视频号

抖音号

哔哩哔哩号

前沿资讯手机看

北京大学化学院教授,主要从事化学和计算驱动的功能蛋白质组学相关研究



分享到微信朋友圈
打开微信,点击底部的“发现”,
使用“扫一扫”即可将网页分享到朋友圈。
引言
准确预测蛋白质-配体(在本文的语境中,配体意指小分子有机化合物)的结合构象是计算生物学中的一项重要任务。一方面,它有助于人们理解蛋白质与内源小分子或药物分子的互作机制。另一方面,在药物设计或药物筛选(无论是单个蛋白-多个配体的正向筛选,还是单个配体-多个蛋白的逆向筛选)的过程中,也离不开对复合物构象的准确预测:这是准确计算蛋白质-配体结合稳定性(亲和力)的必要条件。
对于给定蛋白和给定配体分子,依据是否已知结合口袋(也称配体结合位点)可以将复合物构象预测任务分为两类:口袋未知的盲对接(blind docking)任务和口袋已知的局部对接(local docking)任务。其中,盲对接任务是更普遍的、更富有挑战性的任务。在传统的对接流程中,这一任务又被划分为几个子任务:先借助口袋搜索算法确定可能的结合位点(即,转化为局部对接任务);再利用构象生成(采样)方法生成大量可能的复合物构象,并依据打分函数评价各个构象(即,进一步确定配体的位置、朝向,以及各个键对应的扭转角);挑选出打分值最优者作为最终结果。但是,对接过程面临着打分函数不够准确、构象搜索空间巨大导致计算耗时过长等挑战。
比如,对于后者,文献[1]计算结果表明:对于单个配体复合物的盲对接任务而言,借助GNINA和Glide这两款传统的对接软件,生成百万量级的复合物构象并从中挑选最优者,平均耗时分别约为150 s和1500 s。在巨型分子库的(如ZINC 15数据库,含有约数十亿个化合物分子[2])虚拟筛选过程中,使用传统方法逐一对接每个分子在计算速度上是不可接受的。而数据驱动的机器学习方法为优化各个子任务的准确性和计算效率带来希望。
此外,同样基于机器学习方法,部分研究者发展出“端到端”的、更为直接的对接流程:训练一个拟合自由能面(free energy landscape)的模型,无需将原有的对接任务划分为多个子任务,以蛋白质与配体的三维结构为输入,输出即为可能的复合物构象。参照Anfinsen提出的蛋白质折叠热力学假说,可以设想:如果存在一个能量函数,能够将复合物在三维空间下的所有状态映射到它对应的自由能,那么可以将复合物构象预测问题转化为该能量函数的最优化问题[3]。这是机器学习拟合自由能面的出发点。
与传统的对接方法相比,这样的方法也同样可能在准确性和计算效率等指标上得到提升。本文将针对口袋搜索、构象生成、打分函数、自由能面建模这四个方向:整理相应的评价体系,包括常用的评价指标或测试集;挑选并简要介绍部分具有代表性的机器学习模型;结合模型评估实验讨论当前模型或评价体系存在的不足以及未来可能的发展方向。1.机器学习口袋搜索1.1 评价体系目前存在两种描述蛋白质-配体结合口袋的方式。
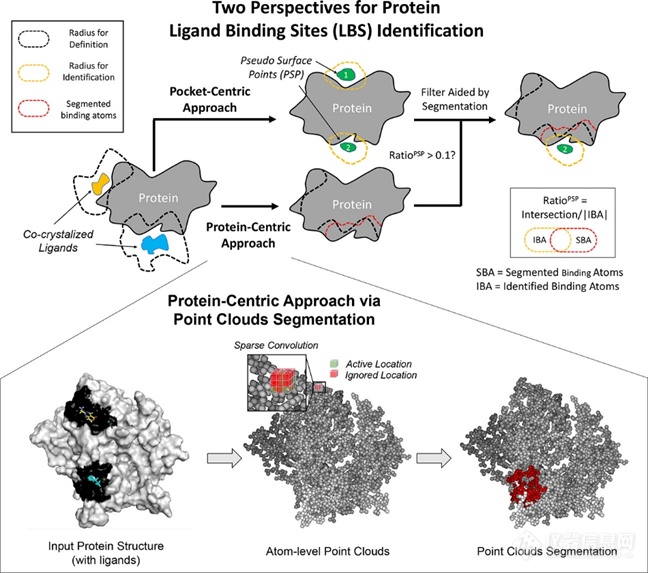
其中一种方法借助蛋白质表面的点云(surface points)来描述。这种方法被称作以配体为中心(ligand-centric)的方法。另一种方法则借助蛋白质上的原子来描述,那些蛋白质上的、与配体重原子距离小于一定阈值的重原子被定义为配体结合原子。这种方法被称作以蛋白质为中心(protein-centric)的方法。
这两种描述方法相应地衍生出两类评价指标。对于前者,通常以配体原子中心距(预测的口袋中心与配体任意原子的距离,distance between the center of the predicted site to any atom of the ligand,DCA)、配体中心距(预测的口袋中心与配体中心的距离,distance between the center of the predicted site to the center of the ligand,DCC)、体素交并比(预测的口袋体积与配体所占据的空间体积的交并比,discretized volume overlap,DVO)等指标来衡量[4][5]。直观上,在这三个指标中:DCA最为宽松(只需大致判断配体位置),DCC更为严格(额外考察了配体大小),DVO最为严格(额外考察了配体构象)。但在多数文献中,常使用DCA和DCC这两个指标,并以4 Å作为预测成功与否的阈值[4]。对于后者,Yan等人以原子水平的交并比(Intersection over Union)来衡量[6]。具体地,计算预测的配体结合原子与标注的配体结合原子的交集与并集的比值。这也是机器学习目标检测任务中的常用指标。1.2 模型方法目前发展的大多数口袋搜索算法,都是以搜索蛋白质表面点云为目标。例如Krivák等人于2015年提出的P2Rank[7]:刻画蛋白质Connolly点云中各个点的物化特征,并使用随机森林模型对每个点进行可靶性预测,最后对点聚类得到口袋预测结果。以DCA为评价标准,P2Rank在多个数据集上表现优于传统方法Fpocket。Krivák等人后续还提供了P2Rank的网络服务[8],并对多种方法口袋搜索方法进行了更加全面的总结和比较,其中包括Jiménez等人于2017年开发的、使用3D网格(体素)刻画结合位点的DeepSite[9]。
值得一提的是,在Krivák等人的结果中(测试数据集为COACH420、HOLO4K,评价指标DCC),DeepSite表现不及Fpocket;而在Jiménez等人的结果中(测试数据集CHEN251,评价指标DCC、DVO),DeepSite表现显著优于Fpocket。最近,Yan等人另辟蹊径,以鉴定蛋白质上的配体结合原子为目标,发表了PointSite方法[6],并声称其在多个数据集上(包括COACH420、HOLO4K、CHEN251等等,以原子水平的交并比为评价指标)取得了SOTA。Yan等人将口袋搜索问题转化为计算机视觉领域的点云分割问题;此处的点以蛋白质上的原子表示,以充分挖掘原子之间的键连特征。此外,作者还指出:将PointSite方法引入到其它口袋搜索方法如FPocket、P2Rank中(具体而言,使用点云分割的结果对后者的预测结果进行过滤),可以进一步提升配体结合位点的鉴定效果。
1.3 讨论从以上几种方法的评估实验中可看出,模型在不同测试数据集上的相对表现可能存在差异,因此需要建立一套统一的、合适的数据集进行测试。另外,在训练数据集的准备过程中,将未鉴定到配体结合的位点直接划分为负样本也值得考量。2.2在具体使用建议上,最近有研究将口袋搜索方法引入到完整的对接流程中[10][11],相较于FPocket、P2Rank等方法,PointSite表现最优。
2. 机器学习构象生成
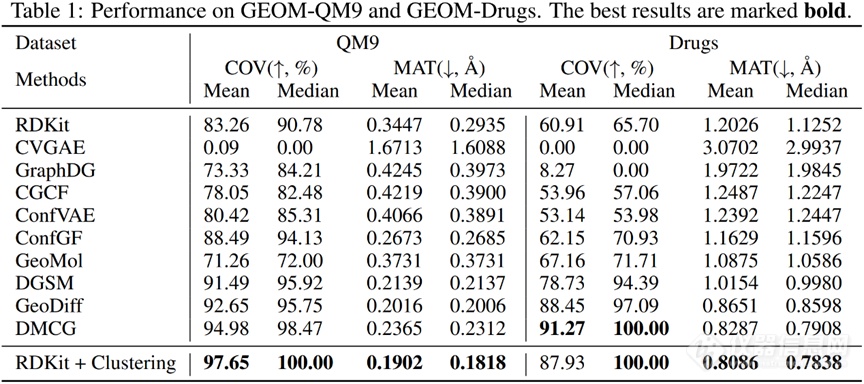
2.1 评价指标对于构象生成模型而言,需要考察所生成构象的多样性和准确性,由此分别衍生出两类指标:覆盖率(COV,coverage)和匹配程度(MAT,matching metrics)。针对测试集中的每一个构象:查看是否能够在生成的构象集合里找到RMSD值小于给定阈值的构象(如果能,则表示该模型能够覆盖当前测试构象),可以计算得到覆盖率;计算生成的构象集合与当前测试构象的最小RMSD值,取平均可以得到匹配程度的值。以上是从测试集的角度衡量模型表现(召回率);相应地,将测试集构象与生成集构象在计算中对调,则可判断模型的准确率。目前常见的测试数据集包括GEOM-QM9和GEOM-Drugs[12]。
2.2 模型方法构象生成模型沿着两个思路发展:直接生成各个原子的3D坐标;先生成原子对距离、二面角等中间参数,再将分子3D坐标还原。对于前者,技术难点在于保证模型对于输入分子的旋转-平移不变性(整体改变分子坐标,得到的构象是相同的,也即对齐过程)。对于后者,则可能生成不合法的中间参数(比如违反三角几何关系),或者中间参数的误差在训练过程中不断累积,影响分子3D坐标重构,需要进行后续的力场优化;但这是目前大多数方法所选取的道路。此处简要介绍最近发展的GeoDiff模型[13]和DMCG模型(Direct Molecular Conformation Generation)[14]。X
u等人于2022年提出GeoDiff,使用扩散模型直接生成原子坐标。GeoDiff在GEOM-Drugs数据集上测试集覆盖率可达约89%、匹配程度约0.86 Å,并且经过力场优化之后可以进一步提升模型表现。作者指出:在逆向扩散过程中(生成过程),如果某一时刻T的密度函数具有旋转-平移不变性,且逆向生成的条件概率函数具有旋转-平移不变性,则T时刻以前任意时刻的密度函数也具有旋转-平移不变性。
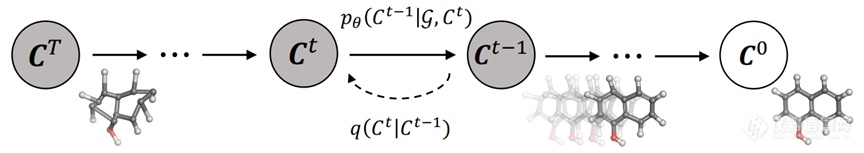
同年, Zhu等人提出基于变分自编码器的DMCG模型,在GEOM-QM9和GEOM-Drugs数据集上均取得最优(对于后者,覆盖率约96%,匹配程度约0.70 Å)。作者指出:模型除了满足旋转-平移不变性外,对于对称结构还应当满足置换不变性(交换对称原子的坐标,得到的构象是相同的)。为此,计算任意旋转-平移操作以及对称原子置换操作下的两个结构的距离最小值,并将其引入损失函数中,以满足以上两种不变性。消融实验表明,如果不考虑这一项损失,则覆盖率将下降约20个百分点,匹配程度将提高约0.3 Å。
2.3 讨论今年3月,Zhou等人[15]基于RDKit设计了一种简单的生成算法:使用RDKit分别采样二面角、采样几何片段、采样能量并生成相应的构象,随后按照能量大小进行聚类。在GEOM-QM9和GEOM-Drugs两个数据集上,与GeoDiff、DMCG等深度学习方法相比,该算法几乎在所有指标上取得SOTA。作者认为,目前的测试基准不足以覆盖实际应用中(如分子对接中)涉及的构象生成任务。

事实上,生成足够的分子构象不会降低测试构象集上的匹配程度和覆盖率(召回率),但可能降低生成构象集的相应指标(查准率)。而Zhou等人(包括Zhu等人的DMCG)并未在文中给出关于后者的模型评价,因此模型的实用性仍有待考察。另外,目前的构象生成方法均以单个配体的势能极小值作为优化目标,针对(已知口袋的)复合物中配体的构象生成模型仍有待开发。最后,GeoDiff模型与DMCG模型的发展也启示我们挖掘任务目标中蕴含的性质(对称性、不变性),在模型训练中引入合适的归纳偏置。
3. 机器学习打分函数
3.1 评价指标Su等人[16]于2019年建立了一套打分函数的基准测试数据集CASF-2016。CASF-2016及其前身CASF-2013已被广泛用于评估打分函数的表现。同时,Su等人设计了四类指标分别考察打分函数的打分能力、排名能力、对接能力和筛选能力。打分能力通常以Pearson相关系数来衡量:考察天然蛋白复合物的计算打分值与实验结合常数的对数之间的线性相关性。排名能力通常以Spearman等级相关系数或Kendall等级相关系数来衡量:对于同一蛋白、不同配体的多个天然蛋白复合物结构,考察计算打分值给出的排名与实验结合常数给出的排名之间的匹配程度。对接能力以对接成功率衡量:对于单个复合物,在天然配体结合构象和一系列计算生成的诱饵分子构象(decoy)中,若计算打分最高者与真实结合构象的RMSD小于2 Å,则认为对接成功;对于多个复合物,进一步计算对接成功率。
筛选能力以筛选成功率衡量:在天然配体和一系列其它配体分子中,计算打分前1%(5%、10%)结果里包含天然配体的比例。由此可见,打分能力直接以实验数据作为参考,是评估打分函数是否可靠的基本测试。排名能力是对打分能力的补充。打分能力越好,排名能力通常也越好;反之则未必成立(存在对实验结合常数进行非线性拟合的打分函数)。对接能力测试将计算生成的构象引入测试集中,因此更贴近实际对接操作、对于打分函数的选择更具参考意义。需要指出的是,对接能力测试给出的结果通常只能代表该打分函数的对接能力上限,在CASF-2016的测试中可能呈现分数虚高的情形(在测试中能够以90%的成功率在top-1中挑选出天然配体构象,但在实际应用中却不能达到这一表现)。这主要归结于实际应用中计算生成的构象不够充分。筛选能力涉及多种配体的对接,因此可视为对排名能力和对接能力的综合考察。
3.2 模型方法针对打分函数的机器学习方法,前人已给出详尽的综述[17][18][19]。本文将展开介绍经典的ΔVinaRF20打分函数[20],以及最近发展的DeepDock[21]和RTMScore[22]。ΔVinaRF20由Wang等人于2016年提出,在CASF-2013、CASF-2016测试集上的各指标中均排名靠前[16][20]。具体地,在CASF-2016测试集上,ΔVinaRF20在打分能力和排名能力两个指标上分别以0.82和0.75取得最优,在对接能力上以89.1%(top1)仅次于Autodock Vina(90.2%),在正向筛选能力以42.1%(top1)取得最优、逆向筛选能力以15.1%(top1)位居第五(次于最优方法ChemPLP@GOLD约2.4%)。Wang等人指出:机器学习打分函数与经典的打分函数在这些指标中各有所长,前者长于打分,后者长于对接和筛选。因此作者拟结合二者优点:一方面对训练集进行数据增强,将计算生成的诱饵结构引入训练集中(同时计算估计亲和力作为标签),以提高机器学习打分函数的对接和筛选能力;另一方面使用随机森林方法对AutoDock Vina中的打分函数进行参数化修正(使用Δ-machine learning方法,类似残差拟合)。Méndez-Lucio等人于2021年提出的DeepDock方法在CASF-2016的正向筛选和逆向筛选能力评估中分别以43.9%和23.9%取得SOTA,但DeepDock给出的打分值与实验结合常数的对数之间不存在相关性(在训练过程中未引入实验结合常数的相关信息)。DeepDock方法使用二维分子图刻画配体、多面体网格点刻画蛋白质口袋(参考了MaSIF的编码框架[23]),分别学习蛋白质口袋与配体原子的节点表示;随后两两组合配体和靶蛋白的节点形成节点对,使用混合密度函数拟合节点对的距离分布(概率密度函数)。作者指出:相较于通过最小化距离误差来学习节点对距离的平均值,混合密度函数能够学习训练集中节点对多个可能的距离,从而更好地刻画构象预测任务中的多值特性。
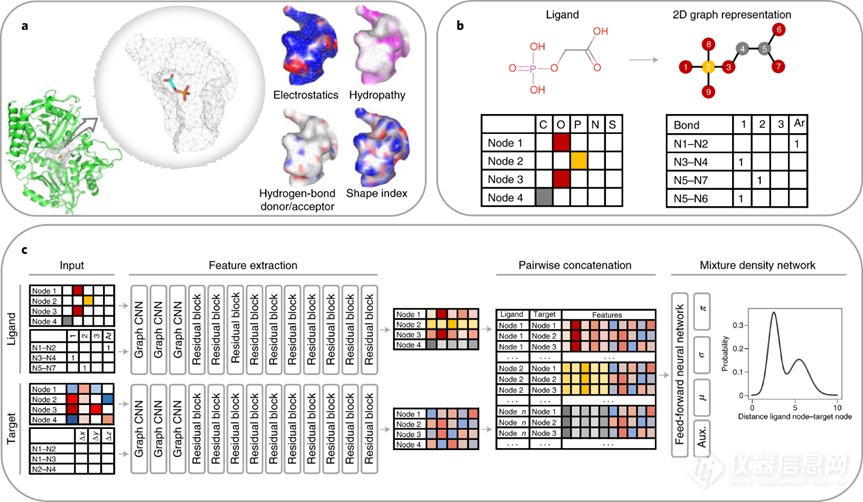
在DeepDock的基础上,Shen等人从两方面进行改进得到RTMScore:一方面,使用无向图编码蛋白质口袋残基,在编码过程中满足对于输入复合物坐标的旋转不变性;另一方面使用Graph Transformer模型以学习更深层次的特征。作者声称RTMScore在CASF-2016测试集上的对接能力和筛选能力达到SOTA,相较DeepDock得到大幅提升:对接成功率达到98.6%,正向筛选成功率达73.7%,逆向筛选成功率达38.9%。
3.3 讨论DeepDock等方法的发展展现了图模型在捕获蛋白质-配体互作的潜力,以及直接拟合蛋白残基-配体原子距离似然函数的有效性。事实上,距离似然函数不仅能作为打分函数评估当前构象,还能够作为某种“势能面曲线”指导构象优化。此外,这些新近提出的DeepDock等方法有待更广泛的测试验证。在其它论文的评估实验中[24],RTMScore在对接能力中依旧表现最优。但考虑到缺少打分能力、排名能力等测试数据,后续仍需要更多的测试评估(尤其是将打分函数整合到完整的对接流程中)以验证这些方法的可靠性。
4. 机器学习自由能面
4.1 评价体系拟合自由能面的模型直接处理盲对接任务,生成复合物构象。其中存在两类评价指标。一类指标评估计算准确性。通常以预测复合物(如果模型给出多个可能的构象,则选取打分值top-1者)中配体重原子RMSD小于2 Å所占的比例来衡量模型对接能力。一般以2 Å作为对接成功与否的判断阈值[10]。还有通过配体质心距离小于2 Å(或5 Å)所占的比例来考察模型是否能够找到正确的结合口袋。此外,为判断生成的配体构象在化学上是否合理,Corso等人额外考察了配体构象中存在位阻冲突(steric clash,配体内部重原子之间的距离是否小于0.4 Å)的比例。另一类指标评估计算效率;这在大型分子数据库的虚拟筛选过程中同样不可忽视。以对接一个分子所需的平均CPU(如果可能,使用并行加速)或GPU时间来衡量。
4.2 模型方法拟合蛋白质-配体自由能面的机器学习方法最近得到逐步发展,代表性的方法包括EquiBind[1]、TANKBind[25]、DiffDock[10]。Stärk等人于2022年提出基于图几何深度学习的EquiBind方法,在机器学习方法直接预测蛋白质-配体结合构象这一问题中做出开创性贡献。该方法以随机的配体分子构象(比如使用RDKit生成的构象)作为输入,无需经过大规模的构象采样即可在约0.1 s的时间内给出复合物结构。由此给出的结构在寻找结合口袋的能力上与传统方法(如QuickVina-W)相当(配体质心距小于2 Å的比例均约40%),但在配体结合构象的预测上却表现不佳(配体RMSD小于2 Å的比例约6%,不及QuickVina、GLIDE等方法所达到的约20%)。虽然可以在该结构的基础上结合传统方法进一步微调配体位置和构象,但将增加预测所需的时间成本至数十秒或数百秒。
同年,Lu等人提出TANKBind。相较于EquiBind,在保留推理速度(约0.5秒)的同时,TANKBind在配体构象预测上取得和传统方法相当的结果(配体RMSD小于2 Å的比例约19%),口袋预测能力则获得较大提升(配体质心距小于2 Å的比例约56%)。不同于EquiBind对整个蛋白质进行编码的方法,TANKBind采用P2Rank寻找口袋位置,随后针对该位置的蛋白质区块(由半径20 Å内的残基构成),拟合蛋白质残基与配体原子的距离。此外,受AlphaFold2的启发,TANKBind将三角几何约束引入残基与配体原子的距离建模中。消融实验表明,三角几何约束可以显著提升模型表现:配体RMSD小于2 Å的比例提升约15%,配体质心距小于2 Å的比例提升约12%。

同年十月, Corso、Stärk等人提出DiffDock模型。该模型在对接准确性上首次实现了对传统对接模型的大幅超越。在holo态的蛋白晶体对接结果中,配体重原子RMSD小于2 Å所占的比例可达到38.2%,约为传统方法的两倍。这一结果对应于40次采样,消耗计算时间约40秒。与DeepDock想法类似,DiffDock使用生成模型来学习构象的概率分布并建立了一套相应的“扩散”方法(构象采样方法)。

4.3 讨论今年2月,Yu等人[11]重新设计实验,考察了DiffDock等机器学习模型在盲对接任务中的哪一阶段领先传统方法、领先到何种程度。作者将盲对接任务拆分为口袋搜索和局部对接两个子任务,设计了三组实验:完全使用DiffDock等模型完成盲对接;使用其它方法搜索口袋(如前文所述的PointSite、P2Rank),使用Uni-dock[26](一种基于AutoDock Vina 1.2的GPU加速对接方法)局部对接;使用DiffDock搜索口袋,使用Uni-dock局部对接。结果表明:DiffDock方法在口袋搜索中效果更佳(相较于PointSite等方法而言,引入了配体分子的结构信息),但与ground truth、即表中的GT pocket相比仍存在提升空间;口袋确定,传统对接手段得到的结果优于DiffDock等机器学习模型。作者进一步指出:给定口袋下预测蛋白质-配体构象的机器学习方法是后续发展的方向(正如机器学习构象生成中所讨论的);对于端到端的模型比较实验中,需要更审慎地评估传统方法的表现。
另外,随着蛋白质结构预测方法的发展,评估模型在apo态蛋白质上的对接表现是有必要的,也是更符合实际情形的。事实上,目前几种模型所使用的训练集均为holo态蛋白(缺乏足够数量的与holo态对应的apo态蛋白结构)。为泛化模型的对接能力至apo态蛋白结构,通常采取折中方案:假定apo态与holo态的主链变动不大,而在模型编码过程中只使用主链碳原子的信息。DiffDock论文中首次评估了各种方法在ESMFold给出的蛋白质结构上的对接能力。结果显示,各模型的对接表现均显著下降(对于DiffDock而言,配体重原子RMSD小于2 Å所占的比例从38.2%下降至20.3%)。
机器学习方法建模对接过程中蛋白质的结构变化仍然道阻且长。将分子动力学模拟过程中产生的动态信息引入模型中也许是一种可能的突破方向[27]。最后,正如AlphaFold2可作为打分函数评估蛋白质结构是否合理[3],DiffDock等拟合自由能面的模型,其在打分函数的各项评价指标中表现如何也值得进一步探究。
参考文献
[1] Equibind: Geometric deep learning for drug binding structure prediction
[2] Artificial intelligence–enabled virtual screening of ultra-large chemical libraries with deep docking
[3] State-of-the-Art Estimation of Protein Model Accuracy Using AlphaFold
[4] A Critical Comparative Assessment of Predictions of Protein-Binding Sites for Biologically Relevant Organic Compounds
[5] Improving detection of protein-ligand binding sites with 3D segmentation
[6] PointSite: A Point Cloud Segmentation Tool for Identification of Protein Ligand Binding Atoms
[7] P2RANK: Knowledge-Based Ligand Binding Site Prediction Using Aggregated Local Features
[8] P2Rank: machine learning based tool for rapid and accurate prediction of ligand binding sites from protein structure
[9] DeepSite: protein-binding site predictor using 3D-convolutional neural networks
[10] DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking
[11] Do Deep Learning Models Really Outperform Traditional Approaches in Molecular Docking?
[12] GEOM, energy-annotated molecular conformations for property prediction and molecular generation
[13] GeoDiff: a Geometric Diffusion Model for Molecular Conformation Generation
[14] Direct Molecular Conformation Generation
[15] Do Deep Learning Methods Really Perform Better in Molecular Conformation Generation?
[16] Comparative Assessment of Scoring Functions: The CASF-2016 Update
[17] Machine-learning methods for ligand-protein molecular docking
[18] Protein–Ligand Docking in the Machine-Learning Era
[19] From machine learning to deep learning: Advances in scoring functions for protein–ligand docking
[20] Improving scoring-docking-screening powers of protein–ligand scoring functions using random forest
[21] A geometric deep learning approach to predict binding conformations of bioactive molecules
[22] Boosting Protein–Ligand Binding Pose Prediction and Virtual Screening Based on Residue–Atom Distance Likelihood Potential and Graph Transformer
[23] Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning
[24]: A fully differentiable ligand pose optimization framework guided by deep learning and a traditional scoring function
[25] TANKBind: Trigonometry-Aware Neural NetworKs for Drug-Protein Binding Structure Prediction
[26] Uni-Dock: GPU-Accelerated Docking Enables Ultralarge Virtual Screening
[27] Pre-Training of Equivariant Graph Matching Networks with Conformation Flexibility for Drug Binding
本文作者:ZF责任编辑:WFZ
[来源:仪器信息网] 未经授权不得转载
2023.07.04
清华胡泽平团队揭示代谢组学结合AI模型在胃癌诊断及预测患者预后中的临床应用潜能
2024.03.08

2024.02.05
品牌合作伙伴






























版权与免责声明:
① 凡本网注明"来源:仪器信息网"的所有作品,版权均属于仪器信息网,未经本网授权不得转载、摘编或利用其它方式使用。已获本网授权的作品,应在授权范围内使用,并注明"来源:仪器信息网"。违者本网将追究相关法律责任。
② 本网凡注明"来源:xxx(非本网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责,且不承担此类作品侵权行为的直接责任及连带责任。如其他媒体、网站或个人从本网下载使用,必须保留本网注明的"稿件来源",并自负版权等法律责任。
③ 如涉及作品内容、版权等问题,请在作品发表之日起两周内与本网联系,否则视为默认仪器信息网有权转载。
![]() 谢谢您的赞赏,您的鼓励是我前进的动力~
谢谢您的赞赏,您的鼓励是我前进的动力~
打赏失败了~
评论成功+4积分
评论成功,积分获取达到限制
![]() 投票成功~
投票成功~
投票失败了~