胨 ,BRSDS| 价格P304956 大豆蛋白胨 ,SDS| 价格P304955 鱼蛋白胨 ,BRSDS| 价格P304928 蛋白酶 来源于灰色链霉菌 ,~7000 units/g proteinSDS| 价格P123670 硫酸鱼精蛋白 ,SDS| 价格P298987 蛋白酶,来源于米曲霉 ,≥1100 LAPU/gSDS| 价格P298992 蛋白酶 来源于解淀粉芽胞杆菌 ,≥0.8 U/gSDS| 价格P298986 蛋白酶,来源于地衣芽孢杆菌 ,≥2.4 U/gSDS| 价格P298996 蛋白酶 来源于芽孢杆菌属 ,≥8 U/gSDS| 价格P298993 蛋白酶 来源于芽孢杆菌 属 ,≥16 U/gSDS| 价格P109033 蛋白酶K ,冻干粉,≥30 units/mg proteinSDS| 价格P274341 蛋白酶K ,生物技术级SDS| 价格P301575 蛋白酶K 来源于林伯氏白色念球菌 ,≥500 units/mL, buffered aqueous glycerol solutionSDS| 价格P128666 蛋白酶K 来源于林伯氏白色念球菌 ,≥20 units/mg dry weightSDS| 价格P301902 蛋白酶抑制剂混合物 ,100×,不含EDTASDS| 价格R141086 重组蛋白A ,≥95%,Mw 25kDSDS| 价格R141084 重组胰蛋白酶 来源于人 ,≥2500 units/mgSDS| 价格R141080 重组胰蛋白酶 来源于猪胰腺 ,≥3800 units/mg proteinSDS| 价格S141078 重组胰蛋白酶 ,≥4500 units/mg,测序级SDS| 价格R128560 逆转录酶,大肠杆菌产生的重组蛋白产生的重组HIV ,≥5,000 units/mg proteinSDS| 价格P196382 非还原型蛋白上样缓冲液 ,5×SDS| 价格N301937 非变性非还原性蛋白上样缓冲液 ,5×SDS| 价格S491421 蛋白质稳定剂 ,STBSDS| 价格T329673 甜蛋白 ,SDS| 价格T302200 牛转铁蛋白 ,95%SDS| 价格T274333 胰蛋白酶 ,1:250,组织培养级SDS| 价格T113170 胰蛋白酶抑制剂 ,试剂级SDS| 价格T105533 胰蛋白酶 来源于牛胰脏 ,potency ≥3000 units/mgSDS| 价格T105531 胰蛋白酶(牛胰脏) ,potency ≥2500 units/mgSDS| 价格T128774 胰蛋白酶 来源于牛胰腺 ,≥180 units/mg proteinSDS| 价格T128775 胰蛋白酶 来源于牛胰腺(0.22 μm,过滤) ,≥180 units/mg protein (10,350 BAEE/3,450 USP/NF units/mg protein)SDS| 价格T128772 胰蛋白酶 来源于牛胰腺(2X,无菌,辐射) ,≥180 units/mg protein (10,350 BAEE/3,450 USP/NF units/mg protein)SDS| 价格T128769 胰蛋白酶 来源于牛胰腺(修饰,测序级) ,≥150 units/mg protein(at least 8,625 BAEE/2875 USP/NF units/mg protein)SDS| 价格T128770 胰蛋白酶 来源于牛胰腺(纯化,测序级II) ,≥150 units/mg protein (at least 8,625 BAEE/2875 USP/NF units/mg protein)SDS| 价格T128771 胰蛋白酶 来源于牛胰腺(TPCK处理) ,≥180 units/mg protein (10,350 BAEE/3,450 USP/NF units/mg protein)SDS| 价格T128773 胰蛋白酶 来源于牛胰腺(TPCK处理,辐射) ,≥180 units/mg proteinSDS| 价格T105532 胰蛋白酶(猪胰脏) ,potency ≥250 units/mgSDS| 价格T118254 糜胰蛋白酶 ,2400:400(6:1)SDS| 价格Z304904 玉米蛋白 ,from CornSDS| 价格C420153 α-酪蛋白(片段90-95)TFA ,97%SDS| 价格G304901 γ-球蛋白 来源于牛血液 ,99%SDS| 价格M301777 甲基丙烯酰化丝素蛋白 ,分子量 10-20KDaSDS| 价格S278688 大豆蛋白胨 ,用于微生物SDS| 价格W293432 水溶性丝素蛋白 ,分子量 6-10kDaSDS| 价格B265994 牛血清白蛋白(BSA) ,新西兰源, 巯基封闭型, ≥ 98.0%SDS| 价格B265991 牛血清白蛋白(BSA) ,新西兰货源, 标准级 pH 7.0, ≥ 96.0%SDS| 价格B265993 牛血清白蛋白(BSA) ,新西兰源,精制级, ≥ 98.0%SDS| 价格B278686 牛骨蛋白胨 ,试剂级SDS| 价格M301779 甲基丙烯酰化弹性蛋白(ElaMA) ,SDS| 价格P301903 蛋白酶抑制剂混合物 ,100×,无动物源性SDS| 价格R283942 重组人纤维连接蛋白OsrhFN ,细胞培养级,≥95%SDS| 价格R283924 重组人血清白蛋白OsrHSA ,冻干粉,99%SDS| 价格R283936 重组人血清白蛋白OsrHSA ,细胞培养级(高辛酸钠),99%SDS| 价格R283928 重组人血清白蛋白OsrHSA ,细胞培养级,99%SDS| 价格C106197 α-糜蛋白酶 来源于猪胰脏 ,800 usp u/mgSDS| 价格C106198 α-糜蛋白酶 来源于猪胰脏 ,1000 usp u/mgSDS| 价格C128653 α-糜蛋白酶 来源于牛胰腺 ,≥45 units/mg proteinSDS| 价格C128654 α-糜蛋白酶 来源于牛胰腺 ,≥35 units/mg proteinSDS| 价格C128695 胶原蛋白酶 来源于溶组织梭菌(纯化) ,≥500 units/mg dry weightSDS| 价格C128702 胶原蛋白酶 来源于溶组织梭菌(类型1) ,≥125 units/mg dry weightSDS| 价格C128703 胶原蛋白酶 来源于溶组织梭菌(类型2) ,≥125 units/mg dry weightSDS| 价格C128708 胶原蛋白酶 来源于溶组织梭菌(类型3) ,≥100 units/mg dry weightSDS| 价格C128709 胶原蛋白酶 来源于溶组织梭菌(类型4) ,≥160 units/mg dry weightSDS| 价格C128710 胶原蛋白酶 来源于溶组织梭菌(无动物源性,A) ,≥150 units/mg dry weightSDS| 价格C128711 胶原蛋白酶 来源于溶组织梭菌(无动物源性,B) ,≥300 units/mg dry weightSDS| 价格C128712 胶原蛋白酶 来源于溶组织梭菌(无动物源性,C) ,≥200 units/mg dry weightSDS| 价格C129076 α-糜蛋白酶 来源于牛胰腺(TLCK处理,序列) ,≥40 u/mgPSDS| 价格C129079 α-糜蛋白酶 来源于牛胰腺(TLCK处理) ,≥45 units/mg proteinSDS| 价格C129080 α-糜蛋白酶 来源于牛胰腺(纯化) ,≥45 units/mg proteinSDS| 价格C140836 α-糜蛋白酶 来源于牛胰腺 ,1000 usp u/mgSDS| 价格L196972 脂蛋白脂肪酶,来源于伯克霍尔德氏菌 ,≥1000 units/mgSDS| 价格M135414 Myoseverin ,≥98%价格
应用实例
2023.05.12

【阿拉丁】积分派“兑” 倍享精彩——购物狂欢,积分翻倍!
企业动态
2023.05.08

抽奖 | 查收阿拉丁五一发货通知,送小米手环8!
企业动态
2023.04.28

阿拉丁质量评定服务再升级——新增【检测服务】
我们很荣幸地宣布,阿拉丁【检测服务】已于3月14日正式上线!我们欢迎所有注册用户前往官网,通过导航栏访问检测页面。为了方便并加快您对产品的化学分析,我们将提供专业可靠的原始数据和图谱。此次官网升级不仅进一步展现了阿拉丁「便捷快优」的一站式服务特色,也将为您的检测体验带来更大的便利。 化学分析是化学领域中非常重要的一个分支,它通过物质的化学反应来测定物质的组成。这种分析方法在化学工业、医药产业以及科学研究等领域都有着广泛的应用。化学分析不仅可以让我们更好地了解物质的特性,还可以为工业生产和科学研究提供可靠的依据。 化学分析的精度往往取决于分析方法的精确程度。为了确保分析数据的准确性,通常会使用许多复杂的检测仪器,例如气相色谱仪、液相色谱仪和质谱仪等高端仪器。除此之外,还需要专业的技术人员来进行精细的操作和数据处理。阿拉丁此次推出的检测服务是一项高价值的服务,旨在通过使用阿拉丁自身拥有的精密仪器,为客户提供专业而高效的原始数据或图谱,使客户能够深入研究检测样品的内部结构以及原子性质等方面,从而提升其化学分析的便利性和准确性。这项服务的核心优势在于我们拥有一支高素质的专业团队和先进的技术设备,能够确保精准的分析结果以及及时的反馈。我们的服务对于需要高质量数据进行分析处理的客户而言,非常具有价值。无论是研究人员、企业还是政府机构,都可以通过阿拉丁的检测服务,快速准确地获取到相关数据,从而为其研究和创新提供有力支撑。我们相信,阿拉丁的检测服务将为客户在化学研究领域提供一条可靠的捷径。以下将为您介绍阿拉丁所提供的检测服务使用的部分精密仪器及检测技术特点: (A)核磁共振波谱检测(NMR)核磁共振波谱法,简称NMR,是一种无损的、高度灵敏的分析方法,其独特的特点在于能够针对样品中的有机化合物以及某些无机物进行定量和定性分析。该检测方法在化学生物学、医药学以及其他领域中的研究中都具有重要价值。600MHz核磁共振波谱仪是阿拉丁购置的一台近千万元级的仪器,具有高分辨率、高灵敏度等优势,且在检测时效性方面也有很大提升。阿拉丁检测仪器:日本电子400MHz/600MHz NMR (B)电感耦合等离子体质谱检测(ICP-MS) 电感耦合等离子体质谱测试(ICP-MS)被广泛认定为一种高度可靠且应用广泛的元素分析测试方法,具有极高的检测灵敏度和精度,能够在非常短的时间内快速、准确地测量痕量金属,检测范围可达ppb级别。ICP-MS不仅在环境科学领域中具有较广的应用,也在生物医学、产品质量控制等多个领域中发挥着重要的作用,对于解决元素循环和健康评估等问题具有重大意义。值得一提的是,ICP-MS检测方法所具备的可靠性和高准确度,是该检测方法在学术界和工业界广泛应用的重要原因之一。阿拉丁检测仪器:安捷伦ICPMS7850 (C)气相色谱质谱检测(GC-MS)气相色谱质谱测试(GC-MS)技术是常用的化学分析方法,可以精确地检测和鉴定样品中的化合物。它通过分离和检测分子量,将混合物中的不同化学成分分离出来,进而进行定性和定量分析。这种分析技术在医药、环保、食品、石油化工等领域得到广泛应用。举例来说,气相色谱质谱测试可用于检测食品中的农药残留以及环境中的各种污染物。相对于其它检测方法而言,GC-MS技术具有高灵敏度、高分辨率、可靠性强等特点,是当今分析领域中不可或缺的技术手段。阿拉丁检测仪器:安捷伦GC7890或GC8890点击跳转至官网【检测服务】详情检测服务的流程一般可分为三个主要步骤,分别是前期准备、样品检测、检测结果处理。①前期准备:涉及到制定检测方案、调配设备、培训人员等。②样品检测:需要对样品使用适当的检测方法和工具,获取关键数据和指标。③检测结果处理:需要将采集的数据和指标分析,以确保所有结果的准确性和可靠性。 以下流程图为阿拉丁所提供的检测服务流程直观展示,您可按照此步骤登录阿拉丁官网,选择相应服务进行操作。 检测服务是现代社会必不可少的重要保障,它能够确保产品和服务的安全可靠,在促进国际贸易和提高质量标准方面也有不可或缺的作用。在此背景下,阿拉丁检测服务的推广和应用,无论是在化学、工业、医疗还是环保等领域,我们都致力于为各行各业提供专业、稳定且可靠的支持,从而更好的促进行业发展并推动社会进步。 随着工业4.0时代的到来,智能化检测及精准检测等新技术将成为检测服务的重要组成部分。这些技术将以更加高效、准确的方式满足各行各业的需求,推动产业升级和发展。在化学检测领域,阿拉丁致力于技术创新和实践探索,不断提升化学检测技术水平,为服务对象提供更为优质和专业的检测服务。我们相信,在未来的探索中,阿拉丁的检测技术将继续推动实现更可持续的社会发展,让我们共同期待阿拉丁的检测技术能为人类带来更美好的未来!
企业动态
2023.04.14

氢化硅烷化反应及氢化硅烷化催化剂
简介氢化硅烷是一类相对稳定的化合物,但在是金属催化剂存在的条件下,可以与不饱和键发生加成反应。该方法已日益发展成为有机硅化合物的合成方法。近年来,在半导体的表面修饰和一些功能材料的开发中发挥着重要的作用。[CP*RU(MECN)3]PF6:一种高效的氢化硅催化剂乙烯基硅烷是多用途有机金属试剂,参与多种反应,例如Tamao–Fleming氧化、烯烃复分解、钯催化交叉偶联、原脱甲硅烷化和环加成。在制备乙烯基硅烷的可用方法中,炔烃的氢化硅烷化是最直接和具有原子经济性的方法(方案1)。同时,已经设计了许多过渡金属催化剂以区域和立体控制的方式执行这些反应(图1)。方案1.炔烃的氢化硅烷化图1.设计了以区域和立体控制的方式进行反应的过渡金属催化剂。末端炔烃的氢化硅烷化方法是在不久前发展起来的,用于制备顺式和反式β-乙烯基硅烷。经典的铂催化(Speier's和Karstedt's催化剂)1-5以及基于铑的催化([Rh(cod)2]BF4和[RhC(nbd)]2)6-9仍然是合成反式β-乙烯基硅烷的有力方法。Wilkinson催化剂也被验证在极性溶剂中产生反式产物,而在非极性介质中以顺式异构体为主。钌催化剂(例如[Ru(benzene)Cl2或[Ru(p-cymene)Cl2]可得到顺式-β-乙烯基硅烷10-13。尽管氢化硅烷化的立体选择性和区域选择性高度依赖于炔烃、硅烷和溶剂,但在某些条件下,第一代Grubbs催化剂也产生顺式产物。尽管存在大量制备线性β-乙烯基硅烷的方法,但直到最近,还没有制备1,1-双取代α-乙烯基硅烷的通用方法。此外,尽管可以实现内部炔烃分子内部的选择性氢化硅烷化,分子间变体的选择性实际上是未知的。斯坦福大学的Trost课题组开发了一种非常稳健的方案,通过[CP*RU(MECN)3]PF6实现末端乙炔的氢化硅烷化以得到α-乙烯基硅烷14。该催化剂还为内部炔烃的区域选择性分子内和分子间氢化硅烷化提供了一种仅生成Z-三取代烯烃的有效方法。分子间氢化硅烷化:末端炔烃类在[Cp*Ru(MeCN)3]PF6的存在下,一组不同的末端炔烃进行快速和温和的氢化硅烷化,以良好到优异的产率和低催化剂负载量得到1,1-双取代α-乙烯基硅烷(方案2)。该反应能耐受多种官能团,包括卤素、游离醇、烯烃、内炔烃、酯和胺。此外,可以在具有良好的可预测性反应中使用大量硅烷。方案2.分子间氢化硅烷化:末端炔烃分子间氢化硅烷化:内部炔烃类如方案1所示,内炔烃的非选择性氢化硅烷化将可能产生四种异构体加成产物。Trost课题组已经证明,内炔烃与[Cp*Ru(MeCN)3]PF6的氢化硅烷化仅产生三取代的Z-乙烯基硅烷,这是硅烷向炔烃反加成结果(方案3)15。方案3.分子间氢甲硅烷化:内部炔烃重要的是,氢化硅烷化反应表现出了高水平的区域选择性。区域选择性可概括如下:(i)2-炔烃的氢化硅烷化导致Z-烯烃的形成,其中硅烷基占据空间要求较低的位置(1&2);(ii)对于炔烃不在2-位的底物,硅烷取代基将占据空间要求较高的位置(4);(iii)对于炔丙基、高炔丙基和高炔丙醇底物,发生氢化硅烷化,使得硅烷基位于Z-烯烃的羟基官能团的远端(5-9);(iv)在α,β-炔基羰基的情况下,硅烷基再次选择性地占据Z-烯烃的远端位置(10–13)15,16。对于游离炔丙基、高炔丙基和高炔丙醇底物,与带有离去基团(例如乙氧基取代基)的硅烷氢化硅烷化形成环状硅氧烷(5&8)。值得注意的是,使用[Cp*Ru(MeCN)3]PF6的氢化硅烷化可以在保持炔基中不对称中心的立体化学完整性的同时进行(9)。最后,尽管非空间分化的炔烃经历立体但非区域选择性的氢化硅烷化(3),但产物混合物的原脱硅提供了单一的反式非对映体。这对在Lindlar还原条件下观察到的顺式选择性提供了有价值的补充。如方案4所示,即使是高活性硅烷也可以以优异的可预测性参与分子间氢化硅烷化反应。生成的烯基氯硅烷与己二烯醇结合,生成硅氧烷键。加热三烯导致分子内Diels-Alder(IMDA)反应,生成具有四个连续立体中心的硅氧烷。然后可以将加合物进行原脱硅或Tamao–Fleming条件处理以分别提供伯醇或二醇。方案4. 高活性硅烷可以以优异的可预测性参与分子间氢化硅烷化反应。在原脱硅或氧化之前处理烯烃也是可行的。例如,乙烯基硅烷容易被m-CPBA以非对映选择性方式环氧化(方案5)。随后的原脱硅提供了相应的合成环氧醇,而Tamao–Fleming氧化提供了合成二醇。因此,这个过程可以作为羟醛缩合的替代物。方案5.乙烯基硅烷容易被m-CPBA以非对映选择性方式环氧化。分子内部氢化硅烷化最后,使用羟基炔烃可以实现分子内氢化硅烷化,如方案6所示的3-hydroxypiperidine alkaloid (+)-spectaline合成方案17。用四甲基二硅氮烷(TMDS)处理高炔丙醇,然后进行区域(远端)和立体选择性(Z)分子内氢化硅烷化,得到环状叠氮硅氧烷。通过Tamao–Fleming氧化,然后还原并伴随环化,以可观的产率得到该产物。方案6. 3-hydroxypiperidine alkaloid (+)-spectaline的合成分子内氢化硅烷化和随后的交叉偶联为在游离羟基位于远端的炔碳上引入新的碳键提供了一种极好的方法(方案7)18。方案7.游离羟基位于远端的炔碳阿拉丁为您提供[Cp*Ru(MeCN)3]PF6,以及其他多种氢化硅烷化催化剂。参考文献1. Speier, J. L.; Webster, J. A.; Bernes, G. H. J. Am. Chem. Soc. 1957, 79, 974. https://doi.org/10.1021/ja01561a0542. Lewis, L. N.; Sy, K. G. ; Bryant, G. L.; Donahue, P. E. Organometallics 1991, 10, 3750. https://doi.org/10.1021/om00056a0553. Denmark, S. E; Wang, Z. Org. Lett. 2001, 3, 1073. https://doi.org/10.1021/ol01567514. Itami, K.; Mitsudo, K.; Nishino, A.; Yoshida, J. J. Org. Chem. 2002, 67, 2645. https://doi.org/10.1021/jo01633895. Kettler, P. B. Org. Proc. Res. Dev. 2003, 7, 342. https://doi.org/10.1021/op034017o6. Ojima, I.; Kumagai, M. J. Organomet. Chem. 1974, 66, C14. https://doi.org/10.1016/S0022-328X(00)93873-77. Dickers, H. M.; Haszeldine, R. N.; Mather, A. P.; Parish, R. V. J. Organomet. Chem. 1978, 161, 91. https://doi.org/10.1016/S0022-328X(00)80914-68. Takeuchi, R.; Tanouchi, N. J. Chem. Soc., Perkin Trans. 1 1994, 2909. https://doi.org/10.1039/P199400029099. Takeuchi, R.; Nitta, S.; Watanabe, D. J. Org. Chem. 1995, 60, 3045. https://doi.org/10.1021/jo00115a02010. Esteruelas, M. A.; Herrero, J.; Oro, L. A. Organometallics 1993, 12, 2377. https://doi.org/10.1021/om00030a05711. Na, Y.; Chang, S. Org. Lett. 2000, 2, 1887. https://doi.org/10.1021/ol005969712. Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2001, 123, 12726. https://doi.org/10.1021/ja012103313.Trost, B. M.; Ball, Z. T. J. Am. Chem. Soc. 2005, 127, 17644. https://doi.org/10.1021/ja052858014. Trost Barry M.,Ball Zachary T.. Intramolecular Endo-Dig Hydrosilylation Catalyzed by Ruthenium:  Evidence for a New Mechanistic Pathway[J]. J. Am. Chem. Soc.,2002,125(1).https://doi.org/10.1021/ja028766h15. Denmark SE, Pan W. 2002. Org. Lett.. 44163.16. Chung LWea. 2003. J. Am. Chem. Soc.. 12511578.17. Trost BM, Ball ZT, Jöge T. 2002. A Chemoselective Reduction of Alkynes to (E)-Alkenes. J. Am. Chem. Soc.. 124(27):7922-7923. https://doi.org/10.1021/ja026457l18. Trost BM, Machacek MR, Ball ZT. 2003. Ruthenium-Catalyzed Vinylsilane Synthesis and Cross-Coupling as a Selective Approach to Alkenes: Benzyldimethylsilyl as a Robust Vinylmetal Functionality. Org. Lett.. 5(11):1895-1898. https://doi.org/10.1021/ol034463w
企业动态
2023.04.13

锂离子电池正极材料研究进展
引言随着1991年锂离子电池(LIB)的出现,消费电子产品得到了迅速发展[1,2]。很快,LIB成为笔记本电脑和手机制造商的首选电源。当智能手机变得越来越小,必须运行更多的能源需求应用程序时,LIB必须跟上步伐。电池制造商已经开发出更精细的方法来组装电池,从而为消费应用提供高能量密度的现代LIB[3,4]。与此同时,科学家已经开发出更好的活性物质,并在不断改进。本文重点介绍目前研究应用最多的钴酸锂(LiCoO2,LCO),锰酸锂(LiMnO2,LMO),磷酸铁锂(LiFePO4,LFP)以及三元正极材料,概述了这些正极材料最新研究进展。钴酸锂:LiCoO2LiCoO2在1980年由John B. Goodenough教授发现,当时被当做是一种锂离子插层材料。与所有其它正极材料相比,LiCoO2具有许多独特的优点,包括高Li+/电子导电性,高压实密度(4.2 g cm−3),以及优异的循环寿命和可靠性。因此,LiCoO2目前仍是锂离子电池正极的主要材料。LiCoO2具备较高的理论比容量(274 mAhg−1)和高放电电压(约4.2 V vs Li−1/Li)。然而,由于LiCoO2在充电电压超过4.2 V时固有的结构不稳定和界面损失,商业化锂离子电池只能提供理论容量的一半。Yao等人开发了一种简便的吹旋合成方法,实现了LiCoO2颗粒的精确掺杂和同时自组装涂层,在现有的LiCoO2正极中取得了创纪录的性能[5]。由于吹丝微纤维的空间约束效应,每批样品都能实现LiCoO2基体中Mn和La的均匀掺杂,以及LiCoO2表面Li-Ti-O的均匀分离。结果表明,Mn和La共掺杂可以悬浮LiCoO2基体的本向不稳定性,提高Li+扩散率。Ti基涂层可以在充电电压高达4.5 V时稳定LiCoO2颗粒的界面。与之前报道的LiCoO2正极相比,所获得的复合LiCoO2正极具有最佳的倍率性能(在2C时为1.85 mAhcm-2)和在2.04 mAhcm-2面积容量下最长的循环稳定性(在0.3C时保持83%的容量超过300次循环)。图1:纺丝技术的原理图,以制备LCO正极精确复合与Mn和La掺杂和Ti偏析诱导纳米涂层锰酸锂:LiMnO2Xia等人设计了一种尖晶石层状LiMnO2 (SPL-LMO),它具有独特的界面轨道顺序,即相对于方向的近似正交关系畴界面处的MnO6八面体[6]。这种异质结构正极是通过简单的电化学转化Mn3O4在原位实现的。在电化学氧化过程中,有少量氧气非水电解质中Mn3O4的损失导致了反常的尖晶石-层状相变。尖晶石层状异质结构显著降低了Jahn-Teller畸变和Mn溶解,提高了LiMnO2的结构稳定性。这种SPL-LMO正极的可逆比容量高达254.3 mAhg−1,对应约90%的锂离子嵌入/脱嵌到LiMnO2中。在2000次循环后达到了90.4%的高容量保留,表明所提出的界面工程方法对稳定Jahn-Teller活性电极材料的结构非常有效。图2:SPL-LMO和SPL-LMO/Mn3O4的合成。a) Mn3O4纳米壁阵列到SPL-LMO纳米壁阵列的电化学转化(CE:对电极;RE:参比电极;WE:工作电极);b) 粉状Mn3O4电极转化为SPL-LMO/Mn3O4粉状电极Komarneni以纳米棒状MnO2为模板和锰前驱体,采用简单经济的原位碳热还原法制备了正交LiMnO2纳米棒[7]。所得产物纯度高,电化学性能优良。当用作正极材料时,其容量为165.3 mAhg−1,在0.1 C时容量损失较小。经过40次循环后,仅损失7.4%的放电容量。此外,在0.05和1.0 C的低速率和高速率下,最大放电容量分别为227.5和95.3 mAhg−1。同样地,Liu等人采用一步通量法直接制备了介孔正交LiMnO2[8]。通过煅烧不同Li/Mn摩尔比的助熔剂LiOH·H2O和Mn2O3混合制备的正交LiMnO2,得益于其独特的介孔结构,具有较好的储锂性能。当介孔正交LiMnO2用作锂离子电池正极时,在0.1 C的电流密度下循环50次后,其最大放电容量为191.5 mAhg-1,可逆容量为162.6 mAhg-1(保留率为84.9%)。这些结果表明了其在高性能锂离子电池中的潜在应用。磷酸铁锂:LiFePO4Hassoun及其同事报道了一种基于石墨烯油墨正极和磷酸铁锂正极的先进锂离子电池。通过仔细平衡电池各组分含量,并在几轮循环中抑制阳极的初始不可逆容量,该电池的比容量可达165 mAhg−1,估计能量密度约为190Whkg−1,并在超过80个充放电循环下能够稳定运行。该电池的组件成本低,并且具有扩展潜力[9]。图3:磷酸铁锂和石墨烯材料SEM图,TEM图及电池性能图磷酸铁锂材料成本在整个锂离子电池中占比较大,因此,许多学者的研究以回收磷酸铁锂为主要目标,提出了各种回收策略。Wang等人提出了一种将磷酸铁锂电池充电机制与浆液电解工艺相结合的简单、绿色、有效的废旧磷酸铁锂回收方法[10]。阴离子膜液电解可分离Li和FePO4,无需添加化学试剂。Li的浸出率可达98%,96%以上的Fe回收为FePO4/C。动力学分析表明,液浆电解过程中,表面化学反应是控制步骤。此外,还通过X射线衍射(XRD),X射线光电子能谱(XPS),电化学阻抗谱(EIS)表征和热力学浸出机理进行了分析。图4:浆液电解示意图三元材料NCM即镍钴锰酸锂三元材料,由于其电化学性能稳定,循环性能好受到人们的广泛关注。不同NCM成分的原材料成本变化不大。因此,提高镍含量是在相同成本下提高产能的有效方法。在前期工作集中于比较平衡的NCM材料之后,镍线出现了明显的下降趋势现在可以看到图5。虽然NCM523已成为商业现实,但较高的镍含量带来了许多阻碍其成功的问题。1. 高活性的Ni4+将在充电结束时占主导地位,导致与电解质溶液发生副反应。这将最终导致活性物质的消耗以及容量的衰退。此外,随着镍含量的增加,材料的高温稳定性将下降,从而导致严重的安全问题。2. 就像在纯LNO中一样,NCM中镍含量高会导致Li/Ni阳离子混合。这将导致尖晶石在表面形成,并最终导致容量衰减。3. 从NCM811开始,长时间的循环将导致二次颗粒沿晶界出现裂纹。这导致表面积的持续增加,因此失去反应更活跃的位点。5:LNO、LCO和LMO三元体系的相图参考文献[1] Erickson E M, Ghanty C, Aurbach D. New horizons for conventional lithium-ion battery technology[J]. The journal of physical chemistry letters, 2014, 5(19): 3313-3324. https://doi.org/10.1021/jz501387m[2] Nagaura T. Lithium-ion rechargeable battery[J]. Progress in Batteries & Solar Cells, 1990, 9: 209.[3] Schipper F, Aurbach D. A brief review: Past, present and future of lithium-ion batteries[J]. Russian Journal of Electrochemistry, 2016, 52(12): 1095-1121. https://link.springer.com/article/10.1134/S1023193516120120[4] Tarascon J M, Armand M. Issues and challenges facing rechargeable lithium batteries[J]. nature, 2001, 414(6861): 359-367. https://www.nature.com/articles/35104644[5] Tian T, Zhang T W, Yin Y C, et al. Blow-spinning enabled precise doping and coating for improving high-voltage lithium cobalt oxide cathode performance[J]. Nano Letters, 2019, 20(1): 677-685. https://doi.org/10.1021/acs.nanolett.9b04486[6] Zhu X, Meng F, Zhang Q, et al. LiMnO2 cathode stabilized by interfacial orbital ordering for sustainable lithium-ion batteries[J]. Nature Sustainability, 2021, 4(5): 392-401. https://www.nature.com/articles/s41893-020-00660-9[7] Zhao H, Wang J, Wang G, et al. Facile synthesis of orthorhombic LiMnO2 nanorods by in-situ carbothermal reduction: Promising cathode material for Li ion batteries[J]. Ceramics International, 2017, 43(13): 10585-10589. https://doi.org/10.1016/j.ceramint.2017.04.158[8] Tong W, Chu Q, Meng Y, et al. Synthesis of mesoporous orthorhombic LiMnO2 cathode materials via a one-step flux method for high performance lithium-ion batteries[J]. Materials Research Express, 2018, 5(6): 065511. https://iopscience.iop.org/article/10.1088/2053-1591/aac9a2/meta[9] Hassoun J, Bonaccorso F, Agostini M, et al. An advanced lithium-ion battery based on a graphene anode and a lithium iron phosphate cathode[J]. Nano letters, 2014, 14(8): 4901-4906. https://doi.org/10.1021/nl502429m[10] Li Z, Liu D F, Xiong J, et al. Selective recovery of lithium and iron phosphate/carbon from spent lithium iron phosphate cathode material by anionic membrane slurry electrolysis[J]. Waste management, 2020, 107: 1-8. https://doi.org/10.1016/j.wasman.2020.03.017
应用实例
2023.04.11

阿拉丁2022年度优秀员工表彰大会
2022年公司所取得的每一点进步和成就,离不开全体员工的辛勤劳作和无私奉献!为增强企业凝聚力,表彰先进树立楷模,激励员工奋发向上。2023年3月31日,上海阿拉丁生化科技股份有限公司隆重举行了2022年度优秀员工表彰大会,金桥总部的同事们现场参与,其他奉贤研发基地及张江生物研发中心的同事们通过视频会议线上参与。这是一场庆祝全体员工共同努力的盛会,也是对于那些在过去的一年中,为公司做出卓越贡献的员工们的表彰和肯定。 表彰大会在公司领导的致辞中正式开始。主持人向全体员工介绍了今年评定的人员基本来源于公司的基层,他们为公司的发展注入了活力,夯实了基础。所谓长江后浪推前浪,每年的表彰台上都涌现出一大批优秀员工,他们兢兢业业、埋头苦干,在各自的工作岗位上坚守付出。阿拉丁因他们而壮大,他们也因阿拉丁而自豪!主持人逐一宣布了获奖者的名单。各位优秀员工在此次大会上受到了高度肯定和充分表彰,这也是对公司价值观“和谐公正、仁爱共享、真诚团结、感恩敬业”的最好诠释。面对着掌声和赞美,每位获奖员工或代表上台领取荣誉奖牌和奖品,他们的脸上洋溢着满足和自豪。 图一:金桥总部颁奖现场 此次表彰大会不仅对员工们的辛勤工作加以肯定和赞扬,更展示了我们团结和谐、有凝聚力的企业形象。在这个特别的时刻,除了表彰,还有优秀员工代表发言。经验丰富的优秀员工代表们讲述了自己在工作和个人成长上的故事和感受,分享了在公司成长的历程和心得,也感恩公司的支持和栽培。对于未来,他们将会更加努力的工作,在个人与公司的关系之间寻求平衡,争取更大的进步。 随后,阿拉丁董事长兼总经理徐久振先生发表致辞,他围绕着公司战略、客户服务、营销策略等话题,与大家共同探讨企业发展的理念,为员工提供了更加详实和深刻的指导,旨在帮助大家改进工作方法、提升创新能力。同时,他向员工们表达了感激之情,并鼓励年轻的管理人才抓住机会,追求卓越。 图二:董事长兼总经理徐久振先生致辞 最后,在参会员工的热烈掌声中,大家共同合影留念,记录下这个难忘的时刻,为今后持续奋斗注入了新的动力。 图三:金桥总部获奖员工与领导合影 图四:奉贤研发基地获奖员工合影 通过此次年度优秀员工表彰大会,阿拉丁再次向员工们传递了激励、鼓舞与关爱,展现了公司对先进员工的认可与赞赏。此次盛会也展示了公司作为一个有凝聚力的团队,其所拥有的文化底蕴和人才实力。我们有理由相信,在未来的日子里,公司和全体员工将会携手并进,再创辉煌!
企业动态
2023.04.07

锂电池隔膜常见改性策略
引言锂电池具有能量密度高、重量轻、灵活性强、自放电速率慢、充电速率高、电池寿命长等优点,是一种集可再生资源和大功率应用于一体的极具发展前景的储能设备[1]。隔膜是可充电锂电池中最重要的部件之一,它不仅能在正负极之间提供物理屏障,防止电路短路,还可以作为锂离子和电解质阴离子的主要传输路径,通过调节其组成和结构,可以达到改善电池的离子传输特性的目的。尽管它们具有良好的化学/电化学稳定性,优异的机械强度和适当的热关闭性能,但最广泛商业化的聚烯烃基(如聚乙烯(PE)(项目号:P434353),聚丙烯(PP)(项目号:P301642)和PP/PE/PP夹层复合材料)微孔隔膜存在热收缩严重、电解质润湿性差、孔隙率低、易燃等问题,不可避免地会引起安全隐患,从而影响锂电池的电化学性能。[2-4]隔膜结构优化采用相转化法和静电纺丝法制备隔膜时,通过调控前驱体和实验参数,可以优化隔膜结构,达到提高隔膜离子传输速率的目的。相转化法是将聚合物溶解在溶剂之中,并通过溶剂交换使聚合物沉淀形成微孔结构[4]。这种方法避免了干法和湿法中的拉伸工艺,可有效防止隔膜多孔结构的热收缩。Wang等人通过相转化法制备了一种海绵状聚磺胺(PSA)/SiO2复合膜,并成功应用于锂离子电池(LIBs)隔膜[5]。与商用聚丙烯(PP)隔膜相比,海绵状PSA/SiO2复合材料具有更好的物理和电化学性能,如更高的孔隙率、离子导电性、热稳定性和阻燃能力。使用海绵状复合隔膜的LiCoO2/Li半电池比使用商用PP隔膜的LiCoO2/Li半电池表现出更好的速率能力和循环性能。此外,海绵状复合隔膜可以保证LiCoO2/Li半电池在90°C的极高温度下正常工作。Ma等人采用静电纺丝-热压法制备了9种不同纤维直径、不同膜孔率的聚丙烯腈(PAN)(项目号:P303200)纳米纤维膜[6]。随后,这些膜被探索作为锂离子电池(LIB)隔膜。研究了在阴极半电池充放电循环和速率能力测试中,纤维直径和膜孔隙率对电解质吸收、锂离子通过膜的传输、电化学氧化电位和作为锂离子隔膜的性能的影响。结果表明,PAN基隔膜具有较小的纤维直径:200-300nm,并且在高压(超过20兆帕)下表现出最佳性能,其最高放电容量(89.5mAh/g-1在C/2速率下)半电池的循环寿命(容量保持率为97.7%)。该研究揭示了热压静电纺丝PAN纳米纤维膜(特别是由薄纳米纤维组成的PAN纳米纤维膜)作为高性能锂离子分离材料的前景。图1:PAN基隔膜在C/2速率下的初始充放电电压分布极性基团接枝锂金属电池性能一直受到锂析出的影响。Wu等人通过简单的涂层聚丙烯酰胺接枝氧化石墨烯(项目号:G405797)分子到商用聚丙烯膜上,开发出了功能化多孔双层复合隔膜[7]。该复合隔膜将毛状聚丙烯酰胺链的亲锂特性和快速电解质扩散途径与氧化石墨烯纳米片的优异机械强度结合起来,从而提高了电极表面的锂离子通量。结果显示,在高电流密度(2mA cm−2)下,锂金属阳极实现了无枝晶的锂沉积,具有高库仑效率(98%)和超长期可逆的锂电镀/剥离(超过2600 h)。值得注意的是,在20mA cm−2的超高电流密度下,锂金属阳极具有超过1900小时的循环稳定性。图2:PP隔膜和GO-g-PAM改性PP隔膜分别组装的电池电极上锂沉积示意图无机粒子涂覆隔膜对锂离子电池的安全性起着至关重要的作用。然而,目前商品化的隔膜主要是基于微孔聚烯烃膜,存在严重的安全风险,如热稳定性。虽然已经做出了很多努力来解决这些问题,但是还不能完全确保电池的安全性,特别是在大规模应用的情景中。Zhao等人通过对聚多巴胺(PDA)进行简单的浸涂工艺,在陶瓷层和原始聚烯烃膜上形成了一层整体覆盖的自支撑膜,使陶瓷层和聚烯烃膜呈现为单一的一面,并进一步改善了隔膜的成膜性能[8]。同时,该隔膜也具备较好的耐热特性。图3:通过简单的浸涂工艺制备复合改性分离器示意图图4:(a)原始PE隔膜、PE-SiO2隔膜和PE-SiO2@PDA隔膜在170℃热处理前后的热收缩率随温度的变化图;(b)原始PE隔膜热处理前后对比图;(c)PE-SiO2隔膜热处理前后对比图;(d)PE-SiO2@PDA隔膜热处理前后对比图;(e) PE-SiO2@PDA隔膜在220℃处热处理前后对比图。聚合物改性Cui等人以贻贝为载体的聚多巴胺包覆层对环保型纤维素微纤维进行改性,采用简便、经济的造纸工艺制备了纤维素/聚多巴胺(CPD)膜[9]。结果表明,CPD膜具有致密的多孔结构、优良的机械强度和良好的热尺寸稳定性,可作为锂离子电池的隔膜。进一步地,采用CPD分离机的电池的交流阻抗在第100个循环后出现了9Ω的微小变化,表明电池具有良好的界面稳定性。这些优异的性能使CPD膜作为高性能锂离子电池隔膜具有广阔的应用前景。图5:(a)采用隔膜、纤维素隔膜和CPD隔膜的LiCoO2/石墨电池的循环性能,(b)速率性能和(c)第一个周期和(d)之后测量的电池Nyquist图锂金属阳极循环稳定性差和安全性问题是阻碍高能量密度锂金属电池商业化的两个主要问题。对此,Leif Nyholm提出了一种新型的三层式隔膜设计,显著提高了锂金属基电池的循环稳定性和安全性[10]。纳米纤维素层平均孔径在20 nm左右,介孔厚度为2.5μm,这为Li+的流动提供了较好的通道,从而稳定了锂金属阳极,提高了循环稳定性。由于三层式隔膜在内部PE层熔化时,即使在200°C也能保持尺寸稳定,并阻断离子通过隔膜的运输,因此隔膜在高温条件下还具备较好的安全性。该纳米纤维素基三层隔膜有望极大地促高能量密度锂金属基电池的实现。 图6:隔膜的孔隙分布对锂电极形态影响的示意图参考文献[1] Ma H, Liu J, Hua H, et al. Facile fabrication of functionalized separators for lithium-ion batteries with ionic conduction path modifications via the γ-ray co-irradiation grafting process[J]. ACS Applied Materials & Interfaces, 2021, 13(23): 27663https://doi.org/10.1021/acsami.1c06460[2] S.S. Zhang, A review on the separators of liquid electrolyte Li-ion batteries. J.Power Sources, 164(1) (2007) 351-364.https://doi.org/10.1016/j.jpowsour.2006.10.065[3] J.H. Parka, J.H. Choa, W. Parka, D. Ryoob, S.J. Yoonc, J.H. Kimc,Y.U. Jeongd,S.Y. Lee, Close-packed SiO2/poly (methyl methacrylate) binary nanoparticles-coated polyethylene separators for lithium-ion batteries. J. Power Sources, 195(24) (2010) 8306-8310. https://doi.org/10.1016/j.jpowsour.2010.06.112[4] Yuan B, Wen K, Chen D, et al. Composite separators for robust high rate lithium ion batteries[J]. Advanced Functional Materials, 2021, 31(32): 2101420. https://doi.org/10.1002/adfm.202101420[5] Wang X, Xu G, Wang Q, et al. A phase inversion-based sponge-like poly-sulfonamide/SiO2 composite separator for high performance lithium-ion batteries[J]. Chinese journal of chemical engineering, 2018, 26(6): 1292-1299. https://doi.org/10.1016/j.cjche.2017.12.010[6] Ma X, Kolla P, Yang R, et al. Electrospun polyacrylonitrile nanofibrous membranes with varied fiber diameters and different membrane porosities as lithium-ion battery separators[J]. Electrochimica Acta, 2017, 236: 417-423. https://doi.org/10.1016/j.electacta.2017.03.205[7] Li C, Liu S, Shi C, et al. Two-dimensional molecular brush-functionalized porous bilayer composite separators toward ultra-stable high-current density lithium metal anodes[J]. Nature communications, 2019, 10(1): 1363. https://www.nature.com/articles/s41467-019-09211-z[8] Dai J, Shi C, Li C, et al. A rational design of separator with substantially enhanced thermal features for lithium-ion batteries by the polydopamine–ceramic composite modification of polyolefin membranes[J]. Energy & Environmental Science, 2016, 9(10): 3252-3261. https://doi.org/10.1039/C6EE01219A[9] Xu Q, Kong Q, Liu Z, et al. Polydopamine-coated cellulose micro fibrillated membrane as highly performance lithium-ion battery separator[J]. Rsc Advances, 2014, 4(16): 7845-7850. https://doi.org/10.1039/C3RA45879B[10] Pan R, Xu X, Sun R, et al. Nanocellulose modified polyethylene separators for lithium metal batteries[J]. Small, 2018, 14(21): 1704371. https://doi.org/10.1002/smll.201704371
应用实例
2023.04.06

小分子抑制剂选择指南
小分子抑制剂是了解蛋白质功能及其在正常生理和疾病病理中的作用最容易获得和最通用的工具之一。为了在询问生物过程或验证新目标方面有价值,这些工具必须满足提供可靠和可重复数据的必要标准。由于有时很难从大量可用的分子中确定最合适的抑制剂,我们收集了一系列在选择过程中应该考虑的参数。 当你在研究中考虑使用每一种新的小分子时,必须注意它的1)化学性质,2)效力,3)选择性和4)使用环境。最好查阅科学文献,了解您所在领域的成熟工具以及某些化合物可能存在的优点和缺点(例如,细胞毒性或脱靶效应)。此外,对于每个产品阿拉丁提供相关的详细生物信息,以帮助您为您的实验选择正确的化合物。通常对于更新颖或相对未鉴定的抑制剂,最好自己进行初步测试,以了解化合物在实验系统中如何反应。虽然并非所有化合物在每种情况下都需要达到以下建议的阈值,但考虑这些参数将帮助您确定探针是否适合您的目的。 化学性质 结构 具有明确的化学结构,其合成方法应该是可复制的。避免常见的毒性部分和pan试验干扰部分(PAINS)。同样避免化学反应基团,除非需要,例如共价加成。 稳定性 应在相关介质中保持稳定性(纯度和化学特性),并注意pH敏感性。活动应保留在文化媒体。分子不应表现出非特异性的化学反应性(例如,氧化还原反应或膜不稳定)。 溶解度 在水介质中的溶解度应足够(例如,IC50的10倍或低% DMSO中的0.05 μg/ml)。溶解度和亲脂性都需要平衡。高电荷,高可溶性化合物可能显示低细胞或组织渗透性。疏水化合物可能表现出高渗透性和效力,同时存在溶解度问题。使用盐形式的疏水分子应提高水溶解度。 渗透性 通过被动或主动运输的渗透性对于细胞活性是必不可少的。血脑屏障穿透对于预期的中枢神经系统(CNS)效果很重要。Caco-2细胞渗透性测定或平行人工膜渗透性测定(PAMPAs)是评估被动扩散的常用测定方法。MDCK-MDR1渗透性可用于评估中枢神经系统渗透,包括p -糖蛋白(PGP)的主动外排。 效力 IC50 和 Ki IC50和Ki是表示抑制剂效力的最常用方法。在酶抑制的背景下,IC50表示在给定的实验条件下将酶反应速率降低50%所需的抑制剂的浓度。Ki表示抑制剂-目标复合物分解(koff)与抑制剂-目标复合物形成(kon)的比值,用于抑制剂与酶的结合。术语Ki是一个热力学平衡常数,因此是一个固定值。另一方面,IC50是在一组确定的条件下的抑制作用的表示,并将根据反应环境中存在的底物浓度等因素而变化。对于竞争抑制剂,IC50可以通过Cheng-Prusoff方程与Ki相关:IC50=Ki*(1+[S]0/Km)。这是一个有用的基于网络的工具,可以使用不同的抑制剂结合模型将IC50值转换为Ki值。 注意,对于酶以外的蛋白质(如G蛋白偶联受体或离子通道),拮抗剂的功效也可以通过Ki或IC50来报告。在GPCRs的情况下,IC50(或通常EC50)是细胞反应抑制的指示(即cAMP浓度的降低),而Ki仅指与天然配体结合在受体上。 体外试验 体外药效基准标准通常为IC50或Ki值在生化测定中为100 nM,在细胞测定中为1-10 μM。酶测定的效力应与细胞中的效力相关。使用尽可能低的浓度,以避免脱靶效应。仅在浓度为10 μM时对细胞有效的抑制剂可能是非特异性靶向蛋白质。 体内试验 体内效力基准标准应达到目标组织中与细胞效力相关的水平。必须注意与化合物的吸收、溶解、代谢或排泄有关的影响。代谢稳定性是关键问题,如果该化合物将推进动物研究。 浓度依赖性 当使用低到高范围的推荐浓度进行实验时,剂量依赖性活性应很明显。在饱和浓度下对目标的近乎完全抑制证实了该化合物的功效。 构效关系 构效关系(SAR)分析可以解释一系列具有不同活性的化合物与一种酶或受体的结合,从而了解目标蛋白结合的要求、允许量和限制。SAR系列覆盖3-4个效能数量级是很重要的。一个“平坦”的合成孔径(SAR),即包含巨大结构差异的整个系列化合物都具有近乎等效的弱效力,可以表明给目标下药的难度增加。 选择性 数据剖析 分析将定义对相关目标的选择性,这通常是一个比效力更关键的因素。通常情况下,在生化分析中,靶标的效力比其他家族成员强10-100倍的因素定义了一种抑制剂对该靶标的选择性。针对你感兴趣的目标设计的选择性抑制剂仍然可以在更高浓度下结合其他蛋白质。重要的是要了解任何与特定化学类相关的额外活动。 阴性对照 可以设计阴性对照实验,以证明该抑制剂在用于抑制所需靶标的浓度下,不会有效地改变任何脱靶蛋白的功能。经过充分考虑的阴性对照,如仅将细胞或蛋白质暴露于溶剂或替换密切相关的非活性结构类似物,如R/S立体异构体,有助于确认抑制剂的效果。 阳性对照 阳性对照实验可用于证明在效果不明显的情况下,抑制剂的行为符合预期。对照组不接受实验治疗,而是接受已知能产生预期效果的其他治疗。 正交探测器 具有相似活性但来自不同化学型和/或作用方式的正交探针也可考虑用作附加对照。如果正交探针产生类似的反应,则更有可能是对靶标的抑制导致了所观察到的表型。如果没有,那么混淆脱靶或毒性作用可能是由抑制剂引起的。 使用环境 作用机理 作用机制对于进一步定义抑制剂的性质非常重要,以便确定天然底物在生理浓度下如何调节抑制剂的功效,并确定与该机制相关的任何漏洞。潜在的抑制事件包括: 不可逆抑制-抑制剂 不可逆抑制-抑制剂通常通过共价附着结合酶,不可逆地使酶失活。虽然不可逆抑制剂通常是共价的,但非共价抑制剂有时可以很长时间地发挥不可逆抑制剂的作用。 可逆抑制剂 ● 可逆抑制可以分解为以下几种:● 竞争抑制-抑制剂和底物竞争自由酶,但每一个都阻碍了对方的结合,因为这种结合事件通常发生在目标的活性位点(正位位点),正是底物也结合的地方。● 非竞争性抑制-抑制剂与游离酶和酶-底物复合物结合得同样好。虽然非竞争性抑制通常发生在变构位点,但非竞争性抑制也可以在结合正构位点时发生,通常在活性位点是双底物位点的情况下(酶与一种底物竞争,但与另一种底物不竞争)。● 非竞争性抑制-抑制剂只能与酶-底物复合物结合,形成无活性的可逆三元复合物。非竞争性抑制是混合抑制的一种特殊情况。● 混合抑制-抑制剂与酶的结合在自由酶和酶-底物复合物中不同。这种类型的抑制剂可能表现出对一种状态(非竞争性抑制)或另一种状态(竞争性抑制)的更大亲和力。● 变构抑制-抑制剂结合到靶上的变构位点而不是活性位点,引起靶酶的构象变化,这是抑制所必需的。这些构象变化会影响通常酶-底物活性位点复合物的形成,破坏过渡态的稳定,或降低降低催化活化能的能力。● 部分抑制-酶-底物-抑制剂相互作用产生的复合物比酶-底物复合物的周转率低。部分活性仍然存在,因为酶-底物-抑制剂复合物的催化中心可能保留一些在底物附近排列和促进催化的能力。● 紧密结合抑制-最初的酶抑制剂复合物经历异构化形成第二个更紧密的复合物。紧密结合抑制剂倾向于显示非竞争性表型,即使它们可以以竞争性、非竞争性或非竞争性的方式与目标酶结合。因为这可以表现为缓慢增加的酶抑制,这是时间依赖的,传统的Michaelis-Menten动力学将给出一个错误的Ki值。通过分析kon和koff速率常数,可以得到更真实的Ki值。● 时间依赖性抑制-抑制剂在酶周转的时间尺度上缓慢地与酶结合,这具有减缓观察到的抑制发作的效果。这可能导致催化剂速率常数(kcat)值变慢。 抑制物-靶点动力学 抑制物-靶点动力学,包括kon、停留时间和koff,为化合物的效力和选择性增加了额外的维度。与靶标缓慢解离的化合物在较低浓度时可能具有较长的活性,从而使剂量水平或剂量频率降低。通常停留时间比热力学效力更能与体内活性相关。 靶标易损性 目标脆弱性是观察到效果所需的目标参与的最低水平的函数。高易损性靶标需要较低水平的靶标占用(即较低水平的抑制剂暴露)才能达到预期效果。以细胞为基础的冲洗实验可以通过帮助确定一旦抑制剂从系统中移除,目标接触的表型后果,从而提供对目标脆弱性的深入了解。 生理环境 必须考虑目标的生理环境和目标交战的下游后果。例如,与该途径中的其他酶相比,破坏一种催化代谢途径中限速步骤的酶可能会产生更大的后果。在抗菌或抗癌的背景下,抑制生化途径的最后步骤,高能量/高成本生化产品的下游,可能被证明对目标细胞特别有毒。 时间范围 在设计试验时,应考虑预期表型效应或触发信号事件级联所需的时间范围,以相应地捕获这一时间长度。 补充实验 使用可用的RNAi或突变体的互补实验,在可用时,有助于就生物系统中给定目标的作用建立共识。 适应性 化合物对目标的应用适应性最终取决于它与所研究假设的生物学背景的联系程度,因为在一个生物系统中使用抑制剂不一定能推断出另一个生物系统。一种适合目的的抑制剂既适用于靶标,也适用于更广泛的科学背景。 可得性 该化合物的可用性是广泛的,并且可以获得后续使用的数量。 探索来自阿拉丁的超过38,000种小分子抑制剂https://www.aladdin-e.com/zh_cn/bioactive-small-molecules.html同时并提供丰富的化合物库用于药物筛选https://www.aladdin-e.com/zh_cn/bioactive-small-molecules/compound-libraries.html
应用实例
2023.04.04
企业名称: 上海阿拉丁生化科技股份有限公司
企业地址: 上海市新金桥路36号财富中心16楼 联系人: 阿拉丁客服 邮编: 201206 联系电话: 400-860-5168转3457
仪器信息网APP

展位手机站