
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转3328
仪器信息网认证电话,请放心拨打

糖是“毒-品”,那么引发对糖的渴求和消费的脑细胞是什么?
大多数人时不时地享受甜食。但是,不加控制的“甜食”会导致对含糖食物的过度消费和慢性健康问题,如肥胖和2型糖尿病。了解控制糖摄入量和甜味偏好的生物学机制对管理和预防这些健康问题具有重要意义。这项新的研究由爱荷华大学医学院神经科学和药理学副教授Matthew Potthoff博士和丹麦哥本哈根大学的Matthew Gillum博士领导,重点研究了一种名为成纤维细胞生长因子21(FGF21)的激素作用。此前据悉,这种激素在能量平衡、体重控制和胰岛素敏感性中起作用。“这是第1个真正确定这种激素在大脑中的作用的研究,它提供了一些非常酷的见解,它是如何调节糖的摄入量的,”Potthoff说。Potthoff和他的同事们先前发现,FGF21是在肝脏中随着糖含量的增加而产生的,它在大脑中起着抑制糖分摄入和甜味偏好的作用。在这一发现的基础上,研究小组现在首次展示了哪些脑细胞对FGF21的信号作出反应,以及这种相互作用如何帮助调节糖的摄入量和甜味偏好。这项发表在《Cell Metabolism》杂志上的研究也揭示了激素如何调节其作用。虽然已知FGF21在大脑中起作用,但由于激素受体的表达水平很低,很难“看清”,因此确定确切的细胞靶点很复杂。利用各种技术,研究人员现在能够精-确识别哪些细胞表达FGF21的受体。通过对这些细胞的研究,表明FGF21靶向大脑中的谷氨酸能神经元,以降低糖的摄入量和甜味偏好。研究人员还表明,FGF21作用于下丘脑腹内侧部的特定神经元,通过增强神经元对葡萄糖的敏感性来减少糖的摄入。一些基于FGF21的改良形式的药物已经被用于治疗肥胖和糖尿病。这项新发现有可能开发出更精-确地针对FGF21控制的不同行为的新药,这可能有助于控制一个人的食糖量。
厂商
2020.07.16

“秃如其来”的希望,干细胞技术让人造皮肤长出毛发
皮肤是一种复杂的多层器官。此前,利用干细胞技术只能获得特定类型的皮肤细胞,无法再生出包含毛-囊、汗腺等附属结构的完整皮肤。而《自然》杂志报道的最-新研究,首次在体外培育出包含这些附属结构的皮肤类器官,并在移植到小鼠背部后长出了2—5毫米的毛发。人的皮肤结构复杂而且极为重要。目前,人们在临床应用的皮肤替代品,只能实现表皮层和简单真皮层的结构性修复,并不包含毛-囊、汗腺、皮脂腺等皮肤附属结构,更难以重建皮肤的体温调节等各项功能。因此,长期以来,重建皮肤及其相关结构是生物医学界的重大挑战之一。不久前,学术期刊《自然》发表重磅论文,介绍人造皮肤领域的新突破——美国哈佛医学院耳鼻喉科助理教授卡尔·科勒,以及研究助理李智运(音译)等人报告,他们在仔细优化类器官培养系统生长条件后,利用人类诱导多能干细胞,培养出能生长毛发的皮肤类器官。最-新突破 再生出毛-囊和神经等附属结构皮肤是一种复杂的多层器官,由多种不同细胞类型构成,包括角质细胞、毛-囊、黑色素细胞、汗腺、神经、肌肉、脂肪、免疫细胞和表皮细胞等。其中真皮含有成纤维细胞、肥大细胞、黑色素细胞和朗格汉斯细胞等。成纤维细胞能产生胶原和弹力纤维等基质,而真皮中又密布着血管、神经、毛-囊、汗腺和皮脂腺等附属结构。皮肤还参与人的体温调节、体液维持、触觉及疼痛感知等各种过程。因此,构建具有附属结构再生性能的新型皮肤替代物具有重要的意义。昆明理工大学灵长类转化医学研究院李天晴教授向科技日报记者介绍,此前,利用人类诱导多能干细胞分化为皮肤特定类型的细胞取得了成功,然而再生出非常完整的皮肤结构,仍在探索中。美国这项新研究采用逐步调节转化生长因子β(TGFβ)和成纤维细胞生长因子(FGF)信号通路的方法,将聚集成球的多能干细胞诱导生成了颅骨上皮细胞和神经嵴细胞。在随后的4至5个月内,球体内的细胞产生皮肤类器官,这些类器官由分层的表皮细胞、富含脂肪的真皮和带有皮脂腺的色素毛-囊组成。与毛-囊相关的神经及其受体、肌肉和脂肪组织也开始生成,最终形成了非常完整的皮肤。“这是第一个将人类诱导多能干细胞分化为完整结构的皮肤类器官,因此,也是诱导多能干细胞再生组织器官的一个重大突破。”李天晴说。在谈到此项研究的创新点时,山东大学干细胞与再生医学研究中心李栋教授说,李智运等人的创新,首先在于细胞间诱导分化模式。他们在仔细优化生长条件后,利用人类诱导多能干细胞,不但形成了多层皮肤组织,而且还包含毛-囊、皮脂腺、脂肪组织和神经等多种附属结构,这是其最主要的突破点。3D培养模式 调控组织细胞分化方向人类诱导多能干细胞是一类具有自我更新、快速增殖和多向分化能力的多潜能细胞。“从某种意义上说,只要给它一定的诱导分化信号,它可以分化成体内所有的细胞类型。”李栋介绍,由于人体就是由细胞和细胞分泌物构成的,所以从理论上讲,人类诱导多能干细胞可以修复任何一种组织细胞损伤。虽然目前普遍认为人类诱导多能干细胞尚不能发育成一个完整的个体,但也足以模拟一些组织或器官的发生过程。因此,人类诱导多能干细胞可以直接在定向诱导为某几种细胞之后,用于组织损伤的修复和器官再生,也可以用于模拟形成皮肤、肝脏等复杂的组织器官,进行功能替代、药物筛选或是靶向肿瘤治疗的研发。最-新研究利用来自干细胞生物学和毛-囊发育领域的技术,来产生接近功能完整的皮肤类器官,并在实验室中完成模仿皮肤的生长发育过程。此前,已有科学家培养出包括肠、肺、肾和脑的各种类器官。而干细胞包括胚胎干细胞和诱导多能干细胞,后者是通过将成年细胞重编程为类胚胎状态而产生的,因此具有形成类器官所需的所有成年细胞类型的能力。李栋介绍,李智运等人利用类器官3D培养模式,诱导出多种细胞类型,结合基质胶(matrigel,由多种细胞胶质蛋白组成的混合物)三维环境,促进细胞极性排列,让细胞间实现通信互动。他们先诱导颅神经嵴细胞(CNCC)的形成,后续采用旋转漂浮培养技术,控制CNCC和表皮细胞间相互影响、诱导和反馈,在类器官中形成具有突出毛-囊的胞囊,协作发育并最终形成功能性皮肤组织,模拟了皮肤组织间相互诱导分化的发育过程。此项研究的另一个重要创新,在于皮肤组织培养的多个关键节点上。他们有序地采用了多种因子和信号通路抑制剂,并精密控制气液界面培养的介入时机,调控组织细胞的分化方向。“这一培养体系也可用于其他组织器官的再生。”李天晴说。“毛-囊和汗腺等皮肤附属结构精细,形态微小,分离纯化过程复杂,制约了人工皮肤领域的研发进展。而这项研究,直接利用诱导多能干细胞分化形成皮肤附属结构中的各种组织细胞,如果能在组织支架材料上得以放大这种培养工艺,将更具产业化价值。”李栋说。仍处于初级阶段 朝皮肤重建和毛发移植迈出重要一步皮肤类器官在用于皮肤烧伤的疤-痕体质患者的皮肤重建以及头发移植等方面,具有潜在的重要价值。《自然》杂志特别邀请美国宾夕法尼亚大学医学院皮肤科主任乔治·科萨雷利斯教授等就这一成果撰写了新闻与观点文章。他们认为,这一成果使人们离“产生无限量的毛-囊更进了一步”,这些毛-囊可以“移植到毛发稀疏或脱落的头皮上”。“此外,如果这一方法应用于临床,那些有伤口、疤-痕和遗传性皮肤病的人将有机会获得革命性的治疗。”“新的研究所形成的细胞诱导顺序、培养试剂体系和方法,如果能够得到重复,而且解决工艺规模化等问题,不但可以用于临床治疗脱发等难题,还可以用于构建含有附属结构的复合皮肤,直接应用于临床。”李栋认为,因为科勒等人将培养的皮肤类器官-移植到免疫缺陷小鼠背部后,55%的移植物上都长出了2—5毫米的毛发,所以这种技术可直接用于毛发移植治疗。同样这个体外培养体系也可以用于筛选脱发治疗的药物,或用于加速遗传性皮肤病和皮肤癌症的药物筛选等研究。论文作者还发现,他们的类器官在基因表达上具有多个部位皮肤的特征。这样通过改变细胞生长的培养条件,可以定制化地生成具有不同身体部位特征的皮肤,用于治疗重度烧烫伤以及机械外伤导致的大面积皮肤损伤。当然,此项研究还处于初期阶段,产生的皮肤类器官缺乏免疫细胞,而且这些结构能否在高等动物以及人体上实现皮肤或毛发的再生仍是未知。“因此,这些移植的皮肤是否具有足够的安全性以及能否像正常体内皮肤一样具有循环生长的能力,都需要去探索。”李天晴说。另外,类皮肤器官产生、移植需要140天的时间,也可能会影响治疗的潜力——例如,烧伤患者不能等那么久才进行植皮手术。未来的研究,应提供策略来加速这个过程,使用分子改变相关的信号通路。但学界仍普遍认为,李智运等的研究是朝着治愈人类脱发迈出的重要一步,并为其他更多类型的治疗提供了可能性。李智运接受媒体采访时表示:“我们的结果表明,新技术在体外可自我组装成几乎完整的皮肤,并可以在体内重建皮肤。我们预计这种皮肤类器官将为人类皮肤发育、疾病建模和重建手术等研究奠定重要基础。”
厂商
2020.07.15

抗生素检定用芽孢悬液的制备及注意事项
管碟法是国内外常用的抗生素微生物检定法,利用管碟法测定抗生素效价,具有准确、直观、重复性好等优点,因而被广泛采用。但是整个试验过程中影响结果的因素很多,任何一个环节操作不当或者忽略就会造成很大误差,导致整组试验失败。其中,芽孢悬液的制备就有很多需要注意的地方。在用管碟法检定抗生素时,需要将菌悬液与培养基混合均匀、覆盖在底层培养基上,以形成抑菌圈。其中,对于菌悬液的制备要求为:将所用菌的斜面培养物用 1~2 ml 灭菌水洗下,形成菌悬液;将菌悬液在营养琼脂平皿上均匀涂布,置于 35~37℃ 条件下培养 7 天;涂片、染色,镜检芽孢率 ≥ 85% 时,用 10 ml 灭菌水将芽孢和菌体的混合物洗下,制成悬液;65℃ 水浴加热 30 min,杀死菌体,形成有活性的芽孢悬液;冷却、冰箱中保存备用。根据实际操作中的经验,对这一环节需要注意的事项总结如下:1 应该严格按照标准操作,将涂好的平皿连续培养 7 天再洗脱芽孢,这样也有助于芽孢悬液的均一性,避免由于操作标准不一造成的试验结果的偏差。有文章报道,将平板培养至 2、3、4、5、6 天与培养至第七天,制成的悬液在抗生素效价测定中无差异;但本人在实际试验中发现,培养 3 天的短小芽孢菌,镜检时芽孢率2 试验菌的菌龄对抑菌圈有一定影响。故检定时应保持菌种及菌液的新鲜。一般菌种一月转种一次,冰箱冷藏保存。对易变异的菌株,如藤黄微球菌等在制备菌悬液前进行单菌分离;其他菌株可半年分离一次。3 试验菌传代最好不超过 5 次,以防止菌种老化变异。芽孢杆菌培养物用灭菌水洗下后,应在 65℃ 加热 30 分钟,使菌体的菌龄一致(有资料显示可 70~75℃ 水浴 30 min 杀死菌体)。4 加入菌悬液的体积,一般不少于 0.3 ml,并且不大于上层培养基体积的 1%(有资料说不大于 2%)。如果加入菌悬液的体积过小,则在同次实验中,不同次加入菌悬液的体积相差较大, 造成实验误差;如果加入菌悬液的体积过大,则会使上层培养基变稀,并且因菌悬液的温度较低,加至培养基中可能造成培养基结块,影响测定结果。对于加入菌悬液体积的控制,可通过调节菌悬液的浓度来实现。
厂商
2020.07.15
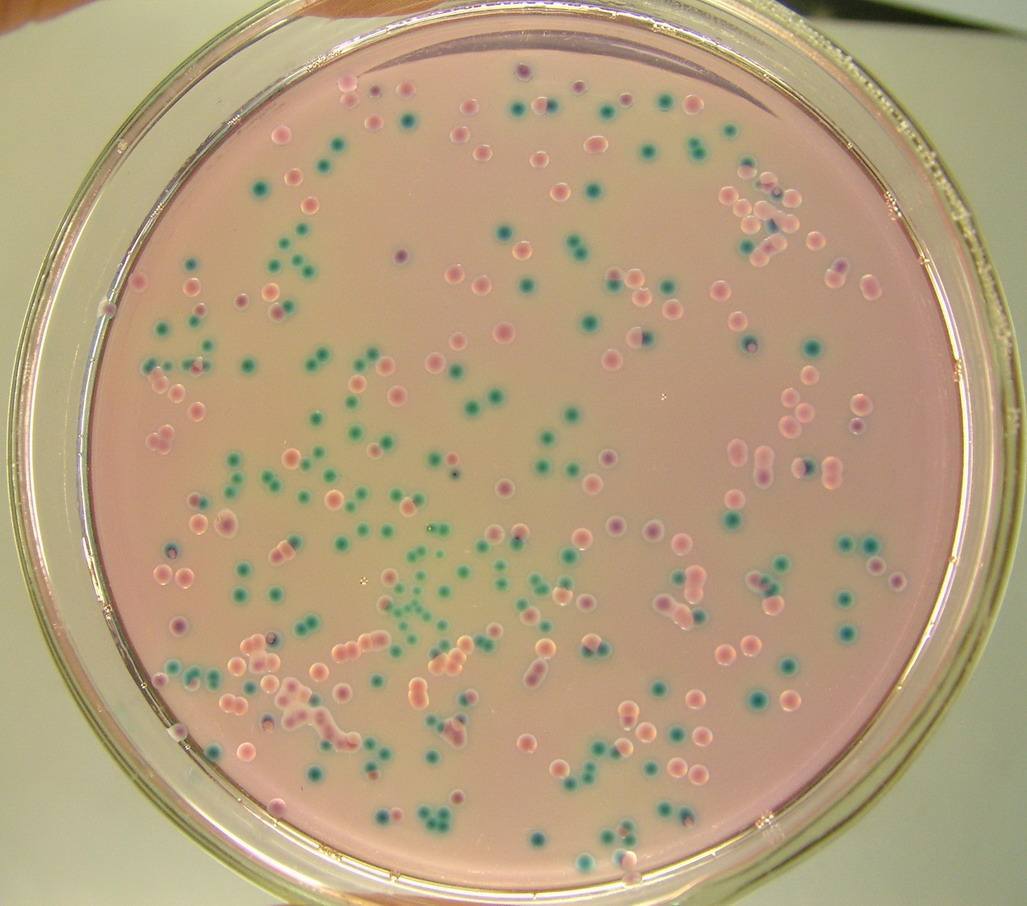
人类皮肤细菌具有抗癌能力
某些居住在皮肤上的微生物可能是抗癌超级英雄,抑制细胞不受控制的生长。这一惊人的发现有一天可能会导致治疗甚至预防皮肤癌的药物问世。这种细菌的秘密武器是一种化学化合物,可以阻止DNA在其轨道上形成。研究人员2月28日在《科学进展》(Science Advances)网络版上发表了一篇研究报告。研究人员在老鼠身上涂满了一种表皮葡萄球菌(Staphylococcus epidermidis),这种细菌在受到破坏性紫外线辐射后,会比那些没有这种细菌的老鼠产生更少的肿瘤。威斯康星大学麦迪逊分校的生化学家林赛?卡兰说,这些发现突出了“微生物群落影响人类疾病的潜力”。葡萄球菌是生活在人类皮肤上的众多细菌中数量最多的一种。理查德·加洛和他的同事们在调查这些细菌的抗菌能力时,发现了一株表皮葡萄球菌,它能产生一种化合物——6- n -羟色胺嘌呤,简称6-HAP——看起来很像DNA的组成部分之一。“由于这种结构,我们想知道它是否会干扰DNA合成,”加州大学圣地亚哥分校的内科科学家加洛说。在试管实验中,6-HAP阻断了构建DNA链的酶,阻止了链的生长。癌细胞生长失控,所以研究人员认为这种化合物可能会抑制这些细胞。果然,6-HAP阻止了实验室培养的不同肿瘤细胞的DNA形成,但这种化合物无法在正常皮肤细胞中实现。研究人员发现,正常皮肤细胞中的某些酶会使6-HAP失活,而被测试的肿瘤细胞似乎缺乏这些酶。加洛和他的同事发现这种化合物在注射和局部使用时都有效果。在注射了皮肤癌细胞的老鼠中,一些老鼠注射了6-HAP,而另一些老鼠注射了假hap。所有的老鼠都长出了肿瘤,但是给了这种化合物的老鼠的肿瘤只有没有这种化合物的老鼠的一半大。然后研究人员将表皮葡萄球菌传播到没有毛发的老鼠背上,使其受到紫外线的照射。一些老鼠得到了6-HAP的菌株;另一些则没有。在定期暴露于紫外线下12周后,第一组老鼠每个只长了一个肿瘤,而第二组老鼠长了4到6个肿瘤。盖洛说,美国表皮葡萄球菌菌株可能已经获得了阻止DNA合成以阻止其他细菌生长的能力。这样,细菌就能保护它们的家园免受其他入侵病原体的侵害。“也许我们进化的目的是为这些生物提供一个安全的避风港,因为当它们这么做的时候,它们也会给我们带来好处。”加洛说,研究人员对来自人类皮肤微生物的现有基因数据进行了一项小规模研究,估计20%的人都有表皮葡萄球菌菌株,在他们的皮肤上制造6-HAP。卡兰说,还需要做更多的工作来了解表皮葡萄球菌如何制造6-HAP,以及有多少化合物在皮肤上。“在我们开始对微生物进行治疗之前,了解微生物如何与宿主相互作用是很重要的。”她说,一种方法是开发针对皮肤的益生菌——添加有益的细菌来抵御感染,甚至可能预防癌症。除了皮肤癌细胞外,6-HAP还能阻断淋巴瘤细胞和癌症免疫系统细胞的DNA合成。现在说还为时过早,但这种秘密武器有可能杀死不止一个恶棍。
厂商
2020.07.15
无损探测技术? 让细胞不再“受伤”
在低分辨率(左)和高分辨率(右)下的小鼠大脑组织中,红外光谱学可将星形胶质细胞与神经元分辨开来。 为了观察细胞,研究人员经常不得不“虐待”它:把它从“家”里揪出来,用有毒的固定液浸泡它,修改它的DNA,强迫它产生可能会扰乱生物化学性质的异质蛋白质。即便这个细胞能够存活下来,它永远也不可能是原来的那个细胞了。但未来有一天,一束强大而温柔的光线或可让研究人员在不伤害细胞并使其存活以用于其他研究的情况下对其进行归类。美国加州劳伦斯·伯克利国家实验室(LBNL)生物物理学家Cynthia McMurray和物理学家Michael Martin带领的一个团队发现,通过用同步加速器产生的红外辐射强光扫描细胞,它们会捕捉到一种可揭示细胞特点的生物化学标记。研究人员在今年6月于英国举行的一次会议上报告了该方法的初步结果,现在,他们在用陈扎克伯格倡议(CZI)为期1年的试验拨款对其进行评估。如果该方法可以起作用,该团队的光谱表型技术将可以为另一个由CZI支持的项目提供工具,这个名为“人类细胞图集”的国际合作项目旨在绘制人体内每个细胞的种类和位置。如果该同步加速器驱动方法适用于其他实验室和医院使用的更加柔和的红外仪器,那么光谱表型技术未来有一天或可帮助诊断疾病、探测导致疾病的细胞变化并了解胚胎的发育。“我们使用的工具将会让这个领域焕然一新。”McMurray预测。熟悉这一未发表成果的科学家称该方法具有前景。英国曼彻斯特大学光谱学家Peter Gardner说:“我很期待看到这项研究的结果。”都柏林理工学院化学物理学家Hugh Byrne对该团队如何彻底测试该方法印象深刻。他表示,“这是证明该技术潜力的恰当研究”。Martin和McMurray喜欢把他们的方法与另一种广泛使用的细胞鉴别技术作对比:荧光标记。为了刺激特定种类的细胞产生诸如绿色荧光蛋白(GFP)的标记,科学家需要给它们装备相关分子基因。但加入DNA的技术会改变细胞,因为GFP对它们来说是外来的——来自于一种水母,且会改变细胞的生理机能。此外,McMurray指出,研究人员通常需要用激光照射荧光标签使其发光,这会损伤或杀死细胞。其他技术也具有一定的侵入性。“如果做标记或染色,就会改变细胞的真正化学性质。”Martin说,“我们想要探索细胞的化学性质,而非对它作出改变后进行测量。”这正是红外光谱技术的用武之地。“红外线没有侵入性,因此,它可被用于未受损伤的组织和活体细胞。”McMurray说。当一个样本被暴露在不同波长的红外辐射下时,它吸收的每个波段的红外光量可以表明其中含有的化学物质群的种类。与荧光标记不同,这种吸收模式通常不会揭示细胞是否在产生一种特殊分子,例如免疫受体CD4或CD8,它们经常被用于界定两类T细胞。但细胞的红外光谱特征的确可以揭示广泛的细胞种类,例如脂肪和蛋白质,从而提供生物化学指纹。因此,“你可以得到更全面的细胞图像”,Byrne说。Martin和McMurray说,标准的红外来源并不能提供他们所需要的敏感性,因此该团队转而使用LBNL的先进光源同步加速器,其产生的红外光束是世界上最亮的光束之一。Martin说:“它可以让我们得到更高的分辨率和保真度。”在6月份于格拉斯哥举行的SPEC2018会议上,McMurray和Martin说,它们可以区别小鼠大脑切片中的两种脑细胞——神经元和星形胶质细胞。在模拟亨廷顿氏病症状的啮齿类动物脑组织中,它们也能检测出表明器官退化的脂肪增多。未来,研究人员计划通过征募机器学习算式选出每个细胞种类的特征,让细胞分辨自动化。McMurray和同事仍然需要决定细胞的光谱标签是否会在体内保持一致,还是会随着位置而变化。作为潜在的医疗用途,他们还希望了解当个人细胞的红外标签发生改变时,这个人是否会生病。然而,到目前为止,研究人员仅仅分析了小鼠的组织。“我们想要确保这种方法的稳健性。”McMurray说。新技术有一个明显的限制,同步加速器体积庞大、成本高昂,而且十分稀少,它们经常有着要等待数月的研究名单。“你不可能把同步加速器带进医院。”Gardner说。但他指出,实验室设备正在迅速向能够产生粒子加速器的红外光强度靠拢。McMurray补充说,在使用同步加速器确定各种细胞类型的独特光谱模式后,研究人员计划发布一个目录,让其他科学家能够比较自己的样本结果,即使是用识别能力较低的实验室设备得到的结果。Gardner期待该项目能有一定影响。“他们有工具、有技术,还有专家,可以让这项工作加速推进。”
厂商
2020.07.14

一年了,科研经费 “包干制” 试点搞得咋样
“去年我在科技界联组会的发言中,说的就是希望尽快启动‘包干制’,很高兴现在看到了实质进展。” 中科院生物物理研究所所长许瑞明委员欣慰地表示。2019 年的政府工作报告提出,进一步提高基础研究项目间接经费占比,开展项目经费使用 “包干制” 改革试点,不设科目比例限制,由科研团队自主决定使用。去年 12 月份印发的《国家自然科学基金委员会 科学技术部 财政部关于在国家杰出青年科学基金中试点项目经费使用 “包干制” 的通知》(以下简称《通知》)指出,将在 2019 年起批准资助的国家杰出青年科学基金项目中试点实行“包干制”。中科院理化技术研究所研究员沈俊是第1批受益 “包干制” 试点的科研人员。“以前科研人员申请项目时,必须提交一份包括不同科目的财务预算,每一个科目都对科研经费使用有一定比例要求,” 沈俊说,“比如你还没开始课题呢,就要让你填写未来需要具体使用多少只试管之类的,而且有时候你做着做着会发现,当时预算中没有填写的设备后来变得需要了,这个时候要调整就很困难,科研经费的使用不灵活,非常不方便。”“现在实行了‘包干制’,就省去了这些烦恼,项目负责人有了geng大的自由度,这geng符合科研规律,因为科学研究本身有一定的不确定性,具有探索未知的特点。” 沈俊表示。中科院生物物理研究所去年有两名科研人员成功申请 “杰青” 项目,该所是如何落实 “包干制” 的?许瑞明介绍道,按照《通知》规定,首先,项目负责人需签署承诺书,承诺尊重科研规律,弘扬科学家精神,恪守科研伦理道德、科研诚信和作风学风要求,认真开展科学研究工作;承诺项目经费全部用于与本项目研究工作相关的支出,不得截留、挪用、侵占,不得用于与科学研究无关的支出。“研究所对项目单独建账,专款专用,独li核算。项目负责人为经费核算账号负责人。开设课题账号不同于其他类别项目的管理,不需要对项目各科目进行预算,只需录入总预算额度。” 许瑞明说。不少科研人员此前表示,在科研事业单位,很难通过正规途径对核心人员进行绩效激励,希望 “包干制” 试点也能在这方面带来改变。对此,许瑞明介绍道,他们目前尝试的做法是项目经费中直接经费和间接经费打通使用。“绩效支出由项目负责人根据实际科研需要和相关薪酬标准自主确定,并报研究所人事处审核后发放。”“包干制” 并非将科研经费一发了之。在去年两会科技界联组会上,科技部部长王志刚就强调,“包干制” 试点是根据科研人员的经费管理、科研成果、科学操守、素养及科研团队的稳定性等前提条件决定的。“如果都‘包干’了,光讲钱和投入,之后什么都不管,这不可能。”许瑞明也强调,在落实中,依托单位尤其注重经费使用的监管和监督。“项目负责人需要遵守国家自然科学基金委员会科研经费及研究所相关管理规定,对经费使用的合规性、合理性、真实性和相关性负责。” 他说,研究所负责组织、协调和督促科研计划的实施,在项目结题时,项目经费决算和项目结题(成果报告)将会在研究所内部进行公示。geng多人在期待 “包干制” 试点范围的扩大。“作为一个研究所所长,操心zui多的往往是科研经费使用的合理合法合规,要做好需要较复杂的程序,处理不合适,经常会影响到科研人员的积极性,‘包干制’能让科研人员感受到政府geng多的信任,希望国家有关部门在总结试点经验的基础上,能尽快推动在大部分科研项目中的实施。” 甘肃自然能源研究所所长周剑平委员表示。“繁琐的财务管理是管理不出科技创新成果的,这个弊端必须革除。” 四川省畜牧科学研究院院长蒋小松代表表示,“‘包干制’为科研人员减负,让科研人员全身心投入科技创新本身。”还有委员分析,“包干制”从 “杰青” 项目开始试点,是因为获得 “杰青” 项目的多为各领域领军人才,他们正需要稳定的支持,也希望摆脱条条框框的束缚,以zui大限度释放创新活力。此外,“杰青”属于人才项目,通常不会涉及多个单位或课题组,相对geng容易检验试点的效果。“这是一个探索给科研人员‘松绑’的很好的开端,不妨多给点时间,看看具体效果。”一位科技界委员建议,geng大范围推行 “包干制” 应综合考虑项目特点、经费监管等诸多因素。
厂商
2020.07.14

凝胶开发用于培养大量神经干细胞
在许多方面,干细胞是生物界的天才。一方面,这些天然的变形器可以将自身转化为体内几乎任何类型的细胞。在这方面,他们承诺能够治愈从脊髓损伤到癌症等疾病。另一方面,材料科学与工程副教授Sarah Heilshorn表示,干细胞就像女主角一样,也是一种善变和困难的工作。“我们只是不知道如何有效地生长大量干细胞并使其保持再生状态,”Heilshorn说。“这使我们无法在创造治疗方面取得更多进展。”到现在为止。在Nature Materials最近的一篇论文中,Heilshorn描述了一种解决方案,即在一种状态下生长和保存神经干细胞的双重挑战,它们仍然可以成熟为许多不同的细胞类型。第一个挑战是大量增加干细胞需要空间。像传统农业一样,它是一种二维的事物。如果你想要更多的小麦,玉米或干细胞,你需要更多的表面积。因此,培养干细胞需要大量相对昂贵的实验室空间,更不用说将其全部拉下来所需的能量和营养。第二个挑战是,一旦它们在实验室培养皿中分裂多次,干细胞就不容易保持在成为其他类型细胞的理想状态。研究人员将这种品质称为“干性”.Heilshorn发现,对于她正在使用的神经干细胞,保持细胞的干性需要细胞接触。Heilshorn的团队正在研究一种特殊类型的干细胞,这种干细胞会成熟为神经元和神经系统的其他细胞。如果产生足够数量的这些类型的细胞,可以产生治疗以修复脊髓损伤,抵抗创伤性脑损伤或治愈一些最严重的神经系统退行性疾病,如帕金森病和亨廷顿氏病。寻求干燥Heilshorn的解决方案涉及使用更好的材料来生长干细胞。她的实验室开发了新的聚合物基凝胶,使细胞能够以三维而不是两种方式生长。这种新的3-D工艺占目前干细胞培养技术所需实验室空间的不到1%。而且由于细胞很小,3-D凝胶叠层只有一毫米高,大约是一角硬币的厚度。“对于三维文化,我们只需要一个4英寸×4英寸的实验室空间,或大约16平方英寸。根据Heilshorn实验室最近的生物工程博士毕业生Chris Madl的说法,二维文化需要四英尺乘四英尺或约16平方英尺的地块,“超过100倍的空间”。
厂商
2020.07.14

追踪 “巨噬细胞” 的来龙去脉
巨噬细胞是人体免疫系统的重要组成细胞。它可以吞噬细胞残片、垃圾,消化病原体,发挥 “清道夫” 的作用,还能像 “哨兵” 一样提醒其他免疫细胞“有敌入侵,准备战斗”,在免疫细胞与病原体激战时,它也常常冲在最前面。随着研究的深入,科学家们发现,它还可以调控器官发育、维持组织细胞间稳态、影响组织再生、参与神经系统修复等。巨噬细胞功能失调与癌症、糖尿病及阿尔茨海默病等多种疾病密切相关。但一直以来人们并不清楚,这种细胞最初来自哪里。日前,一项研究揭示了人胚胎巨噬细胞的多重起源及发育过程,解析了组织驻留巨噬细胞特化过程的关键分子特征。相关研究成果在线发表于《自然》,由解放军第五医学中心刘兵课题组、暨南大学基础医学院兰雨课题组等合作完成。寻找巨噬细胞来源的 “金钥匙”除了为人熟知的免疫功能,巨噬细胞也发挥着一些重要的非免疫作用。比如,胚胎早期,巨噬细胞参与卵黄囊脂质代谢、造血微环境形成、骨和神经系统发育等过程。对成年个体来说,新生成的血液和免疫细胞一般被认为是由造血干细胞分化而来。但新近研究发现,小鼠体内有一些巨噬细胞比造血干细胞出现得还早,它通过自我更新,稳定持续地存在着。也就是说,这类巨噬细胞并非由造血干细胞分化而来。已有研究发现,巨噬细胞早在小鼠肝脏造血发生前就在卵黄囊中发育分化了,在趋化因子诱导下,它随着血液到达脑部、表皮、肝脏等各个器官或组织中 “长期驻扎”,形成组织驻留型巨噬细胞。“小鼠中的发现,让我们自然想探究人巨噬细胞是否也存在多种起源。” 该研究第一作者、暨南大学基础医学院博士后边志磊说。但是,由于胚胎样本获取不易、发育早期的巨噬细胞极其稀少,加之无法追踪标记等客观原因,该领域研究一直无法取得突破性进展。“直到最近几年,单细胞组学技术的出现为解决这一科学难题提供了契机。” 边志磊告诉《中国科学报》,“利用这项技术,课题组在造血发育研究领域不断积淀,终于掌握了探究巨噬细胞来源的‘金钥匙’——单细胞转录组测序。”从卵黄囊中来,到全身去研究人员首先从 8 个 3~8 周人胚卵黄囊、头、肝、循环血、皮肤、肺等组织或器官中,按照发育时间进行定期间隔取样,而后利用高精度的单细胞转录组测序技术,得到 1231 个细胞的高质量测序数据,并据此绘制了人胚造血细胞发育图谱。边志磊介绍,巨噬细胞最早来自于卵黄囊,且分两拨产生,一拨在卵黄囊中直接产生,而后分布到胚胎各处;另一拨是由卵黄囊中的髓系偏向祖细胞迁移到肝脏后,先分化为单核细胞,再分化成巨噬细胞。“这两种发育路径与此前在小鼠中发现的高度相似,表明巨噬细胞发育具有相当的保守性。” 他说。接下来,研究人员分别解析了这两种起源的巨噬细胞的发育过程。头部的小胶质细胞属于第一种起源,在受孕约 34 天后,部分头部巨噬细胞开始出现小胶质细胞分子特征(SALL1 基因);在受孕约 41 天后,伴随着 C3、P2RY12 等标志性基因表达,头部巨噬细胞逐渐特化为典型的小胶质细胞。肝脏单核细胞则属于第二种起源,卵黄囊中的髓系偏向祖细胞迁移到肝脏后,分别向单核细胞和粒系特征两个方向进行特化,肝脏产生的单核细胞也可进入组织并最终分化为巨噬细胞。为人体巨噬细胞研究奠基为了验证第二种起源的准确性,研究人员还进行了单细胞体外培养实验。边志磊介绍,利用流式检测,他们分离出了 184 个人体最早的、卵黄囊起源的造血祖细胞,进行培养后,最终有约 36% 的单细胞出现增殖和分化。他们随机抽取其中 39 个细胞进行流式检测后发现,大部分细胞都能分化成单核细胞、粒细胞、红细胞等,具有多系分化的潜能。不管是分子特征还是分化能力,这些细胞都更倾向单核及粒细胞特征。“有些巨噬细胞正是从单核细胞分化而来的,这再次说明卵黄囊产生的造血祖细胞的确具有在肝脏分化为单核巨噬细胞的能力。” 边志磊说。该研究通讯作者、暨南大学基础医学院教授兰雨告诉《中国科学报》,“我们的研究解答了组织驻留型巨噬细胞‘从哪里来、到哪里去’的核心问题,从单细胞层面解析了其逐渐发育及特化的分子过程,这可能为巨噬细胞相关疾病的诊断和治疗提供启发。”香港科技大学生命科学部教授温子龙评价说,“该研究跨越人体胚胎的多个发育阶段、覆盖多个组织器官,是目前为止对人体胚胎巨噬细胞发育最系统完整的研究。”“此外,通过胚胎样本获得的在体实验数据比体外实验数据更真实可靠,这为将来人体巨噬细胞的研究奠定了坚实的基础。”
厂商
2020.07.13

有时,一个蛋白便可决定生死康健
5月19日,《细胞-报告》在线发表了中科院上海营养与健康研究所研究员章海兵团队的研究成果,该研究揭示小鼠RIPK3蛋白突变可导致RIPK1-RIPK3相互作用缺失,从而抑制体内、外细胞程序性坏死的发生。此外,研究人员发现,在FADD敲除小鼠模型中,RIPK3蛋白突变还会导致严重的自身免疫性淋巴细胞增生综合征。章海兵表示,这为相关疾病的治疗提供了理论基础和潜在靶点。神奇的蛋白让细胞又死又活自身免疫性淋巴细胞增生综合征是一种以淋巴细胞增生为特征的自身免疫疾病。该疾病的发病原因是淋巴细胞的死亡通路被阻断,导致淋巴细胞的发育稳态被破坏,表现为淋巴结肿大、脾脏肿大、淋巴瘤以及其它自身免疫疾病的发生。章海兵解释,当机体正常的细胞凋亡和程序性坏死均被破坏时,这种疾病便会发生。细胞程序性坏死是一种新型的细胞炎性死亡方式,以胞质内蛋白形成坏死小体,细胞膜形成孔破裂释放内容物为主要特征。坏死小体主要由受体相互作用蛋白激酶1和3(RIPK1、RIPK3)组成,RIPK1与RIPK3通过功能域RHIM形成多聚蛋白复合体。目前RIPK3蛋白RHIM功能域的生理功能和作用机制尚不清楚。章海兵团队通过体外细胞实验发现,过表达人源和鼠源的RIPK3蛋白能诱导细胞发生细胞凋亡,通过突变RIPK3蛋白RHIM功能域上的重要位点,不仅能阻止RIPK1/RIPK3蛋白多聚体的出现,也可以阻止细胞凋亡和细胞程序性坏死的发生。因此,体外实验结果表明该突变位点能调控RIPK3蛋白的RHIM功能域,从而影响细胞死亡功能。“也就是说,该人源和鼠源位点都可以破坏RHIM介导的RIPK3多聚引起细胞死亡的功能。”章海兵告诉《中国科学报》。小鼠存活但却患病了为研究RIPK3蛋白RHIM功能域的体内功能,研究人员通过CRISPR-Cas9构建了特定点突变的小鼠模型。免疫共沉淀实验显示,点突变后的RIPK3蛋白不能与RIPK1发生相互作用。为进一步证实该点突变抑制细胞坏死的作用,研究人员在FADD基因敲除小鼠模型中引入该突变。结果发现,本应胚胎致死的小鼠活了下来,并且活到了成年。章海兵解释,FADD敲除小鼠在胚胎发育的10天左右时,会因过度细胞坏死而胚胎死亡。而RIPK3的点突变可以挽救FADD敲除导致的小鼠胚胎致死并存活到成年。“这证明,RIPK3蛋白RHIM功能域在体内介导细胞程序性坏死中起关键作用”。值得注意的是,本应胚胎死亡的FADD敲除小鼠虽然因RIPK3点突变活下来了,但却患有系统性淋巴细胞增生。有趣的是,在FADD敲除小鼠的基础上,与敲除RIPK3基因相比,RIPK3点突变小鼠的疾病geng严重了。“换句话说,FADD敲除基础上,RIPK3点突变蛋白能介导炎症反应促进小鼠系统性淋巴细胞增生疾病。”章海兵推测,这可能提示,RIPK3触发的细胞炎症反应是依赖于RIPK3的蛋白骨架,而不依赖于点突变。“因此,RIPK3点突变后,蛋白骨架还在,炎症反应仍会发生,疾病表现geng为严重。”两种蛋白仍有“料”可挖此外,在以上实验基础上,研究人员发现,进一步敲除RIPK1则可以缓解系统性淋巴细胞增生。这表明RIPK3的RHIM功能结构域不仅在介导细胞程序性坏死信号通路中具有关键作用,而且通过与RIPK1的相互作用,该功能域可调控淋巴细胞发育及免疫稳态。“这为治疗相关自身免疫性疾病提供了理论基础和新的靶点。”章海兵表示。RIPK3不仅介导细胞程序性坏死,同时也介导炎症反应。对此,中科院上海巴斯德研究所研究员王海坤对《中国科学报》表示,该研究通过构建不同于之前报道的RIPK3蛋白RHIM结构域突变小鼠,揭示了RIPK3的RHIM结构域在细胞死亡及系统性淋巴细胞增生疾病中的作用。“这也为该领域进一步研究关键分子RIPK3不同结构域的生物学意义,提供了新的动物模型。”王海坤说。
厂商
2020.07.13

多吃水果、蔬菜和全麦食品可以降低患糖尿病的风险
《The BMJ》杂志今天发表的两项研究表明,水果、蔬菜和全麦食品的大量消费与患2型糖尿病风险较低有关,作为健康饮食的一部分,即使适度增加这些食物的摄入量,也有助于预防2型糖尿病。在第一项研究中,一组欧洲研究人员检测了血液中维生素C和类胡萝卜素水平与患2型糖尿病风险之间的关系。维生素C和类胡萝卜素水平是比水果和蔬菜摄入调查问卷更可靠的指标。他们的发现是基于9754名新发2型糖尿病的成年人和在8个欧洲国家参加欧洲癌症与营养前瞻性调查(EPIC)互动研究的340234名受试者中13662名没有糖尿病的成年人。在调整了糖尿病的生活方式、社会和饮食风险因素后,将血液中维生素C和类胡萝卜素水平的升高以及它们的总和结合到“复合生物标志物评分”中,与患2型糖尿病的风险较低相关。与复合生物标志物得分最低的人相比,生物标志物得分在人口前20%的人患糖尿病风险低于50%。研究人员计算出,水果和蔬菜总摄入量每增加66克,患2型糖尿病的风险就会降低25%。在第二项研究中,美国的研究人员检查了全麦食品的总摄入量和个人摄入量与2型糖尿病之间的关系。他们的研究结果是基于没有糖尿病、心脏病和癌症,并且正在参加护士健康研究、护士健康研究II和健康专业人员后续研究的158259名女性和36525名男性。在调整了2型糖尿病的饮食因素后,全谷类总消费量最高的参与者与最低的人群相比,2型糖尿病的发病率低29%。对于全麦食品,研究人员发现,与每月食用少于一份相比,每天食用一份或多份全麦冷早餐麦片或黑面包的人患2型糖尿病的风险较低(分别为19%和21%)。对于其他平均摄入量较低的全谷类食品,每周食用两份或两份以上的这些食物,与一个月不到一份相比,燕麦粥让患糖尿病的风险降低了21%,添加麸皮的降低了15%,糙米和小麦胚芽则降低了12%。这些风险的降低似乎与稳定的一天两次左右的全麦摄入量有关。这两项研究都是观察性的,因此无法确定原因,而且有些结果可能是由于未测量(混淆)因素造成的。然而,这两项研究都考虑到了一些众所周知的生活方式风险因素和饮食质量的标志,并且这些发现支持了其他将健康饮食与健康联系起来的研究。因此,这两个研究小组都表示,他们的研究结果为当前的建议提供了进一步的支持,即增加水果、蔬菜和全麦食品的消费量,作为预防2型糖尿病的健康饮食的一部分。对于水果和蔬菜,研究结果还表明,在通常食用量较低的人群中,即使适度增加食用量,也有助于预防2型糖尿病。
厂商
2020.07.13

远慕生物:血红蛋白测定哪些方法?
1.氰化高铁血红蛋白HiCN测定法:除SHb外推荐参考方法,具有操作简单、显色快、结果稳定可靠、读取吸光度后可直接定值等优点。致命的弱点是氰-化钾(KCN)试剂有剧-毒,使用管理不当可造成公害。氰化高铁血红蛋白测定法操作(1)直接测定法①加转化液:试管内加5ml?HiCN转化液②采血与转化:取全血20μl,加到盛有转化液的试管底部,用上清液反复冲洗吸管3次,充分混合,静置5min。③测定:以符合WH0标准的分光光度计,波长540nm处,光径(比色杯内径)1.000cm,HiCN转化液或蒸馏水调零,测定吸光度(A)。④计算:根据样本的吸光度(A)直接计算出血红蛋白浓度(g/L)(A为测定管吸光度,44为毫摩尔消光系数,64458/1000为1mol/L Hb溶液中所含Hb克数,251为稀释倍数。)(2)HiCN标准液比色法测定HiCN参考液(50g/L、100g/L、150g/L、200g/L),分别测得540nm处的吸光度,以参考液血红蛋白含量为横坐标,吸光度为纵坐标,绘制标准曲线或求出K值。①标准曲线绘制和K值计算②样本吸光度③通过标准曲线查出样本血红蛋白浓度,或用K值计算,血红蛋白浓度Hb(g/L)=K×A。 注意事项(1)HiCN贮存:转化液贮存在棕色有塞玻璃瓶中,不能贮存在塑料瓶中,否则会使CN-丢失,测定结果偏低。HiCN转化液在4℃保存一般可数月,不能在0℃以下保存,因为结冰可使高铁氰-化钾还原,试剂失效。(2)标本:异常血浆蛋白质、高脂血症、白细胞数超过30×109/L、脂滴等可产生浊度,干扰Hb测定。(3)HiCN转化液是一种低离子强度、pH近中性的溶液(7.2±0.2)。样本中白细胞过高或球蛋白异常增高时,HiCN比色液会出现浑浊。(4)氰-化钾试剂是剧,测定后的废液应收集于广口容器中,首先以水稀释废液(1:1),再按每升上述稀释液加次氯酸钠35ml,充分混匀,敞开容器,放置15h以上,使CN-氧化成C02和N2挥发,或水解成C032-和NH4+,再排入下水道。废液不能直接与酸性溶液混合,因为氰化-钾遇酸可产生剧毒的氰氢酸气体。2.十二烷基-硫酸钠血红蛋白SDS测定法:具有操作简单、呈色稳定、准确性和精-确性符合要求、无公害等优点。但由于摩尔消光系数尚未最后确认,不能直接用吸光度计算Hb浓度,而且SDS试剂本身质量差异较大,会影响检测结果。3.HiN3最大吸收峰542nm,显色快,结果稳定。
厂商
2020.07.10
细菌是如何形成天然产物的?
许多活性药物都来自天然产物,之所以这么叫,是因为通常只有微生物才能产生复杂的结构。类似于工厂的生产线,大型酶复合物将这些活性剂分子聚合在一起。慕尼黑理工大学(TUM)和法兰克福歌德大学(Goete University Frankfurt)的一个研究小组已经成功地剖开了其中一个分子工厂的基本机制。许多活性药物都来自天然产物,之所以这么叫,是因为通常只有微生物才能产生复杂的结构。类似于工厂的生产线,大型酶复合物将这些活性剂分子聚合在一起。慕尼黑理工大学(TUM)和法兰克福歌德大学(Goete University Frankfurt)的一个研究小组已经成功地剖开了其中一个分子工厂的基本机制。许多重要的药物,如抗生素或抗癌活性剂,都是由微生物(如细菌或真菌)合成的天然产物。在实验室里,这些天然产物往往根本无法生产出来,或者需要付出很大的努力。许多这类化合物的起始点是聚酮类化合物。在一个微生物细胞中,如发光光杆状菌(Photorhabdus luminescens),它们是通过聚酮合成酶(PKS)产生这类化合物的。在PKSⅡ型系统的第一阶段,为了逐步建立起所需的分子,四种蛋白质协同工作。在第二阶段,它们被进一步的酶修饰成所需的天然产物。以这种方式生产的细菌天然产物的例子包括临床上使用的四环素抗生素或抗癌药物阿霉素。跨学科合作虽然第二阶段的改性步骤得到了很好的研究,但到目前为止,对第一阶段的一般功能几乎没有任何见解,在这些分子工厂中,高活性聚酮中间产物与酶复合物结合并受到保护,使其不能自发反应。慕尼黑技术大学生物化学教授迈克尔?格罗尔(Michael Groll)和法兰克福歌德大学(Goete University Frankfurt)分子生物技术教授海尔格?博德(Helge Bode)两个工作组合作,现已弥合了这一差距,这些成果发表在著名的科学期刊《Nature Chemistry》上。这些发现对活性剂的新合成有启发作用Michael解释说:“在这项工作的背景下,我们第一次能够借助X射线结构分析来分析II型聚酮合酶的不同伴侣蛋白复合物,更详细地了解了整个催化循环。”“基于这些发现,将来有可能以有针对性的方式操纵中枢生化过程,从而改变基本结构,而不是局限于修饰酶,”Helge补充道。尽管开发改良抗生素和其他药物还有很长的路要走,但两个课题组都乐观地认为,现在分子工厂缺失部分的结构和机制可以解释了。“我们已经获得了合成新蛋白质复合物的潜力数据。”
厂商
2020.07.09

RNA是决定干细胞命运的关键吗?
深入观察我们的细胞,你会发现每个细胞都有一个完全相同的基因组——一套完整的基因,但是如果每个蓝图都是相同的,为什么眼睛细胞的外观和行为与皮肤细胞或脑细胞不同呢?干细胞——我们器官和组织细胞的原材料——怎么知道会变成什么样?在7月8日发表的一项研究中,科罗拉多大学博尔德分校的研究人员离回答这个基本问题又近了一步,他们得出结论,分子信使RNA(核糖核酸)在细胞分化中起着不可或缺的作用,在我们的基因和所谓的“表观遗传”机制之间起着桥梁的作用。研究人员在《Nature Genetics》杂志上发表报告称,当这座桥缺失或有缺陷时,干细胞在成为心脏细胞的道路上永远不会学会如何跳动。虽然这项研究还很年轻,但它最终可能会为新的RNA靶向疗法的发展提供信息。“并非所有的基因都在所有细胞中一直表达。相反,每一种组织类型都有自己的表观遗传程序,它决定了哪些基因在任何时候被打开或关闭。我们非常详细地确定了RNA是这种表观遗传沉默的主要调节器,并且在没有RNA的情况下,这个系统无法工作。这对生命至关重要,”本文共同通讯作者Thomas Cech说。2006年,这篇新论文的共同通讯作者,John Rinn,现在是博尔德大学的生物化学教授,他首次提出RNA可能是关键。在《Cell》杂志出版的一篇里程碑式的论文中,Rinn指出,在细胞核内,RNA附着在一个折叠的蛋白质簇上,这种蛋白质被认为可以调节基因的表达。许多其他的研究也发现了同样的现象,并补充说不同的RNA也与不同的蛋白质复合物结合。一个备受争议的问题:这对决定细胞的命运真的很重要吗? 自那以后,已经发表了不少于502篇论文。有些人认为RNA是表观遗传学的关键;另一些人则认为其作用充其量是无关紧要的。因此,2015年,Cech实验室的生物化学家和博士后研究员Yicheng Long开始使用最-新可用的工具再次提出这个问题。在生物前沿研究所(BioFrontiers Institute)的休息室Yicheng Long偶然碰见了Rinn实验室的计算生物学家Taeyoung Hwang。“我们利用数据科学的方法和高性能的计算来理解分子模式,并以一种新颖的、定量的方式评估RNA的作用,”Hwang说,他和Long是这篇新论文的共同第1作者。在实验室里,研究小组随后用一种简单的酶来去除细胞中的所有RNA,以了解表观遗传机制是否仍能通过DNA使基因沉默。答案是“不能”“RNA似乎扮演着空中交通管制员的角色,引导蛋白质复合物到达DNA上的正确位置,降落并使基因沉默,”Long说。第三步,他们利用基因编辑技术CRISPR培育出一系列注定要成为人类心肌细胞的干细胞,但其中的蛋白复合物PRC2不能与RNA结合。从本质上讲,就好像飞机无法与空中交通管制系统连接,迷失了方向,整个过程崩溃了。到第7天,正常的干细胞已经开始看起来和行为像心脏细胞。但是突变细胞没有跳动。值得注意的是,当PRC2恢复正常后,它们的行为开始更加正常。“我们现在可以明确地说,RNA在细胞分化过程中起着关键作用,”Long说。先前的研究已经表明,人类的基因突变会破坏RNA与这些蛋白质的结合能力,从而增加患某些癌症和胎儿心脏异常的风险。最终,研究人员设想有一天,RNA靶向疗法可以用来解决这些问题。“这些发现将开创一个新的科学阶段,展示表观遗传学和RNA生物学之间不可分割的联-系,”Rinn说。“它们对今后理解和处理人类疾病具有广泛的意义。”
厂商
2020.07.09

凝胶电泳实验操作中的小技巧
1、让凝胶电泳变得更快,更漂亮方法改进:将电泳电压进行定时变化,例如可以在开始时将电泳电压调节至 100V,大约 15min。使条带的确可以因为自身片段大小不同而产生较大差别的泳动速度,从而将片段分离,然而现在的分离或许会间距较小,从而图片很不漂亮,或者不易观察.可以紧接着进行 120V-130V 的电压进行较小差 异电泳,但是由于电泳电压较大,可以避免过大的片段残存在胶孔不易泳动的情况.结果:这样两个电压进行配合电泳,便可以得到非常漂亮的电泳条带,并且可以节省 1/5-2/5 左右 的时间.2、RNA 电泳如何得出漂亮的条带方法改进:1.电泳槽,制胶器,梳子等的清洗:去污剂浸泡过夜——>自来水冲洗干净——>ddH20 冲洗——>3%H2O2 灌满浸泡过夜——>灭活的 0.1%DEPC水冲洗干净——>超净台内紫外线照射过夜. 2.烧瓶,烧杯,药匙,量筒等制胶器械的清洗:0.1%DEPC 水浸泡过夜后高压消毒灭活,烘箱烘干.或 者 ddH20 清洗干净, 超净台内紫外线照射过夜. 3.电泳缓冲液必须是 RNase free . 4.预电泳 5-10min 减少了非特异 RNA 条带的出现,有利于分离和纯化,同时可根据电泳仪是否冒泡判断电 泳仪装置是否有误. 5.样品是在电泳缓冲液液略低于胶表面而不是在高过胶面时加进齿槽,避免了加样 时 RNA 的扩散,加样后 RNA 从齿槽逸出造成 RNA 的弥散及定位不良等现象. 6.电泳 3-5min 让 RNA 进入凝胶后再加电泳缓冲液液高过表面,确保了加到每个槽中的 RNA 量及定位的准确性,从而有利于 DNA 的鉴定和纯化.3、如何提高 SDS-PAGE 的分辨率方法改进:借鉴 Tricine-SDS-PAGE 中添加甘油或者尿素来提高分辨率的成功经验,在普通的 SDS-PAGE 中加入约 13%的甘油,同样可以提高分辨率,有效防止小分子量蛋白的弥散.只要把原来 配方中的水换成 60%的甘油,就可以了.结果:加入甘油之后,条带较细,分得更开.4、改进一点点,我们能得到更加美观的 SDS-PAGE 胶方法改进:所做的改进很简单却很有效,加完分离胶后,用移液枪吸取酒精(浓度没有特别要求, 干净无污染就好)加到分离胶上至覆盖界面,静置片刻后放到 37℃恒温箱中可加速胶的凝固.待到分 离胶完全凝固之后倒去上层的酒精,就可以看到齐平漂亮的界面啦!改进二:脱色 背景:给染色结束的 SDS-PAGE 胶脱色往往需要比较长的时间,否则会由于脱色不完全而导致条 带不清晰,影响到拍照的效果.方法改进:改进的方法很简单易行——取一张我们常常随身携带的面巾纸,打个结放入盛有脱色液 的大培养皿里放到脱色摇床上,这样一来,原来过夜脱色达到的效果现在只需要短短的 3,4 个小时就 可以轻松实现了. PS:希望大家这个时候用的面巾纸是质量比较好不容易掉屑的...
厂商
2020.07.08
蛋白提取操作时应注意的细节!!!
避免蛋白降解:总体规则:蛋白酶普遍存在,在提取蛋白质的步骤中,早些抑制,经常抑制分装蛋白溶液,储存于不同的温度和不同的缓冲液中,测蛋白活性,找出最适储存条件。冷藏,并加入稳定剂。这些稳定剂一般都能充分保持蛋白的生物学活性。将蛋白溶液储于50%(V/V)的甘油或悬于硫酸铵(3.2M)中,避免冻存冰结晶会造成物理剪切及蛋白变性。避免酶的反复冻融,如果酶需要冻存,分装成小管后冻存。蛋白质的储存:蛋白样品在液氮中速冻,拿出后加入10-20%的甘油,长期储存整个实验过程中,都要将蛋白样品置于冰上从母管中吸取不同的蛋白时,都要用新鲜的枪头不要将未用的溶液倒回母管戴手套操作,避免蛋白交叉污染,或是蛋白酶的污染避免剧烈的震荡,避免溶液中混入蛋白变性剂实验的过程中应避免灰尘进入,灰尘包含60%的肌氨酸操作过程中:操作时总是小体积小量的操作,避免污染,并且动作要快a:此类蛋白酶在活性中心包含丝氨酸和组氨酸。b:此类蛋白酶在活性中心包含半胱氨酸(巯基,-SH-)。c:此类蛋白酶在活性中心包含金属离子(如:n2+,Ca2+,Mn2+)。d:此类蛋白酶在活性中心包含天冬氨酸族。
厂商
2020.07.08

细胞传代是否一定要等24小时呢?
问:我养的是RAW264.7细胞,不知道为什么长得这么快?而且消化也很容易,好像和园子里其他养RAW细胞的很不一样。一般传代(1传2)后大概16-20个小时左右就能够长到90%,请问是应该一定等到24小时再传代,还是可以等长到90%(答:很多试验中要求分盘后24h时再开始实验(如转染),除了让细胞充分贴壁,还有一个重要原因就是使细胞达到正常代谢水平。从你的描述来看,如果细胞贴壁还比较快的话,个人认为可以进行传代,若细胞密度过高达接触抑制,细胞比较容易老化。还有建议最好传代时采用更低一些的稀释度(1:3或者更低)。 感觉1:2太高了,建议到ATCC上查找一下,会找到最佳的subcultivation ratio。以便保证细胞的最佳状态,不要舍不得扔细胞,subculture的时候把细胞全用上,你的细胞长得快,频繁的subculture,细胞不但不能优化还可能会发生变异。问:请问1:2太高了是什么意思啊?是不是要1:3以上传代呢?因为如果传得太稀了,细胞长起来比较慢,可能有群集效应。所以一般每天传代,到底传代频繁是能够改善细胞状态还是像你所说得会造成变异呢?答:RAW 264.7,Subcultivation ratio: A subcultivation ratio of 1:3 to 1:6 is recommended。Medium renewal: Replace or add medium every 2 to 3 days。所以说你1:2传太高了,是这个意思。建议加大传代比率,1:3或1:4传。反复传代劳命伤财,对细胞也有害,特别是细胞未到24h。细胞传代的比率要适当。细胞太少,生长慢,因细胞生长需相互间有信号传导。细胞太多,也不好,因细胞太多会有“接触抑制”。我也从ATCC上看过这个,不过由于我们养的细胞不可能和他们的条件完全一样,所以对于传代就不可能完全按照它们说的。不过看来还是有一定的道理哦。现在觉得,其实1:3传代可能更适合现在的细胞生长速度,长得太快太慢了,细胞都挤的变圆了。细胞太少的时候会抑制它们的生长速度。问:请教一下,为什么细胞太少的时候会抑制它们的生长速度呢?答:细胞生长需要一定的辅因子,而这些不仅要求血清供给,还有细胞在培养过程中生成的,若细胞太少,会导致这类因子不足。有时候会在培养细胞时候,放一些伴生长细胞是这个原因,好像叫保姆细胞。我养的是骨肉瘤细胞,生长速度也很快。我一般等长满了就传代,不是特别关注时间。你在传代分瓶的时候多分几瓶。但也不能太多,因为细胞太少的时候会抑制它们的生长速度。看是什么细胞了,如是生长比较旺盛的细胞,只要贴壁情况良好就可以传。我培养的是hela细胞。复苏后不到24小时的换液以后,过了-6小时就可以传代了。但是好像对其他多数细胞建议还是24小时后传。
厂商
2020.07.08

噬菌体的检查及效价测定 远慕新闻
1、目的:1.1 了解噬菌体效价的含义及其测定原理。1.2 学会检查噬菌体的方法。1.3 掌握用双层琼脂平板法测定噬菌体效价的操作技能。2、原理:噬菌体是一类专性寄生于细菌和放线菌等微生物的病毒,其个体形态极其微小,用常规微生物计数法无法测得其数量。当烈性噬菌体侵染细菌后会迅速引起敏感细菌 裂解,释放出大量子代噬菌体,然后它们再扩散和侵染周围细胞,最终使含有敏感菌的悬液由混浊逐渐变清,或在含有敏感细菌的平板上出现肉眼可见的空斑──噬 菌斑。了解噬菌体的特性,快速检查、分离,并进行效价测定,对在生产和科研工作中防止噬菌体的污染具有重要作用。检样可以是发酵液、空气、污水、土壤等(至于无法采样而需检查的对象,可以用无菌水浸湿的棉花涂拭表面作为检查样品)。为了易于分离可先经增殖培养,使样品中的噬菌体数量增加。采用生物测定法进行噬菌体检查,约需12h左右,因而不能及时判断是否有噬菌体污染。通过快速检查可大致确定是否有噬菌体污染,以采取必要的防治措施。根 据正常发酵(培养)液离心后菌体沉淀,上清液蛋白含量很少,加热后仍然清亮;而侵染有噬菌体的发酵(培养)液经离心后其上清液中因含有自裂解菌中逸出的活 性蛋白,加热后发生蛋白质变性,因而在光线照射下出现丁达尔效应而不清亮。此法简单、快速,对发酵液污染噬菌体的判断亦较准确。但不适于溶源性细菌及温合 噬菌体的诊断,对侵染噬菌体较少的一级种子培养液也往往不适用。噬菌体的效价即1mL样品中所含侵染性噬菌体的粒子数。效价的测定一般采用双层琼脂平板法。由于在含有特异宿主细菌的琼脂平板上,一般一个噬菌体产生一个 噬菌斑,故可根据一定体积的噬菌体培养液所出现的噬菌斑数,计算出噬菌体的效价。此法所形成的噬菌斑的形态、大小较一致,且清晰度高,故计数比较准确,因 而被广泛应用。3、材料:3.1菌种:敏感指示菌(大肠杆菌)、大肠杆菌噬菌体(从阴沟或粪池污水中分离)。3.2培养基:二倍肉膏蛋白胨培养液,上层肉膏蛋白胨半固体琼脂培养基(含琼脂0.7%,试管分装,每管5mL),下层肉膏蛋白胨固体琼脂培养基(含琼脂2%),1%蛋白胨水培养基。3.3仪器和器具:无菌的试管、培养皿、三角瓶、移液管(1、5mL),恒温水浴锅,离心机、721分光光度计等。4、流程:4.1噬菌体的检查4.2噬菌体效价的测定5、方法:5.1 噬菌体的检查:5.1.1 样品采集将2~3g土样或5mL水样(如阴沟污水)放入灭菌三角瓶中,加入对数生长期的敏感指示菌(大肠杆菌)菌液3~5mL,再加20mL二倍肉汤蛋白胨培养液。5.1.2 增殖培养30℃振荡培养12~18h, 使噬菌体增殖。5.1.3 离心分离将上述培养液以3000rpm离心15~20min, 取上清液,用pH7.0,1%蛋白胨水稀释至10-2~10-3,用于噬菌体检查及效价测定。5.1.4 生物测定法5.1.4.1双层琼脂平板法5.1.4.1.1 倒下层琼脂融化下层培养基,倒平板(约10mL/皿)待用。5.1.4.1.2倒上层琼脂融化上层培养基,待融化的上层培养基冷却至50℃左右时,每管中加入敏感指示菌 (大肠杆菌) 菌液0.2mL,待检样品液或上述噬菌体增殖液0.2~0.5mL,混合后立即倒入上层平板铺平。5.1.4.1.3恒温培养30℃恒温培养6~12h观察结果。5.1.4.1.4 观察结果如有噬菌体,则在双层培养基的上层出现透亮无菌圆形空斑──%26not;%26not;%26not;%26not;噬菌斑。5.1.4.2 单层琼脂平板法省略下层培养基,将上层培养基的琼脂量增加至2%,融化后冷却至45℃左右,如同上法加入指示菌和检样,混合后迅速倒平板。30℃恒温培养6~16h后观察结果。5.1.5 离心分离加热法(快速检查)取大肠杆菌正常培养液和侵染有噬菌体的异常大肠杆菌培养液,4000rpm离心20min,分别取两组发酵液的上清液(A1),一部分于721分光光度计上测定OD650光密度值,另外各取5mL上清液于试管中,置水浴中煮沸2min(A2),检测A2溶液OD650光密度值,记录结果。5.2 噬菌体效价的测定:5.2.1 倒平板将融化后冷却到45℃左右的下层肉膏蛋白胨固体培养基倾倒于11个无菌培养皿中,每皿约倾注10mL培养基,平放,待冷凝后在培养皿底部注明噬菌体稀释度。5.2.2 稀释噬菌体按10倍稀释法,吸取0.5mL大肠杆菌噬菌体,注入一支装有4.5mL1%蛋白胨水的试管中,即稀释到10-1,依次稀释到10-6稀释度。5.3 噬菌体与菌液混合:将11支灭菌空试管分别标记10-4、10-5、10-6和对照。分别从10-4、10-5和10-6噬菌体稀释液中吸取0.1mL于上述编号的无菌试管 中,每个稀释度平行做三个管,在另外两支对照管中加0.1mL无菌水,并分别于各管中加入0.2mL大肠杆菌菌悬液,振荡试管使菌液与噬菌体液混合均匀, 置37℃水浴中保温5min,让噬菌体粒子充分吸附并侵入菌体细胞。5.4 接种上层平板:将11支融化并保温于45℃的上层肉膏蛋白胨半固体琼脂培养基5mL分别加入到含有噬菌体和敏感菌液的混合管中,迅速搓匀,立即倒入相应编号的底层培养基平板表面,边倒入边摇动平板使其迅速地铺展表面。水平静置,凝固后置37℃培养。5.5 观察并计数:观察平板中的噬菌斑,并将结果记录于 计算公式:N=Y/V%26#8226;X(N:效价值,Y:平均噬菌斑数/皿,V:取样量,X:稀释度)例如:当稀释度为10-6时,取样量为0.1 mL/皿,同一稀释度中3个平板上的噬菌斑的平均值为186个,则该样品的效价为:N=186/0.1%26#215;10-6=1.86%26#215;109。6、结果:6.1、噬菌体检查6.1.1离心分离加热法处理方法OD650光密度值正常发酵液(对照) 异常发酵液(试验)离心上清液(A1)离心上清液加热煮沸后(A2)A2/A16.1.2 绘出平板上的噬菌斑检测结果,指出噬菌斑和宿主细菌。6.2、噬菌体效价测定6.2.1 平板上噬菌斑数目噬菌体稀释度10-4、10-5、10-6 对照噬菌斑数(个)/皿平均每皿噬菌斑数目6.2.2 计算噬菌体效价(即噬菌斑形成单位pfu, plague-forming unit).
厂商
2020.07.07

RNA的琼脂糖凝胶电泳实验原理和步骤
一、实验目的掌握植物总RNA非变性胶电泳的原理和方法。二、实验原理RNA电泳可以在变性及非变性两种条件下进行。非变性电泳使用1.0%--1.4%的凝胶,不同的RNA条带也能分开,但无法判断其分子量。只有在完全变性的条件下,RNA的泳动率才与分子量的对数呈线性关系。因此要测定RNA分子量时,一定要用变性凝胶。在需快速检测所提总RNA样品完整性时,配制普通的1%琼脂糖凝胶即可。三、实验材料、器具及药品蘑菇的总RNA溶液。 电泳仪,电泳槽,电子天平,移液器,枪头,微波炉,紫外透射检测仪等。 琼脂糖,1XTAE电泳缓冲液,0.5μg/ml溴化乙锭(EB)10X载样缓冲液。四、实验步骤(1)用1×TAE电泳缓冲液制作琼脂糖凝胶,加1×TAE电泳缓冲液至液面覆盖凝胶。(2)在超净工作台上,用移液器吸取总RNA样品4μl于封口膜上。在实验台上再加入5μl 1×TAE电泳缓冲液及1μl 的10X载样缓冲液,混匀后,小心加入点样孔。(3)打开电源开关,调节电压至100V,使RNA由负极向正极电泳,约30min后将凝胶放入EB染液中染色5min,用清水稍微漂洗。在紫外透射检测仪上观察RNA电泳结果。RNA的变性琼脂糖凝胶检测试剂:(1)MOPS缓冲液(10*):0.4mol/L 吗啉代丙烷磺酸(MOPS) (Ph7.0),0.1mol/L NaAc, 10mol/L EDTA。(2)上样染料:50%甘油,1mmol/L EDTA ,0.4%溴酚蓝,0.4%二甲苯蓝。(3)甲醛。(4)去离子甲酰胺。v电泳槽清洗:去污剂洗干净(一般浸泡过夜)——水冲洗——乙醇干燥——3%H2O2灌满——室温放置10分钟——0.1%DEPC水冲洗。操作:(1) 将制胶用具用70%乙醇冲冼一遍,晾干备用。(2) 配制琼脂糖凝胶。①称取0.5g琼脂糖,置干净的100ml 锥形瓶中,加入40ml蒸馏水,微波炉内加热使琼脂糖彻底溶化均匀。②待胶凉至60--70 ℃,依次向其中加入9ml 甲醛、5ml 10X MOPS缓冲液和0.5ul 溴化乙锭,混合均匀。③灌制琼脂糖凝胶。(3) 样品准备:① 取 DEPC处理过的500ul小离心管,依次加入如下试剂: 10x MOPS缓冲液2ul,甲醛3.5ul,甲酰胺(去离子)10ul,RNA 样品 4.5ul,混匀。② 将离心管置于60℃水浴中保10分钟,再置冰上2分钟。③ 向管中加入3ul 上样染料,混匀。(4)上样。(5)电泳:电泳槽内加入1XMOPS缓冲液,于7.5V/ml 的电压下电泳。(6)电泳结束后,在紫外灯下检查结果。
厂商
2020.07.07

影响电泳的因素有哪些?
不同的带电颗粒在同一电场中泳动的速度不同。常用泳动度(或迁移率)来表 示。泳动度是指带电 颗粒在单位电场强度下泳的速度。影响泳动度的主要因素有:1、首先决定于带颗粒的性质,即颗粒所带净电荷的数量,大小及形状。 一般说来,颗粒带净电荷多,直径小而接近于球形,则在电场中泳动速度快,反 之则慢。泳动度还与分子的形状,介质粘度,颗粒所带电荷有关,泳动度与颗表面电 荷成正比,与介质粘度及颗粒半径反比。带电荷的高分子在电解质溶液中把一些带有 相反电荷的离子吸引在其周围,形成一离子扩散层。加以电场时,颗粒向符号相反的 电极移动即带阳电荷颗粒移向负极,带阴电荷颗粒移向正移;离子扩散层由于带有过 剩的与颗粒符号相反的电荷,可以向相反方向移动。结果颗粒与离子扩散之间的静电 引力使颗粒的泳动度减慢,另外,高分了颗粒表面有一层水,在电场影响下,它与颗 粒一起移动,可以认为是颗粒的一部分。2、电场强度 电场强度也称电位梯度,是指单位长度(每一厘米)支持物体上的电位降,它对 泳动度起着十分重要的作用。例如纸电泳。测量25厘米长纸条两端电压为250伏特, 则电场强度为250伏特/25厘米=10伏特/厘米。 一般,电场强度越高,带电颗粒移动速度越快。根据电场强度大小,可将电泳分 为常压电泳和高压电泳,前者的电压在100-500伏特,电场强度一般是2-10伏特/厘 米;后者电压可高达500-1000伏特,电场强度在200-2000伏特/厘米。常压电泳分离 时间需数小时至数天,高压电泳时间短,有时仅几分钟即可,高压电泳主要用于分离 氨基酸,肽,核苷酸,由于电压升高,电压也随之增大,故需冷却装置。3、溶液的pH值 溶液的pH值决定了带电颗粒解离的程度,也决定了物质所带净电荷的多少,对于 蛋白质,氨基酸等两性电解质而言,溶液pH值离电点越远,颗粒所带净电荷越多;电 泳速度越快;反之则越慢。例如血清中几种主要蛋白质的等电点各不相同,白蛋白等 电点是4.0。α2球蛋白等电点是5.06,β球蛋白等电点是5.1,γ球蛋白等电点是7.1。 当在pH8.6的电泳缓冲液中电泳时,这些蛋白质都带负电荷,它们的泳动速度是白蛋 白β球蛋白>γ球蛋白。因此,当要分离一种蛋白质混合物时,应选择 一种能扩大各种蛋白质所带电荷量差异的pH值,以利于各种蛋白质分子的解离。同 时,为保持电泳过程中溶液pH恒定也须使用缓冲液。4、溶液的离子强度 溶液的离子强度是另一个重要选择的条件,在保持足够缓冲能力前提下,离子强 度要最小。溶液的离子强度越高,带电颗粒泳动速度越慢,反之越快。通常缓冲液离 子强度选择在0.05?FONT FACE="宋体">0.1M之间。浓度大的缓冲液在 合适电压下,有较低的电阻和较大的电流,产热较高。如果这种额外的热可以驱散, 高浓度的缓冲液扩散常数较低,可以产生较窄细的电泳区带。离子强度很高时要用低 电压,避免过多产热,这样又导致长的电泳时间,以致增加变性和区带分离困难。5、电渗作用 在支持物的电泳中,影响电泳的另一个重要因素是电渗作用。所谓电渗就是指在 电场中液体对固体支持物的相对移动,例如在pH8.6时,血清蛋白质进行纸电泳时, γ球蛋白与其他蛋白质一样带负电荷,应该向正极移动,然而它却向负极方向移动, 这就是电渗作用的结果。滤纸的纤维间具有大量孔隙带有负电荷,如一束毛细管,进 行电泳时蛋白质通过这些孔隙向前移动。纸的孔隙带有负电荷,与带电颗粒一样可以 吸引正离子,因此介质沿管壁相对地带的较多的正电荷。在电场中,带负电荷的蛋白 质向正极移动,而介质却向反向阴极移动,从而对蛋白质颗粒的泳动起阻碍作用。由 于γ球蛋白分子颗粒大,净电荷较少,移动速度慢,电渗作用大于颗粒向前泳动的 力,结果γ球蛋白向后退。而其他血清蛋白质,因分子较小,净电荷较多,电渗作用 小于分子向前移动的力,因此分子向前移。所以,血清蛋白质电泳后球蛋白反而在点 样位置之后。
厂商
2020.07.07

远慕分享:细胞传代、培养的方法
实验原理:将动物机体的各种组织从机体中取出,经各种酶(常用胰蛋白酶)、螯合剂(常用EDTA)或机械方法处理,分散成单细胞,置合适的培养基中培养,使细胞得以生存、生长和繁殖,这一过程称原代培养。细胞在培养瓶长成致密单层后,已基本上饱和,为使细胞能继续生长,同时 也将细胞数量扩大,就必须进行传代(再培养)。传代培养也是一种将细胞种保存下去的方法。同时也是利用培养细胞进行各种实验的必经过程。悬浮型细胞直接分瓶就可以,而贴壁细胞需经消化后才能分瓶。实验仪器培养箱(调整至37℃)、培养瓶、青霉素瓶、小玻璃漏斗、平皿、吸管、移液管、纱布、手术器械、血球计数板、离心机、水浴箱(37℃)实验试剂: 1640培养基或DMEM培养基、胎牛血清、0.25%胰酶、Hank′s液、碘酒、75%酒精实验步骤:一、原代细胞培养步骤1、准备:取各种已消毒的培养用品置于净化台面,紫外线消毒20分钟。开始工作前先洗手、75%酒精擦拭手至肘部。2、布局:点燃酒精灯,安装吸管帽。3、处理组织:把组织块置于烧杯中,用Hanks液漂洗2-3次,去除血污;如怀疑组织可能污染,可先置于含有青链霉素的混合液中30-60分钟。4、剪切:用眼科剪把组织切成2-3毫米大小的块,以便于消化。加入比组织块总量多30-50倍的胰蛋白酶液,然后一并倒入三角烧瓶中,结扎瓶口或塞以胶塞。5、消化:或用恒温水浴,或置于37℃温箱消化均可,消化中每隔20分钟应摇动一次,如用电磁恒温搅拌器消化更好。消化时间依组织块的大小和组织的硬度而定。6、分离:在消化过程中见消化液发混浊时,可用吸管吸出少许消化液在镜下观察,如组织已分散成细胞团或单个细胞,立即终止消化,随即通过适宜不锈钢筛,滤掉尚未充分消化开的组织块。低速(500-1000转/分)离心消化液5分钟,吸出上清,加入适量含有血清的培养液。7、计数:用计数板计数,如细胞悬液细胞密度过大,再补加培养液调整后,分装入培养瓶中。对大多数细胞来说,pH要求在7.2-7.4范围,培养液呈微红色,如颜色偏黄,说明液体变酸,可用NaHCO3调整。8、培养:置于37℃温箱培养,如用CO2温箱培养,瓶口需用纱布棉塞或螺旋帽堵塞,纱布塞易生霉菌,每次换液时需要换新塞。二、原代组织块培养法1、剪切:把组织小块置于小烧杯或青霉素小瓶中,用Hanks液漂洗二三次以去掉表面血污,吸静Hanks液,用眼科剪反复剪切1mm块为止。2、摆布:用弯头吸管吸取若干小块,置于培养瓶中,用吸管弯头把组织小块摆布在培养瓶底部,小块相互距离以0.5cm为宜,每一25ml培养瓶底可摆布20-30块。3、轻轻翻转培养瓶,另瓶底向上,注意翻瓶时勿另组织小块流动,塞好瓶塞,置37℃温箱培养2小时左右(勿超过4小时),使小块微干涸。4、培养:从微箱中取出培养瓶,开塞,46度斜持培养瓶,箱瓶底脚部轻轻注入培养液少许,然后缓缓再把培养瓶翻转过来,让培养液慢慢覆盖附于瓶地上的组织小块。置温箱中静止培养。待细胞从组织块游出数量增多后。再补加培养液。三、贴壁细胞传代的操作步骤:1、吸除培养瓶内旧培养液。2、向瓶内加入胰蛋白酶和EDTA混合液少许,以能覆满瓶底为限。3、置温箱中2-5分钟,当发现细胞质回缩,细胞间隙增大后,立即终止消化。4、吸除消化液,向瓶内注入Hanks液数毫升,轻轻转动培养瓶,把残余消化液冲掉。注意加Hanks液冲洗细胞时,动作要轻,以免把已松动的细胞冲掉流失,如用胰蛋白酶液单独消化,吸除胰蛋白酶液后,可不用Hanks液冲洗,直接加入培养液。5、用吸管吸取营养液轻轻反复吹打瓶壁细胞,使之从瓶壁脱离形成细胞悬液。6、计数板计数后,把细胞悬液分成等份分装入数个培养瓶中,置温箱中培养。四、悬浮型细胞传代1、吸出细胞培养液,放入离心管中,离心1000rpm5分钟。2、吸掉上清液,加入适量之新鲜培养基,混和均匀后,依稀释比例转移至新的培养瓶中,以正常培养条件培养。注意事项1、操作前要洗手,进入超净台后手要用75%酒精或0.2%新洁尔灭擦试。试剂等瓶口也要擦试。2、点燃酒精灯,操作在火焰附近进行,耐热物品要经常在火焰上烧灼,金属器械烧灼时间不能太长,以免退火,并冷却后才能夹取组织,吸取过营养液的用具不能再烧灼,以免烧焦形成碳膜。3、操作动作要准确敏捷,但又不能太快,以防空气流动,增加污染机会。4、不能用手触已消毒器皿的工作部分,工作台面上用品要布局合理。5、瓶子开口后要尽量保持45℃斜位。6、 吸溶液的吸管等不能混用。
厂商
2020.07.06
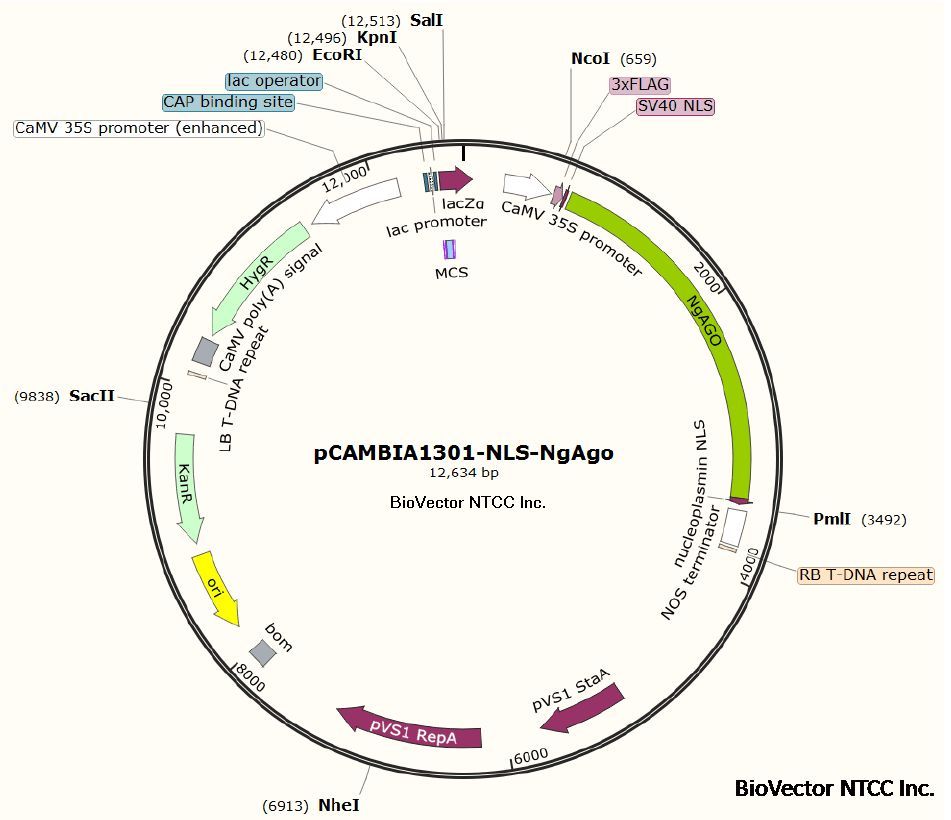
远慕技术:质粒提取实验步骤
实验原理现在较常用的质粒提取方法有三种:碱裂解法、煮沸法和去污剂裂解法,前两种方法较为剧烈,适用于较小的质粒(<15Kb),而去污剂裂解法则比较温合,一般用于分子量较大的质粒(>15Kb)。碱裂解法是一种最广泛使用的制备质粒DNA的方法。其原理为:染色体DNA远大于质粒DNA,且染色体DNA为线状分子,而质粒为共价闭合环状分子。当pH值为 12.0-12.6,碱性环境中,线性的大分子的染色体DNA完全变性且无法复性,而共价闭环质粒DNA在将PH调至中性后即可恢复其天然构象;在高盐浓度存在的条件下,染色体DNA和蛋白质在去污剂SDS的作用下形成沉淀,而质粒DNA为可溶状态,通过离心可除去大部分细胞碎片、染色体DNA、RNA及蛋白质,质粒DNA仍在上清中。残留的杂质可以通过酚氯仿处理掉。实验过程1、实验试剂(1)溶液Ⅰ 50mMl 葡萄糖 / 10mMl EDTA / 25mMl Tris-HCl,pH=8.0;(2)溶液Ⅱ 0.2Ml NaOH / 1% SDS;(3)溶液Ⅲ 3Ml 醋酸钾 / 2Ml 醋酸;(4)异丙醇,无水乙醇,75%乙醇应放-20℃冰箱预冷备用;酚氯仿放4℃冰箱预存;(5)摇菌,2-3ml相应抗性的LB培养液,在37℃温度下过夜培养。2、质粒提取(1)菌液12000rcf离心5min,弃掉上清,加入0.2ml溶液I(已加入RNase),充分混匀后,加入0.25ml溶液II,颠倒混匀,室温放置5min,加入0.4ml 溶液III,颠倒混匀后,13000rcf 离心30min。(2)上清转入新的1.5mlEP管中,加入等体积的酚氯仿混合液,充分混匀。10000rcf 离心10min。(3)取上清,转入新的EP管,加入等体积、预冷的异丙醇,充分混匀后,13000rcf 离心 6min。(4)倒掉上清液,用1ml预冷的75%乙醇,颠倒后,13000rcf 离心 1min。(5)倒掉上清液,沿壁加入1ml预冷的75%乙醇,直接倒掉。(6)沿壁加入1ml预冷的乙醇,直接倒掉。倒扣5min后,室温下放置10min。待管内液体会发干净后,向管内加入100ul水,55℃孵育5min。(7)所得质粒溶液可以放入-20℃中长期保存。注意事项若菌株为革兰氏阳性菌,该菌有一层细胞壁,必须加入溶菌酶才能使之溶解。摇床速度不宜过高。达到OD600 1.5即可。溶液I加入RNase后需要放在4℃中保存,以防酶失活。溶液II和溶液III使用前需注意观察是否有沉淀,如有沉淀可37℃水浴处理。溶液II处理菌体时间应控制在5分钟以内。常见问题及解决方法常见问题原因解决办法质粒浓度低溶液处理不充分增加溶液I/II/III的加入量,或降低菌液体积。质粒拷贝数低增加摇菌量,并降低最后的溶解体积菌体老化重新质粒转化或划线后挑取单克隆摇菌乙醇残留乙醇的存在会影响质粒的溶解和回收,须延长乙醇挥发时间洗脱液不合适可配置合适的EB(10mM Tris-HCl, 1mMEDTA,pH8.5)洗脱体积较小在保证质粒浓度的前提下,增加洗脱液的体积质粒有杂带菌液污染重新质粒转化或划线后挑取单克隆摇菌试剂污染质粒提取溶液重新配置,最后溶解液高压灭菌后使用
厂商
2020.07.06

生物安全实验室的分级与分类
生物安全实验室的分级生物安全实验室可由防护区和辅助工作区组成。根据实验室所处理对象的生物危害程度和采取的防护措施,生物安全实验室分为四级。微生物生物安全实验室可采用BSL-1/BSL-2/BLS-3/BLS-4表示相应级别的实验室;动物生物安全实验室可采用ABSL-1/ABSL-2/ABSL-3/ABSL-4表示相应级别的实验室。生物安全实验室的分级如下:一级,低个体危害,低群体危害,是对人体、动植物或环境危害较低,不具有对健康成人、动植物致病的致病因子。二级,中等个体危害,有限群体危害,是对人体、动植物、或环境具有中等危害或具有潜在危险的致病因子,对 成人、动植物和环境不会造成严重危害。有有效的预防和治疗措施。三级,高个体危害,低群体危害,是对人体、动植物或环境具有高度危害性,通过直接接触或气溶胶使人传染上严重的甚至是致命疾病,或者对动植物和环境具有高度危险的致病因子。通常有预防和治疗措施。四级,高个体危害,高群体危害,是对人体、动植物或环境具有高度危险性,通过气溶胶途径传播或传播途径不明,或未知的、高度危险的致病因子。没有预防和治疗措施。生物安全实验室的分类生物安全实验室根据所操作致病性生物因子的传播途径可分为A类和B类。A类指操作非经空气传播生物因子的实验室;B类指操作经空气传播生物因子的实验室。B1类生物安全实验室指可有效利用安全隔离装置进行操作的实验室;b2类生物安全实验室指不能有效利用安全隔离装置进行操作的实验室。
厂商
2020.07.06

蛋白生产的放大,可不仅仅是增加培养瓶
放大蛋白生产可不仅仅是将细胞培养瓶的数量增加一倍。无论你是打算将你的基础研究提升一个档次,还是准备开展动物研究或i期临床试验,你都需要更加严肃认真地对待蛋白生产。随着项目越来越大,高效利用时间和经费就显得更加重要。选择最有效的方式来进行这一切,从细胞培养到收集和纯化终产物,绝对物有所值。那么,现在就让我们与“马马虎虎”告别,拥抱优化的蛋白生产吧。贴壁细胞fred schendel领导了明尼苏达大学生物技术资源中心(brc)的大规模蛋白纯化工作。他认为:“在研究中,一切都在变小、变小。新技术的出现,让你不再需要大量蛋白去做基础研究。”schendel表示,由于成本高,需求低,brc已经淘汰了哺乳动物细胞培养的设备。不过,对于那些需要几百毫克蛋白的实验室而言,还是有很多方法能提高产量。比如,schendel建议的外包。那么,在实验室中你该如何放大?许多实验室习惯使用多孔板、培养皿和培养瓶来培养贴壁细胞。换成更大的容器似乎很简单,或者尝试一下多层细胞培养瓶,或者将它们相互连接和堆叠有时,对蛋白的需求可能超出了实验室的能力。那么,一种选择就是购买更多的培养箱,或扩展到滚瓶或其他系统,当然这需要投入资金和人员。在sanford-burnham医学研究所,“他们大多数时间都尝试悬浮培养,因为这更容易放大,”主管darrin kuystermans谈道。有时,这也意味着他们要改造细胞系来生产感兴趣的蛋白。不得已,他们也会使用微载体,通过悬浮培养的方式来培养贴壁细胞。内还是外?另一种促进蛋白生产的方式是让蛋白分泌到培养基中,这样下游处理就没那么困难。“我们只需要从培养基中纯化它,而不需要裂解细胞,并在蛋白纯化的过程中处理其他所有蛋白,”kuystermans解释道。偶尔,细胞也没那么听话。即使有了信号肽,它也不分泌蛋白。在这种情况下,kuystermans可能改造纯化标签,以便更容易地从细胞裂解液中捕获蛋白。kuystermans建议先在小的摇瓶系统中检验,而不是一开始就采用较大规模的系统。搅拌引入了额外的氧气,会引起剪切并形成其他压力,这可能导致细胞开始结块,或不再生长。对于那些有疑问的细胞或蛋白,你可以试试瞬时转染,而不要花几个月的时间来筛选稳定的细胞株,然后才发现系统有问题。不断优化当你知道表达系统没问题时,下一步就是优化表达条件。最重要的培养参数包括温度、溶氧(do)、ph和葡萄糖,这些可利用探头和传感器来监控,通过手动或自动控制。一种优化方式是利用“微型反应器”系统,比如applikon biotechnology的micro-matrix或m2p-labs的biolector,也可以利用一组摇瓶。当然,这一切并非线性放大。例如,开发工作往往是分批培养,而生产是以更高密度的灌注模式开展的,利用中空纤维反应器。在这种情况下,一些培养的特征是不同的,意味着结果也可能不同。同样地,液体表面体积比往往也不能直接放大,这会影响do和co2,从而影响ph水平。优化下游(收集和纯化)过程同样重要。你需要筛选填料,以便实现最佳的分离效果。其他考虑因素包括填料在分离条件下的稳定性,树脂的刚性和柱尺寸。据kuystermans介绍,为了确保放大过程中目标蛋白在柱中的停留时间恒定,柱的直径可以增加,但柱床的高度应保持不变。尽情摇摆悬浮培养物的放大可以在可重复使用的生物反应器中进行,kuystermans称之为“clean and play”的系统。这主要指的是搅拌瓶或罐,能够监测和控制各种参数。
厂商
2020.07.03
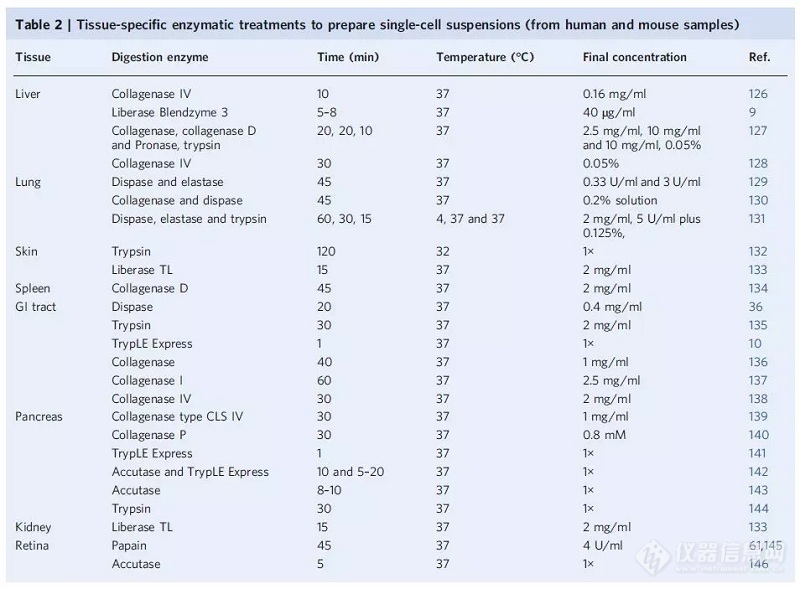
单细胞测序前的3大关键问题,您都处理对了吗?
在无数实战经验中,制备高质量的单细胞悬液被公认为重中之重。实验过程中,如单细胞活性不高,细胞数过低,以及污染和杂质等问题,都会影响到有效单细胞数的产出、单细胞核酸的质量等,最终得到的单细胞数据分析及统计结果不可信。那么,组织如何解离?需要选什么酶?还需要注意什么?科技君根据相关权-威文献整理了单细胞悬液制备的方法和小贴士,让我们一起来解决单细胞研究方案中的拦路虎。实体组织解离关键——酶的合理选择 实体组织解离两大步骤,包含机械分离和酶消化处理。首先,组织需要通过物理切割或刀片切碎,然后通过酶消化来分离细胞。特定的组织消化酶及消化时间不同,相关建议可参考如下表格:人和小鼠的部分组织类型——肝脏、肺、皮肤、脾脏、消化道、胰腺、肾脏、视网膜等[1]。 表1 人鼠各类型组织酶解单细胞悬液方法总结[1]除此之外,常用酶的类型还包括:Accutase™、弹性蛋白酶和胶原酶,以及商业酶混合物,如 TrypLE Express 和 Liberase Blendzyme 等[1],另外,科技君也总结已发表文献中常见实体组织解离所采用的酶,供大家参考,步骤详见参考文献:小鼠心脏肌肉组织:Collagenase IV and Dispase II[2]上皮组织:dispase(Corning) -商业化试剂[3]小鼠胚胎组织:TrypLE Express[4]乳-腺癌及其癌旁组织: Liberase TL (Sigma) -商业化试剂[5]小鼠主动脉血管:Collagenase type II (C6885, Sigma Aldrich) 和 Elastase (LS002292, Worthington Biochemistry)[6]小鼠脑垂体:Collagenase type II, trypsin, DNase I,amphotericin B 混合[7]血液处理成关键——离心稳定操作[1]样品经过密度离心(例如使用Ficoll-Paque或Histopaque-1077技术),可以直接用于外周血单核细胞(PBMC)捕获[1]。建议不少于5mL EDTA 抗凝血,且不要使用肝素抗凝管收集血液;同时注意在合适转数离心操作后,管中内容物分为三层,上层为血浆(内含细胞碎片),中间层为分层液,底层为红细胞,在上、中层液体界面处可见到乳白色混浊的单核细胞层( 白膜层,薄)。此时,需使用无菌吸管小心沿离心管壁周缘吸取界面层单核细胞后,再加入HBSS /PBS重悬。更多注意事项请参见华大科技单细胞送样建议。单细胞悬液制备的8条建议[1]1. 建议采用无菌样品处理方式,包括使用不含核酸酶的试剂和耗材。2. 为降低对细胞的损伤,移液和离心应保持在最低程度。在一定的离心速度、时间和温度下,细胞浓度和大小直接影响制备的效率。3. 在进行细胞清洗和重悬过程中,使用具有合适大小的器皿,避免高浓度导致细胞集聚和结块,请注意选择。4. 应使用适当大小的细胞过滤器过滤悬浮液,孔径大于细胞直径,以去除团块和碎片。5. 细胞清洗和复苏,推荐使用含牛血清的磷酸盐缓冲盐水(不含钙和镁),减少细胞损失和聚集的白蛋白。6. 细胞裂解升高可导致细胞团块形成,在细胞分离过程中,DNase I可减少细胞团块形成。7. 细胞团块会导致自动细胞计数器低估单个细胞的有效浓度,因此制备后应尽快处理悬浮液,最-好在30分钟内处理。8. 总之,在单细胞制备中,尽可能减少细胞聚集物、死亡细胞、非细胞核酸和逆转录(RT)抑制剂是非常重要的。为了在最大限度地提高不同细胞类型的纯度和无偏回收率的同时,最小化这些污染物,可能需要应用优化,例如,调整洗涤步骤的数量、洗涤溶液的组成、离心条件和/或过滤器类型。
厂商
2020.07.03

RNA测序,真的有那么准确吗?
RNA测序是遗传学家工具箱中的一种常用工具。利用RNA测序,研究人员能够定量地检测各种生物体的基因表达,从而更好地了解细胞中正在发生的事情以及特定基因的功能。由于在药物发现、疾病诊断和基因鉴定中具有潜在作用,RNA测序的应用似乎无极限。RNA测序的准确性早在2014年,《Nature Biotechnology》上发表了三篇文章。它们属于RNA测序质量控制(SEQC)项目的一部分,旨在评估新一代测序平台的性能以及RNA分析的优势和局限。研究人员测试了三个常用RNA测序平台的可靠性、准确性和信息内容,希望确定平台的使用范围并寻找变化的来源。在整个研究过程中,三个研究机构都产生了超过10亿个核苷酸的数据。他们还研究了30个不同的RNA测序实验室所使用的技术以及数百名研究人员使用的生化方法。总的来说,他们认为RNA的提取和分析可以跨机构圆满完成,即使样本已严重降解,但是遗传数据仍然可靠。这些结果让研究机构以及医生患者放心,RNA测序是准确且可靠的。一项研究的负责人、梅奥诊所的E. Aubrey Thompson评论道:“患者的护理决策似乎受到基因组数据的影响,而这些数据来源于患者样本的RNA和DNA测序。如今我们知道了这些分析可在多大程度上依赖某个实验室。”生物实验的重复性危机如今,我们正处于所谓的重复性危机中,越来越多的研究变得难以重复,甚至不可能重复。据估计,单单就美国而言,每年花在无法重复的生物研究上的费用高达280亿美元。除了经济成本之外,不可重复的研究还造成药物开发的延误,阻碍疾病疗法的开发。在设计实验和决定使用哪种技术时,重复性仍然是人们主要考虑的因素。可靠的方法通常是许多人的首-选,比如RNA测序。然而,以色列特拉维夫大学的研究人员开展的荟萃分析表明,RNA测序的数据分析过程中存在技术偏倚,可能造成数据的错误解释和大量错误结果1。RNA测序无法重复?在分析了35个公开的RNA测序数据集之后,研究人员注意到某些基因反复呈现基因表达的变化。这些数据来自近年发表的人类和小鼠研究,覆盖了多个生物过程。他们对此感到困惑,想了解这一现象背后究竟是真实的生物学现象,还是实验过程引入的人为误差。在其中30个数据集中,研究人员发现特别长或特别短的基因往往表现出最-明显的基因表达水平变化。大多数短基因编码了组成核糖体的蛋白质,而许多长基因则编码了与细胞外基质有关的蛋白质。经过进一步的研究,他们发现这种现象源于实验的人为因素,而不是天然的生物反应。在比较相同条件下的重复样本后,他们发现这种模式是由于技术偏倚,似乎与基因的长度有关。此外,若统计分析中存在缺陷,人们往往将检测到的偏倚错误地标为细胞反应,特别是与核糖体或细胞外基质有关的反应。在许多常用的数据归一化方法中,这种偏倚并未得到校正,因此可能已包含在许多数据集中。这种效应被称为样本特异性长度效应(sample-specific length effect),之前已在文献中提到过。许多研究人员已经意识到这一问题,但没有积极地解决它。在此次分析的数据集中,未经校正的数据比例仍然很高。不要低估统计分析的重要性尽管乍一看让人担忧,但结果似乎也不是那么严重。在文中,研究人员还介绍了如何克服和消除这种偏倚,从而过滤掉错误的结果,保持生物学上的相关性。将基因长度视为样本特异性的协变量,可以明显减少假阳性结果的数量。目前还不清楚哪些因素能够淡化样本特异性的长度效应,可能还需要进一步研究。作者建议研究人员在自己的研究中注意这种偏倚,并将建议的数据归一化方法作为默认步骤,应用在RNA-seq数据分析的标准实践中。美国北卡罗来纳大学教堂山分校的生物统计学家Michael Love表示:“这篇文章很好地证明了质量控制的重要性。”他指出,还有一些偏倚也会影响RNA测序数据的质量,比如GC含量偏倚,各个研究团队在分析过程中应始终考虑这些因素。他本人并未参与此项研究。这项研究强调称,所有技术都不是100%可靠的,因为偏倚的可能性始终存在。随着各个研究领域的重复性危机浮出水面并获得关注,这项工作强化了准确开展统计分析的必要性。
厂商
2020.07.03
游离核酸抽提方法选择指引
人体细胞在凋亡的过程中,DNA断裂并分泌至细胞外。健康细胞,胎儿细胞,肿瘤细胞以及移植的细胞都可以通过这种方式释放游离DNA至血液中。同时cfDNA(cell-free DNA) 是肿瘤和产前诊断研究中,被证实有用的标记物。随着液体活检技术的不断发展,对这些的游离核酸检测有望成为替代组织活检的重要方法。大家比较熟悉的游离核酸检测手段有定量PCR、NGS等传统方法和正在迅猛发展的数字PCR。但cfDNA在体液中的含量非常低,通常血清中含量低于10ng/mL。 如何从含量如此低的样本中,抽提出适合检测手段的游离核酸?液体活检技术的兴起,使得游离核酸样本的数量大增,高通量形式的核酸抽提成为了工作者们的追求,如何低成本解决通量问题?那就从下方的描述中找到适合您的游离核酸抽提方式吧。市面上的游离核酸抽提试剂盒大多是基于柱膜法的原理、还有一部分是基于磁珠法的抽提原理。有单管离心、多管真空抽滤、96孔板真空抽滤、磁珠法高通量等形式。*常用的为多管真空抽滤的方式。多管真空抽滤采用真空抽滤的方法,在真空抽滤装置上,可同时提取24个1-10mL血清、血浆、尿液等样本, 结合&洗涤过程无需使用离心机。大多客户用此方法来抽提2-4mL血浆,得到的游离核酸用于后续的PCR、NGS、甲基化研究。整个抽提过程需要约45min-3h不等,具体的操作时间要根据使用的品牌来定。应用数据:从5mL EDTA血浆中纯化cfDNANucleoSnap® DNA Plasma kit 和竞争对手(Competitor Q)比较结果从Cell-Free DNA BCT® (Streck)提取cfDNA 3个不同Cell-free DNA BCT® 保存的样品,分别使用 NucleoSnap® DNA Plasma (MN) 和a vacuum-processed kit of a competitor (Q)提取cfDNA.使用Bioanalyzer™ 2100 with a High Sensitivity DNA Kit (Agilent)检测,分离到的游离核酸在170bp附近有典型的峰(箭头标记)10mL尿液中分离cfDNA电泳图抽提的cfDNA使用 Bioanalyzer™ 2100 with a High Sensitivity DNA Kit (Agilent)分析,在170bp附近有典型的片段分布
厂商
2020.07.02

做转染实验,别忽视质粒中内毒素
什么是内毒素?细菌内毒素是革兰氏阴性菌细胞壁上的一种脂多糖(LipopolySaccharide,LPS)和蛋白的复合物,当细菌裂解时便会大量释放出来。内毒素单位为EU。内毒素对试验的影响内毒素存在极大程度地影响转染效率。同时内毒素还可能激活免疫细胞的非特异反应,造成实验中的假阳性。因此,为了保证高转染率和转染细胞高存活率,必须从质粒制备的过程中将内毒素去除。内毒素和质粒的关系由于内毒素两亲性和负电荷性,性质类似于DNA,因此在质粒纯化中会伴随质粒一同被纯化出来。质粒中内毒素含量的高低常用EU/μg质粒表示。按照EU/μg大小,质粒被分为三个级别:测序级别(>50EU/μg)、转染级别(再次强调当你提取的质粒DNA是为了转染原代细胞、悬浮细胞或者敏感细胞;进行基因沉默研究、细胞注射显微操作;进行基因治疗或者DNA疫苗等要求极为严格的实验,就应该考虑Endotoxin-Free的质粒。2017年3月,张锋课题组在《Nature Protocols》发表的《Genome-scaleCRISPR-Cas9 knockout and transcriptional activation screening》中,Endotoxin-Free的质粒获得被列为 Critical Step,同时推荐该厂家的NucleoBond Endotoxin Free Plasmid DNA kit,可避免病毒包装和细胞培养过程中减少了内毒素残留的影响。
厂商
2020.07.02

测序污染有多危险,如何防范与去除
一些研究表明,目前已经公布的基因组存在多种污染,随着这个问题越来越突出,我们需要找出方法来应对Supratim Mukherjee在进行数据分析的时候,发现数以百计的微生物基因组中会重复出现同一种噬菌体序列,这令他感到很惊讶。这位来自劳伦斯伯克利国家实验室的生物信息学家最开始是为了比对这些微生物的代谢途径,但后来他发现了几乎无处不在的序列,“我以为我们发现了一些新的东西,”他回忆道,“在这些不同的微生物中,这整个噬菌体基因组是完整地保留下来的。”但当Mukherjee一开始分析这个噬菌体序列时,他就知道这就是 PhiX 序列,一种Illumina公司测序试剂盒中用做标准品的噬菌体。PhiX 本来是作为一种质控检测指标,用于追踪每个测序过程中出现的错误率的,但在上百个案例中,Mukherjee发现研究人员都没有从其公布的基因组序列中剔除Phi X的序列。并不是只有Mukherjee一人发现此种情况,最近大量的报告表明,发表的基因组出现污染要不之前想象的多得多。那么这些污染是如何出现的呢?我们有能做些什么,避免这些情况的出现呢?就此The Scientist杂志请教了几位研究人员,他们分享了他们的一些Tips,可以检测和预防出现“流氓序列”。广泛的基因污染在Mukherjee 研究组意识到 PhiX 污染可能会出现了多个公布的微生物基因组中之后,这一研究组觉得量化其出现频率。通过分析调查,Mukherjee等人发现在已出版的1.8万个细菌和古细菌基因组(Integrated Microbial Genomes database)中,超过1000个序列被PhiX 序列污染。今年Mukherjee等人已经将这一发现公布在Standards in Genomic Sciences上。而这些其中的10%也出现在了同行评审的期刊杂志中。PhiX 污染还只是冰山一角——现在问题呈指数级增长,NCBI总监David Lipman说,他也正在筛选过去五年间,呈递到GenBank中的数据。“我们检测到2012年细菌和古细菌的污染情况还只有2%-3%,” Lipman说,“但之后就急速攀升,到2014年,已经接近了10%。今年到目前为止,这一比率达到23%”。Sanger研究所的科学家们也发现,DNA提取试剂盒、化学试剂和实验室环境中的杂菌很容易造成污染,影响微生物组分析的结果。研究人员发现,没有污染的话对照样本应该只有一种菌,但有时却出现了270种不同的细菌。与高生物量的样本相比(粪便样本),来自血液或肺部的低生物量样本尤其容易受到污染。“现在的DNA测序技术允许人们进行深度测序,被广泛用于稀少微生物群体的分析。我们发现,这类样本很容易被其他来源的DNA污染,要么在收集样品的时候,要么在DNA提取和扩增过程中。污染会对研究结果产生很大的影响,这一点需要研究者们给予足够的重视,”Sanger研究所的Alan Walker博士说。而且微生物也不是唯一出现这么多污染的研究领域,去年伦敦大学学院的计算机专家William Langdon发现,千人基因组计划中至少7%受到了支原体遗传物质的污染(BioData Mining, 7:3, 2014),因此如果说你对污染的基因组感到头疼的话,放心,你不是唯一一个。污染从哪里来?来自圣地亚哥州立大学的生物信息学家Rob Edwards说,污染出现的来源很多,“首先就是实验室成员可能混淆了两个样品,不小心给文件或者样本贴上了错误的标签。这些都可以通过加强实验室管理,提高实验记录保存制度等很容易解决。”另一方面,污染也有可能来自其它本不应该出现在样品中的外来遗传物质,又或者来自培养细菌周围的环境,Edwards说。即使你认为自己测序的是单一培养产物,但是在一个测序循环中出现多个物种的情况,并不少见。此外,如果正在测序来自人类肠道的微生物,那么样品中自然会出现人体细胞,还有即使你只想要测序某个生物体的细胞核基因,也会出现细胞内线粒体和叶绿体基因,这些也都是污染。这些污染当然很难完全避免,但是可以采取一些措施:在测序之前清理样品,或者在测序结果中剔除污染的序列。Edwards的研究组聚焦于来自环境样品的宏基因组测序,他表示其研究组就常常利用过滤设备,根据大小对病毒和细菌混合物进行分离。如果他们推测样品中存在人体DNA的污染,那么就会先剔除这些序列,只留下微生物的基因样本。同样如果需要清除系统中的污染,比如PhiX 对照序列,目标基因序列扩增测序用的引物和测序接头,还有克隆载体等,也可以采用相类似的方法。考虑完这些,还有一个容易忽略的问题,那就是设备机器在实验过程中留下的污染,清楚了解这些污染的来源,可以帮助研究人员在测序后选择方法剔除他们,Edwards说,如果污染重复出现,那么也许就需要改变方法或调试机器了。然而污染的另一个来源是脏之间实验,出血,通过让基因由事先测序运行出现在下一次的机器。爱德华兹说,只被察觉这种污染可能存在于你的实验可以帮助您选择将其删除后测序的方法。或者,如果它反复出现,您可以尝试geng改协议或故障排除您的机器。如何检测?毫无疑问,在实验过程中越早剔除污染物越好,“这些污染会增加实验直接的成本,”来自爱丁堡大学的Dominik Laetsch 说,出现污染,“每分钱理论上你得到的核苷酸信息就越少,”因为需要花时间处理和分析不需要的序列。但也有个好消息——即使序列中充满了 PhiX、引物、载体和不想要物种的基因,还是能在别人看到你最终公布的基因组之前剔除它们。Laetsch就开发了这样的一个工具,帮助数据分析之前进行序列清除,这个工具叫Blobtools-light,是目前的最新版本,能将你的contigs(组装成最终序列中的测序DNA重叠部分)与NCBI数据库中的已知序列进行比对,然后软件还会通过可视化方式来解释这种比对——来自相似生物物种的序列会突出来。“我们利用这作为初步筛选工具,”Laetsch说,她正在进行病原细菌的相关研究。此外,还有一个类似的程序:ProDeGe (Protocol for fully automated Decontamination of Genomes,全自动净化基因组协议)(ISME, doi:10.1038/ismej.2015.100, 2015).与Blobtools一样,ProDeGe采用的也是公共数据库,可以检测一个基因组中的污染,然后将contigs分组归类到“无污染”组和“污染”组。比价而言,Blobtools-light可以提供可视化序列图表,ProDeGe则能帮助研究人员识别并鉴定污染物是什么。“这种方法比较简单,不用了解太多”,Mukherjee说,“因此对于不擅长此类工具的研究人员来说比较合适。”当然还有其它方法,如NCBI的VecScreen,这是一种可以快速识别序列中污染载体的方法,晚些时候NCBI网站还将公布geng多geng先进的工具。不过所有用来检测污染物的工具都必须把握住特异性和敏感度之间的平衡,也就是精确识别出污染物,而不删除靶标序列。因此了解清楚你的整体数据就显得额外重要,比如说,如果你分析的是新的基因组,那么程序肯定会提示了污染物水平高,因为已有数据库并未包含你的序列数据。又或者,如果你知道会出现高污染细菌基因组,那么就能列出污染物清单,Edwards说,“我推荐多运行几个工具,比对结果。”如何去除污染一旦找到了污染物和污染源,那么就可以开始进行数据清理了。这其中有多种工具可以选择,如Edwards研究组开发的DeconSeq,与其它自动化污染筛选程序不同,DeconSeq需要用户输入污染物的物种属性,然后再自动剔除基因组组装内容里的属于这一物种的序列。如果跳过了这一步骤,也许就会引起麻烦。Lipman研究组在NCBI系统中就运行一个针对每个呈递到GenBank中序列的外源污染物筛选,他希望当筛选出一个序列标记为污染物时,科学家们能将其认为是了解数据的一个机会,并且了解技术的弱点,在未来避免出现这个问题。“如果你只是说‘好吧,我的呈递出现了问题,我现在就修改它’,那么这个问题还是不断出现,”Lipman说。但是如果是在论文公布后发现基因组中出现污染呢?比如说之后进行geng多实验的时候发现了错误,那么重点是尽早修改错误,以防其他人将这些错误的成果用于自己的研究中。在某些情况下,这也许就意味着与杂志取得联系,看看能不能进行勘误。“大家需要对自己的序列数据负责,”Mukherjee说,“如果你发现了问题,那么就要撤回它进行修改,然后再重新发布。”如何改善基因组污染问题随着测序技术的进步,也许未来许多污染源会自动消失,这确实可能,Laetsch说,“随着组装过程越来越容易,读长越来越长,肯定要找出污染也会变得容易,”但是研究人员不能将这作为停止筛选污染物的借口,“你放入的样品越好,测序机器就会做的越好。”而随着基因组数据变得越来越庞大,要想获得干净的序列也越来越难,这有赖于每个学者都尽其所能确保自己基因组序列不出现污染,“我认为科学界都知道污染物是个大问题,但是这还需要geng多的努力”,Mukherjee说。GenBank中污染物出现频率猛增,Lipman也赞同这个问题的共识性,为何会出现越来越的污染呢,Lipman对这个问题表示,“越来越多的实验室都可以进行测序研究了,这本身是个令人高兴的事情。”
厂商
2020.07.02
钱要花在刀刃上,基因组测序如何省钱?
坦率地说,基因组测序中最昂贵的部分是测序本身。测序仪的消耗品成本(包括测序芯片和试剂)明显盖过了测序的其他成本,更不用提一开始的资本支出。当然,钱要花在刀刃上,无论我们是拥有自己的仪器还是借用别人的仪器,都有办法来削减成本。在此,我们梳理了测序的整个流程,从样本制备到数据分析,看看可以在哪些地方减少开支以及这样做的隐性成本有哪些。一般来说,通量较低的Illumina测序仪的购买和维护成本比较低,而耗材成本也相应较低。从另一个角度来看,高通量的测序仪能够产生大量的数据,使得每Mb或每个样本的成本降低。GenapSys的CEO Hesaam Esfandyarpour指出,无论采取哪种方式,由于每次运行的耗材成本是固定的,因此让测序仪满负荷运行才是上乘之选。这样,样本不多、通量较低的研究人员就可以先选择浪费容量(其实也就是浪费钱),等待样本量逐渐上去。或者,他们也可以选择外包服务。外包当然更省事,但需要服务费,从送样到拿回数据还需要一段时间。其实,大家在仪器上还有更多选择,比如最近上市的GenapSys测序仪。这台测序仪在美国的售价低于一万美元,且每次运行的耗材成本仅为几百美元。它的平均读长超过150 bp,>80%的碱基质量高于Q30。利用16M芯片开展测序,每次可产生1.2-2 Gb的数据。Esfandyarpour认为,每个研究人员都可以拥有DNA测序仪,并自行决定运行样本的方式。选择外包?外包也是一门艺术。就外包服务而言,它有许多不同的切入点。GE Healthcare的技术经理Andrew Gane表示,有些人会直接提供样本,而有些人会提供制备好的文库,让供应商上样到仪器中。对于那些愿意自己动手的研究人员而言,节省成本的机会出现了。某些实验达人甚至会自行开发平价的解决方案。他们会考虑,操作方案中的哪些步骤是必需的,有没有办法跳过一些?实验过程中是否需要保真度最高的酶和最好的磁珠来进行PCR扩增和DNA片段筛选?有些研究人员,特别是开展单细胞测序(scRNA-seq)的研究人员,可能会选择同时分析多个样本。加州大学旧金山分校Genomics CoLab主任Walter Eckalbar指出:“在10x Genomics平台上开展单细胞基因组学分析,每次样本制备的成本约是1,500美元,而bulk RNA-seq的成本大约是100美元。”Gane表示:“目前有多种方法可以在多重分析之前或分析期间对样本进行归一化,从而减少质量控制的时间,”不过这些方法也会带来其他成本。Eckalbar认为,在确定特定工作流程的实际成本时需要考虑多个因素。其中包括总共有多少个步骤,是否有附加的步骤(比如在Bioanalyzer上进行QC),是否需要购买额外的磁珠,甚至是否符合标准的工作日安排。自动化操作通过自动化,实验小量化(miniaturization)也更容易实现,而这又是节省试剂的一种方式。“如今,我们不再按照试剂盒操作手册上规定的体积来操作,而是利用液体处理系统进行1/2或1/4体积的反应,有时甚至是1/10,这在手动移液时很难做得到。”他说。“而且,自动化系统可以实现更高通量的操作,比如384孔板或更高。”“小量化也存在缺点,在大多数情况下,你会牺牲一些数据质量,从样本中检测到的基因数量可能会减少,”Eckalbar说。“不过好处是,在预算不变的情况下,你可以分析更多的样本,从而具有更好的统计能力。”数据解读在测序实验完成后,你还需要处理数据。处理数据的方式有很多,包括测序公司提供的云端分析app(免费或付费)、第三方提供的商业化数据分析解决方案,以及提供数据分析服务的供应商。具体选择哪个,这可能要取决于问题的复杂程度和研究人员的熟练程度。Eckalbar指出,许多免费和付费的解决方案可能都存在限制,出于种种原因,你的数据可能不适合。这时候,你不得不将数据交给专业的机构去分析。不过,“许多硕士生和博士生都在努力学习这些生物信息学工具,以便亲自上阵”。
厂商
2020.07.01

酵母计数和活性怎么测? 用它!
酵母菌简介:酵母菌是基因克隆实验中常用的真核生物细胞,也是食品工艺中常用的微生物,如酿酒酵母。酵母菌细胞宽度(直径)约2~6μm,长度5~30μm,有的则更长,个体形态有球状、卵圆、椭圆、柱状和香肠状等。由于其尺寸较小,形状不规则,因此采用明场法无法进行准确的计数,更不易进行酵母活性的检测!采用双荧光通道设计的细胞计数仪,可更加准确地进行细胞计数和细胞活性分析。接下来我们看一下它是如何进行酵母计数和活性检测的吧?步骤一:FDA/PI染色取1?l FDA试剂、1?l PI试剂加入到18?l酵母菌液中,避光孵育10min。FDA/PI试剂染色原理介绍:FDA是一种非荧光性疏水性荧光素衍生物,它可以穿透细胞膜进入细胞,通过细胞内酯酶催化水解二乙酸酯基团产生具有高荧光强度的荧光素。因此活细胞具有绿色荧光。无活性的死细胞被PI着色,发出红色荧光。因此,可以很好的辨别死细胞与活细胞,准确的进行细胞活性的检测。步骤二:上样用移液枪吸取10?l 样品加入到细胞计数板中。将细胞计数板插入到LUNA FL中。步骤三:检测直接点击内置的酵母检测App在明场下进行调节焦距,然后点击Count即开始细胞检测,30秒内即可完成。步骤四:结果查看、分析、导出结果查看:我们可以得到Total /Live /Dead细胞数量、细胞活性、细胞平均尺寸等数据。结果分析:我们可以设置细胞分析的尺寸范围以及聚团情况等。结果导出:点击Save/Print即可将结果导出,您可以选择保存3个通道的原始图片、加了Tag的图片、包含了该次检测所有信息的PDF文件。
厂商
2020.07.01
企业名称: 上海远慕生物科技有限公司
企业地址: 上海浦东新区 联系人: 安小姐 邮编: 201600 联系电话: 400-860-5168转3328
仪器信息网APP

展位手机站