
视频号

抖音号

哔哩哔哩号

前沿资讯手机看

热分析&电镜&表面分析,分享最新国内外仪器技术成果进展



分享到微信朋友圈
打开微信,点击底部的“发现”,
使用“扫一扫”即可将网页分享到朋友圈。
2021年,《高分子学报》邀请了国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。
更多专题内容详见:高分子表征技术专题

高分子表征技术专题前言
孔子曰:“工欲善其事,必先利其器”。 我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!
从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读. 期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来.
高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献. 借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意!


小角X射线散射技术在高分子表征中的应用
Typical Applications of Small-angle X-ray Scattering Technique in Polymer Characterization
作者:吕冬,卢影,门永锋
作者机构:
中国科学院长春应用化学研究所 高分子物理与化学国家重点实验室,长春,130022
作者简介:
门永锋,男,1973年生. 1995年获东南大学理学学士,1998年获中国科学院长春应用化学研究所理学硕士,2001年德国弗赖堡大学物理系获自然科学博士. 2002~2005年,德国BASF公司高分子研究中心,博士后、Physicist. 2005年加入中国科学院长春应用化学研究所开展工作. 2005年入选中国科学院百人计划,2014年入选科技部中青年科技创新领军人才,2016年入选第二批万人计划科技创新领军人才,2015年获国家基金委杰出青年基金、英国皇家学会牛顿高级学者基金. 目前担任高分子物理与化学国家重点实验室主任、中国晶体学会小角散射专业委员会主任、IUPAC商用聚合物结构与性能分会主席. 主要从事高分子结构与性能方面研究工作.
摘要
小角X射线散射(SAXS)技术是表征高分子材料微观结构的一种重要手段. 当X射线穿过材料时,在材料不均一的电子云密度分布作用下,发生散射并形成特定的散射图案,使得我们可以根据特定的模型来反推材料的微观结构,并计算相关结构参数. SAXS特有的对微观结构的统计平均及无损探测使其成为了一种不可或缺的高分子材料微观结构分析手段. 本文首先简述了SAXS技术的基本理论,在此基础上根据测试中的实际问题给出了测试时可采取的实验技巧. 最后,结合典型实例,概述了高分子材料中可用SAXS技术表征的微观结构及其相应的理论模型. 希望本文能作为入门文献,帮助初学者更好地理解SAXS技术的原理,并结合实际需求迅速了解SAXS技术的适用范围及相关实验技巧,高效地完成相关实验.
Abstract
Small-angle X-ray scattering (SAXS) technique is one of the most significant methods for determining the micro-structures of polymeric materials due to its statistical average and nondestructive detecting feature. Usually, a monochromatic parallel beam of X-rays is used for scattering experiments. When passing through a sample, the oscillating electromagnetic field (mostly the electric part) of X-rays interact with electrons, making the electrons secondary sources of X-rays of the same frequency. Those secondary X-rays interfere with each other to form a specific pattern deviating from the primary beam path depending on the actual locations of the electrons in the sample. Mathematically, such interferences can be obtained by a summation of all secondary X-ray waves. As the number of the electrons within the sample is very large, an integration is used to represent the summation mentioned above. Because of the wave nature of the X-rays, the amplitude of the scattered X-rays determined by the above integration is just a Fourier transformation of the electron density distribution within the scattering volume. Due to the limitation in detection technique, the complex value of amplitude of scattered X-rays with real and imaginary parts cannot be recorded. It is the intensity rather than the amplitude that is recorded during experiments resulting in a loss of the phase information. Therefore, obtaining exact structural information (electron density distribution) becomes not easy and must be based on specific model fittings. Besides structures, SAXS intensity distribution can be used to investigate sample’s gross properties such as fraction of phases or local properties such as fractal dimensions of interfaces between phases. This work began with an introduction of the fundamental theories of the SAXS technique, followed by practical suggestions on performing the experiments and brief summaries of models developed for different structures. The authors wish this review could help the beginners to comprehend the elements of the SAXS technique and serve as an instruction manual for valid data acquisition.
关键词
高分子表征; 小角X射线散射(SAXS); 片晶; 微观结构
Keywords
Polymer characterization; Small-angle X-ray scattering (SAXS); Lamellae; Micro-structure
1
1小角X射线散射原理简述
X射线是波长介于紫外与γ射线之间的电磁波,其波长范围涵盖了10-8~10-12 m,相应的频率范围为10 16~1022 Hz. 人们通常利用单一波长(单色)的X射线进行散射与衍射实验,例如:实验室中通常使用波长为0.154 nm的CuKα线特征辐射作为入射光源开展实验,而在同步辐射光源则可以根据需要选择合适的波长. X射线散射通常是指一束近乎平行的单色X射线穿过样品后产生的偏离入射光方向散射光强的现象. 当X射线通过物质时,其电磁波中的高频电场迫使物质中的电子发生同频震荡,产生次级波,这些次级波在空间中传播叠加. 不同位置的电子发出的次级波到达空间特定位置时具有不同的相位,因此,最终在不同位置的散射光的振幅取决于样品中电子的空间分布[1~3]. 由于物质中电子的数量极其巨大,上述各个位置振幅的叠加过程可以简化为积分,也就是:

其中ρ(r)是样品内部电子密度分布函数,r是样品内电子的坐标,V是X射线照射的体积,q是散射矢量,定义为:

其中S0和S分别为入射光及散射光方向的单位矢量.q的大小为:

其中2θ是入射光与散射光之间的夹角,也就是散射角. 可见,X射线散射实验获得的散射光振幅在q空间的分布只与样品内部电子密度分布函数相关,利用不同波长X射线进行测试获得的散射光振幅分布具有不同的角度依赖性,但换算成q空间分布则是唯一的. 观察公式(1)可以发现A(q)其实就是ρ(r)的傅里叶变换. 如果我们可以直接测量A(q),便可以直接进行反傅里叶变换获得期待的ρ(r),也就是样品内部的微观结构. 然而,如前所述,X射线的频率非常高,目前的电子学技术不能有效测量A(q),在测量过程中会丢掉相位信息,只能测得强度信息,也就是:

公式(1)所展示的代表实空间结构的ρ(r)与A(q)代表q空间散射光振幅分布函数显然具有倒易性,即,实空间中尺度越大的结构将在q空间中小q区呈现强的散射光. 可见,根据公式(3)及通常使用的X射线的波长(在0.1 nm量级),几纳米至几百纳米的微观结构将在较小的q(2θ)处产生散射信号. 因此,探测纳米至微米尺度微观结构的X射线散射技术被称为小角X射线散射(SAXS). 尽管通过散射强度I(q)不能直接得到体系的电子云密度分布函数ρ(r),但是ρ(r)的自相关函数Γρ(ρ)恰巧是散射强度的反傅里叶变换. 因此,代表体系微观结构的ρ(r)、散射光振幅A(q)、可测量的散射光强度I(q)及ρ(r)的自相关函数Γρ(ρ)之间就具有了图1所示的关系. 这一物理量间相互转化的关系是SAXS技术的基础[4,5]. 值得注意的是,由Γρ(ρ)不能直接反推样品体系的电子云密度分布情况. 在实际数据分析中,我们还需结合体系的电子云密度分布特性进行讨论. 因此,对于体系电子云密度分布的描述十分重要,目前主流的数据分析发展趋势主要是集中于如何选取简化的模型,推导出具有特征形状的自相关函数Γρ(ρ)[6],来间接描述体系的电子云密度分布. 模型确定后,就可以根据散射强度分布来计算一些特定的结构参数. 篇幅所限,这里只给出了极简版的SAXS原理以及获得结构信息的大致思路,想要深入地了解SAXS技术的原理的读者可以参考文末所列出的经典教科书[1~4,7~11].
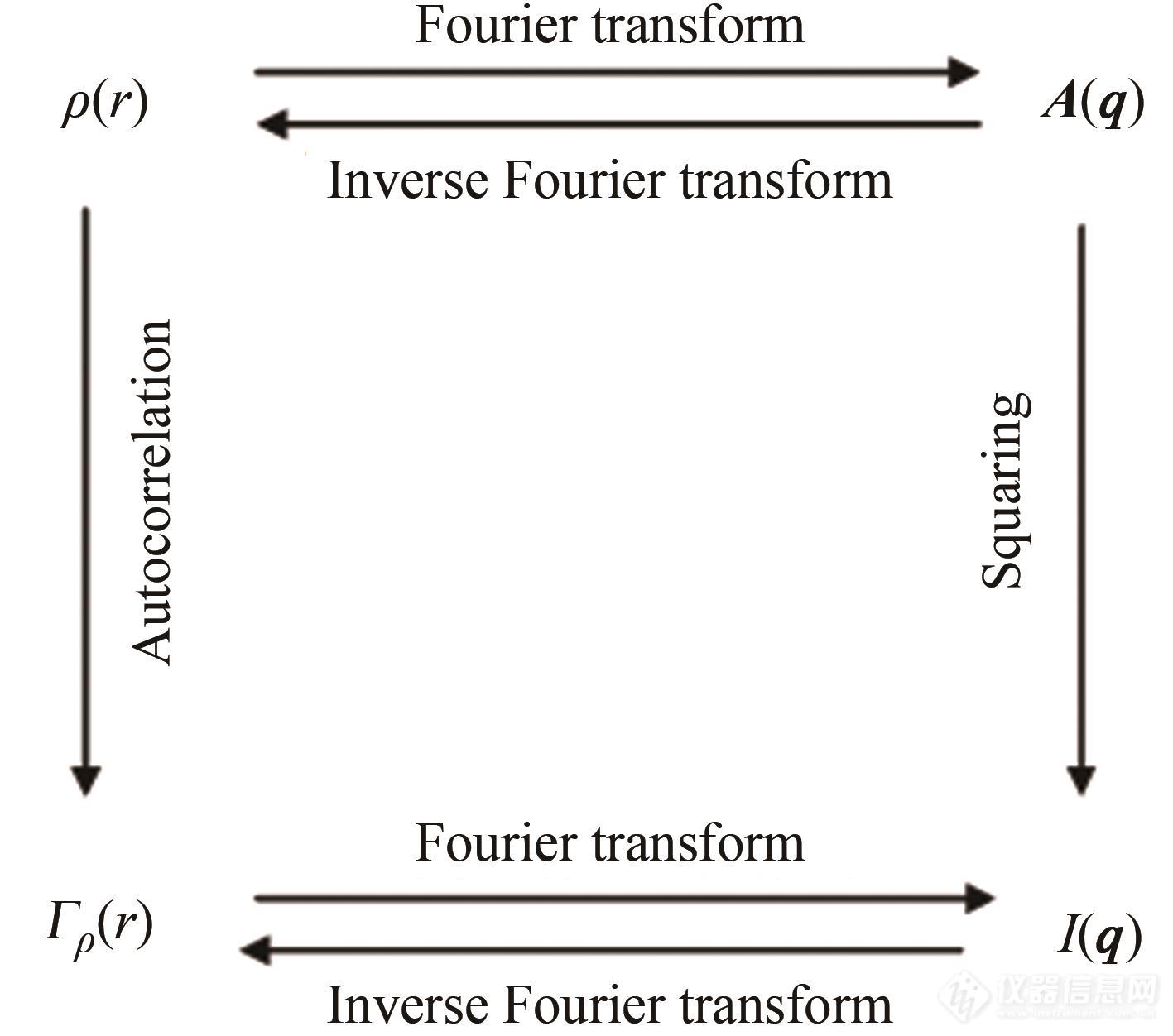
Fig. 1Relationships amongρ(r),A(q),Γρ(r) andI(q)[5].
SAXS技术的测试结果不直观,倒空间的散射信号还原成实空间中材料的微观结构的过程中,涉及到大量的数学运算及相应理论模型的拟合,稍有不慎极有可能得出错误的结果. 因此,在利用SAXS分析材料微观结构时,常常需要扫描电子显微镜(SEM)、透射电子显微镜(TEM)、示差扫描量热仪(DSC)等实验来辅助验证分析结果. SAXS技术的优点则在于适用于多种材料体系,对测试样品不需要进行前期预处理,测试过程中也不改变样品的结构性质,属于无损测试. SAXS测试结果为体系的统计平均值,更能代表材料的整体信息. 此外,多数SAXS仪器与其他仪器兼容性好,可以实现SAXS技术与各种小型设备,例如拉伸仪、剪切仪、热台、注塑机、模拉仪器等多种仪器的联用[12],从而在线观测材料在各种条件下的微观结构演变及服役行为. 因此,虽然SAXS技术对于初学者来说门槛略高,但由于其多方面的优势,在高分子材料结构表征领域中仍扮演着不可替代的角色.
2实验技巧
从上述X射线散射的基本原理可知SAXS实验方面相对简单,只需利用成熟的商用仪器或同步辐射线站将待测样品置于X射线光路之中特定的位置即可. SAXS也可以通过以很小的角度掠过表面来测试薄膜样品,此时的SAXS实验被称为掠入射SAXS (GISAXS),其基本原理与SAXS相近,本文受篇幅限制不对GISAXS进行讨论. 除了简单的静态样品SAXS测试,还可以利用SAXS测试样品在不同外场下的微观结构演化过程,例如高分子加工成型条件下的相变与结晶、服役环境中的形变与破坏等. 为实现最优化的SAXS实验,在开始实施之前确实需要做一些必要的准备. 以下是一些常见的需要注意的事项.
2.1谱仪参数选择
任何SAXS设备都只具备有限的可测量q范围,这就决定了可观测的微观结构尺寸必然是有限的,因此,一个初步的判断,甚至是初步的实空间实验通常是需要的. 很多SAXS设备具有多段可调q范围,初步的实验有助于选择合适的仪器参数实现相关尺度微观结构的统计平均测试.
目前主流的实验室SAXS设备及同步辐射SAXS实验站都可提供X射线波长、光斑尺寸、样品到探测器距离等参数的选择. 配备2种金属靶(例如铜及钼)的实验室SAXS设备逐渐成为一个很好的选择,其提供的铜或钼的特征辐射具有不同的波长,也就是具备不同的穿透能力,在具有环境腔窗口的情况下可根据不同的窗口材料选择合适的光源. 光斑尺寸的选择原则是在宏观的谱仪尺度上可被看成是点光源,但在微观结构尺度上又能实现足够大的覆盖以实现统计平均. 简而言之,如光斑尺寸过大则会对SAXS数据造成明显的模糊效应,也就是探测器同一个像素点采集到了样品不同位置散射的本应是不同q处的信号. 尽管可以利用光斑形状的数据对最终获得的SAXS数据进行去模糊处理,通常我们还是应该避免这一步骤,原因是去模糊过程不可避免地带来计算的简化及误差,进而影响实验数据的精度. 同样的道理,如果光斑尺寸过小则会造成统计平均不足的问题,特别是在先进的同步辐射实验站,当光斑尺寸接近待测微观结构尺寸时就会丧失应有的统计性及小q区数据的可靠性. 这一点可从公式(1)来理解,其中的积分体积V是X射线照射的样品体积,因此,光斑的尺寸的倒数就决定了可探测的最小q值.
取决于通常的二维面探测器像素点尺寸,我们在实验之前可估算能实现的q空间分辨率,也就是2个相邻像素代表的q之差Δq. 常见的误区是只关注能实现的最小q而忽视Δq. 这种情况在探测目标微观结构尺寸较大时尤为突出. 根据SAXS谱仪的结构,可通过改变样品到探测器之间的距离实现Δq的合理选择,该距离越大则对应的从样品位置出发的2个相邻像素点对应的角度也就越小,从而实现更小的Δq.
2.2样品尺寸选择
样品的宏观尺寸对获得优质的SAXS数据也很重要,通常选用足够大的样品以使入射X射线全部照射在样品上而不触及样品边缘,这样做的目的是尽量避免边缘光滑表面可能带来的对掠过的X射线的反射,这种反射将会污染实际的SAXS数据. 这种情况在测试直径小于光斑尺寸的纤维样品时比较突出,通常的做法是利用与纤维密度相仿的液体浸润一束纤维以消除纤维与空气的界面影响.
X射线与物质相互作用除了散射以外还包括被吸收,因此,在确定样品沿X射线传播方向的最佳厚度时就需要考虑吸收和散射的平衡. 基本的思想非常简单,散射强度依赖于X射线照射到的总电子数,也就是和厚度成正比,吸收则相反,厚度越大则吸收越严重,因此,对特定的样品在特定的X射线波长下一定存在一个最佳的厚度进行SAXS实验. 根据计算,通常实验室SAXS设备利用的CuKα线下聚乙烯样品的最佳厚度为2 mm左右. 因吸收系数是X射线波长的函数,具体到特定条件下最优样品厚度的寻找需借助工具书或进行实测[7].
2.3数据处理
从上述讨论可知,与显微学手段(如电子显微镜、原子力显微镜等)相比,SAXS实验实现起来相对简单,但SAXS数据是在q空间呈现的,远没有显微学实验获得的结果直观,并且实验测得的原始数据还需校正才能使用.
首先,任何SAXS谱仪都不可避免会有背底散射,也就是在没有加载样品时也会有一定程度的散射信号可被探测器记录,这些背底散射来自光路中可能的窗口、气体分子及探测器的电子学噪音. 因此,正确扣除这部分背底散射非常重要. 目前主流的SAXS设备和同步辐射SAXS实验站都配备了标准的流程进行背底散射的扣除. 这里需要注意的是正确计算加载样品之后的背底散射,考虑到样品对入射X射线的吸收,在加载样品后通过样品后光路中的X射线总量减少,因此,在没加载样品条件下测得的背底散射数据实际上是被高估了,需要进行样品吸收校正. 背底散射扣除后的SAXS数据已经可以用于体系微观结构参数的计算,但其散射强度还只具有任意单位,需要进行进一步的数据处理才能获得绝对散射强度[10,13]. 绝对散射强度包含了体系微观结构的所有信息. 通常可以利用已知绝对散射强度的样品(例如纯水)作为标准进行比对,获得所测样品的绝对散射强度分布. 上述介绍的强度校正基本可以满足一般需求,但在精确的计算中还涉及到更多信号校正,这里不再一一展开说明. Pauw在综述中关于信号校正的种类,校正对信号影响大小,及应用各类校正的先后顺序进行了详细的阐述[14].
按照正确步骤得到散射曲线后就可以进行数据分析. SAXS数据中散射强度与散射矢量之间一般具有幂率关系,也就是I(q)~q−ν ( ν是正自然数),因此,SAXS曲线通常用双对数坐标表示以方便获得幂律关系,这就要求我们不能对散射强度进行加减操作,以免改变应有的幂律关系. 有时为图示清晰,可对SAXS数据进行乘除常数的操作,获得曲线在双对数坐标下的上下平移,达到合适的视觉效果而不影响通过幂指数规律进行数据分析.
在高分子领域,利用SAXS对结晶高分子体系片晶-非晶区叠层结构长周期的研究极为广泛和成功,其中常见对散射强度(I(q)versusq)数据做洛伦兹校正,既将I(q)乘以q2之后对q做图,然后利用I(q)q2versusq曲线探讨体系结构参数[7,9]. 这里的洛伦兹校正是考虑到片层体系的特殊几何结构而进行的对测得的散射强度的必要修正,具有其他几何形状的微观结构产生的散射强度不能直接套用该校正. 首先,我们考虑一个在空间中固定的片层结构,其宽度和长度远大于厚度,这就意味着该片层产生的散射强度将集中在片层法向方向,在q空间形成一个细棒状分布,所以,理论上该片层在法向方向以外都不产生散射信号. 然而,实际体系中由于片层结构会沿不同方向平均化,这主要是因为在液体分散体系中的片层会高速运动(旋转、平动),使得测量时间尺度范围内本应是细棒状的强度分布平均分不到整个三维q空间,那么对于任意q而言,强度就被稀释了以q为半径的球壳面积倍,也就是4πq2倍. 所以,测得的散射强度需要按q2校正. 在结晶高分子体系,尽管片晶不能旋转,但是众多片晶在空间中沿不同方向分布,其实际效果和上述分散体系类似,因此也需对测得的I(q)进行q2校正. 根据上述讨论,其他形状的散射体,例如球状散射体因其理论上的散射强度在不同q处就应该是均匀分布的,不存在稀释的问题,所以是不能盲目进行洛伦兹校正的.
常用的SAXS数据处理软件有Fit2d[15],GNOM[16],SASfit[17],SasView[18],Scatter[19],ATSAS[20],McSAS[21],BioXTAS RAW[22]等,可实现二维SAXS散射图到各类一维散射曲线的转换,并且部分软件兼具简单的数据拟合功能. 各个软件有其特定的侧重点,需要根据自己的实际需求来选择. 此外,也可使用Matlab、Python等程序语言,自行编辑所应用的公式及选择相应拟合模型,对自己的体系进行个性化处理. 但值得注意的是,曲线拟合完美并不一定代表结果的真实可靠,在得出正式结论前一定要三思.
3小角X射线应用实例
在这一节中,首先介绍不依赖于具体微观结构模型的散射不变量Q的相关应用以及如何利用散射峰位置确定微观粒子的排列模式,之后再按照微观结构分类,分别给出对应结构的拟合公式及实际应用. 由于篇幅所限,本文选取了有限的应用实例而非面面俱到,对特定结构测定有兴趣的读者可参考文末所列的基本SAXS经典教科书[1~4,7~11].
3.1散射不变量Q
散射不变量Q,以两相结构体系为例,取决于样品体系内部各散射体间电子云密度差及各散射体的体积分数[4,7]:

其中,ρ1和ρ2分别为两相的密度,v和(1−v)则分别代表两相的体积分数.
从定义可知,Q只取决于样品本身的性质. 理论上,Q是散射强度在整个q空间的积分. 但在实际测量中,我们对探测器采集到的信号进行积分后得到的是与Q成一定比例关系的Q':
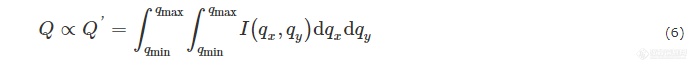
Q′会受到X射线强度、样品信号吸收和数据处理方式等影响,因此,在利用Q′代替Q对样品体系进行讨论时,需要考虑到上述条件的影响,并酌情对Q′值进行归一化处理[23,24].
Q或Q′受到散射体间的密度差及体积分数的影响,也就是说Q可以用来反馈任何涉及到反差或构成变化的过程. 对于高分子体系而言,散射不变量的一种十分重要的应用就是判断体系内部空洞化的发展. 由于空洞-材料本体间的电子云密度差比晶区-非晶区之间的电子云密度差大2个数量级左右[25],体系中一旦出现空洞,Q值就会明显上升[26~31]. 此外,还可以根据不同方向积分散射强度的比值来判断空洞的平均取向程度. 如图2,潘鹏举团队为了比较PBAT/PLA共混物在不同方向的空洞化情况,分别沿不同方向积分计算空洞强度,平行拉伸方向的空洞强度记为IMR,垂直拉伸方向的空洞强度记为IEQ[25]. 对于PLA含量为0.3的共混物来说,IMR/IEQ的比值在形变量εH=0.5时到达峰值并随后逐渐减小. 峰值代表着空洞调整方向的起始点,随后这个值又降到了1,说明大部分的空洞已经重新沿拉伸方向排列.
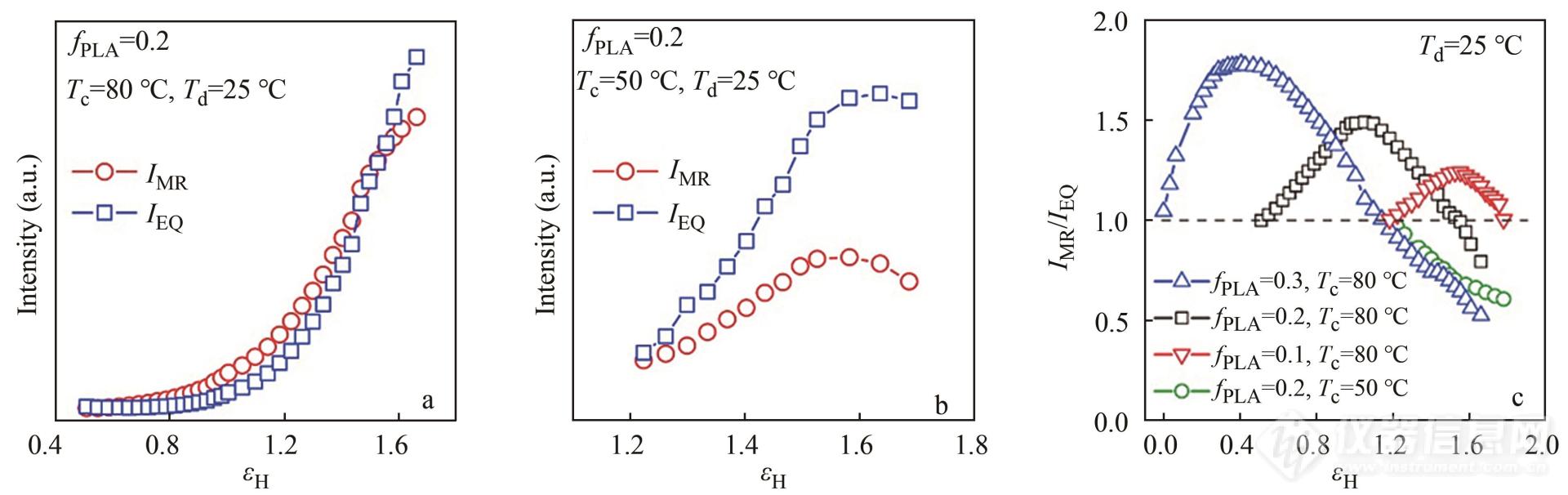
Fig. 2SAXS intensities of cavity signals of PBAT/PLA blends collected during stretching: (a) cavity intensity of PBAT/PLA-0.2 blend (Tc=80 °C,Td=25 °C) in the meridian and equator directions; (b) cavity intensity of PBAT/PLA-0.2 blend (Tc=50 °C,Td=25 °C) in the meridian and equator directions; (c) strain-dependentIMR/IEQ values of PBAT/PLA blends (Reprinted with permission from Ref.[ 25]; Copyright (2020) Elsevier).
除对体系内空洞的表征外,Q还能用来描述乳胶体系干燥、结晶熔化及相转变等过程[4,24,32~34].
3.2有序微观结构
周期性排列的微观结构会以特定比例形成较规整的散射峰,根据实验中测得的衍射峰峰位置的相对关系可以判断出体系内部相区的排列方式[12,35~40]. 表格1中给出了各种结构所对应散射峰峰位置间的比值[41].
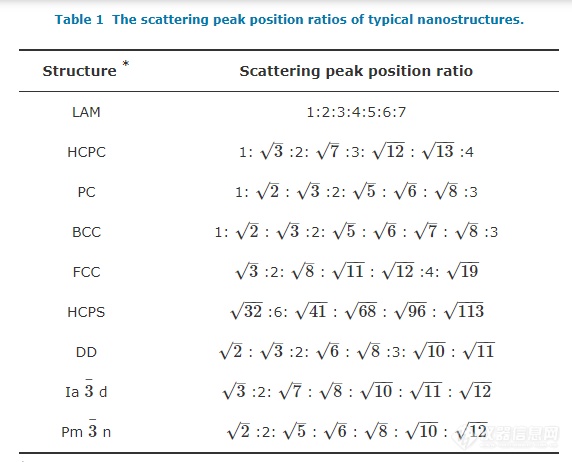
*LAM: lamellar, HCPC: hexagonally close packed cylinders, PC: primitive cubic, BCC: body-centered cubic, FCC: face-centered cubic, HCPS: hexagonally close packed spheres, DD: double diamond, Ia3¯3¯d, and Pm3¯3¯n cubic morphologies belonging to these space groups (Reprinted with permission from Ref.[41]; Copyright (2002) Taylor& Francis).
类比广角X射线衍射的原理,由散射峰峰位置q可算出相对应散射体的尺寸d[42~45]:

但d所对应的尺寸的具体物理含义还需要根据体系内部的结构来判定[46~50].
Hickey团队观测了聚苯乙烯-聚丁二烯嵌段共聚物/苯乙烯在反应过程中的实时相转变过程[51],如图3所示.图3(a-i)为样品初始散射信号,属于典型的片层结构的散射. 反应开始后,体系首先向无规状态转换,之后散射峰又变窄(图3(a-iii)). 随着反应的进行,图3(b)中出现了比例为1.14的2个散射峰,但此时体系相结构仍难以确定.图3(c)中出现了更高级的散射峰,峰位置之间的比例为1.14:2:2.64.
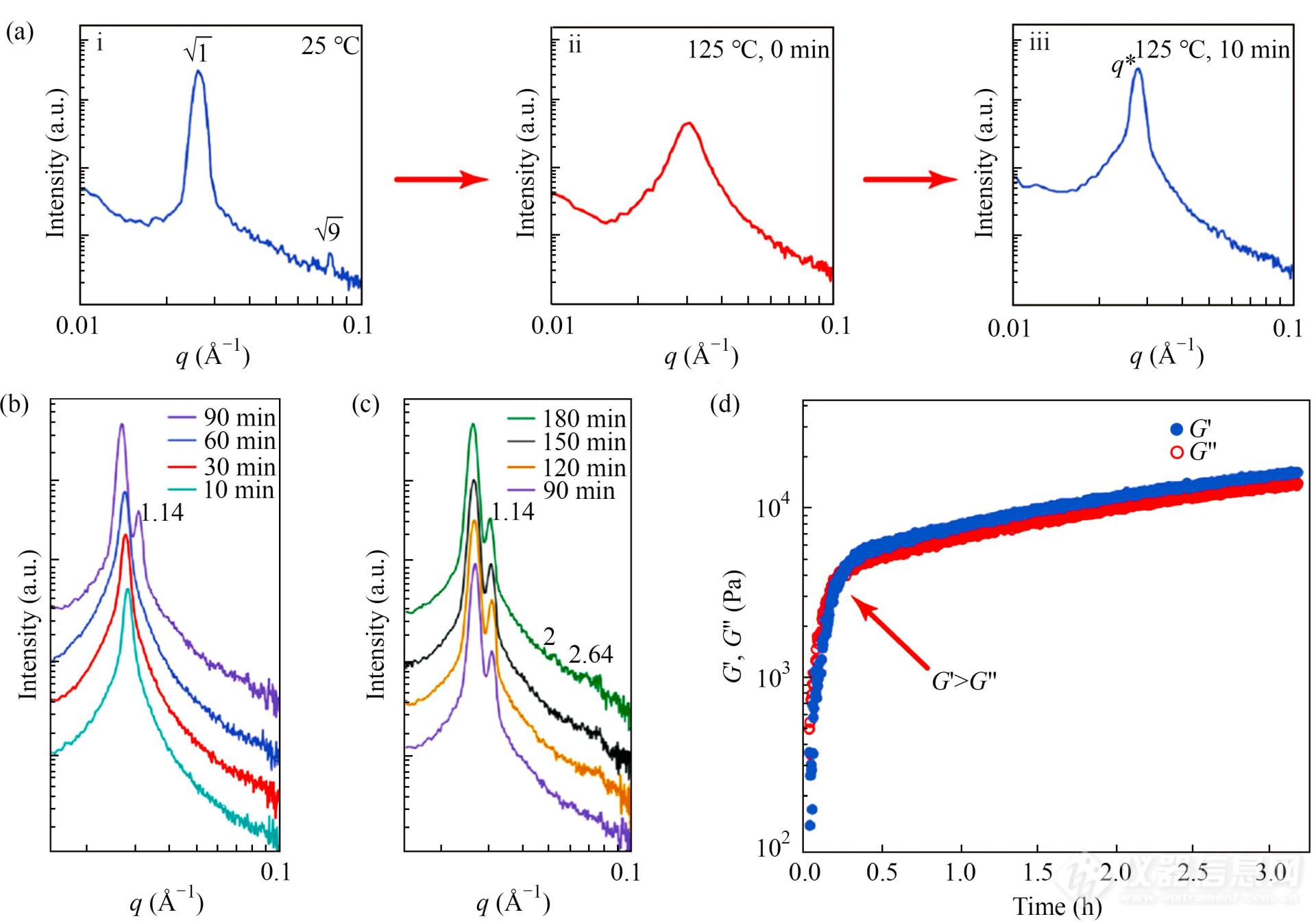
Fig. 3In situ SAXS and oscillatory shear DMS measurements to determine the morphology evolution during polymerization-induced nanostructural transitions for a ϕPS‑PBD=60% PS-PBD/styrene mixture. (a) One-dimensional SAXS patterns forϕPS‑PBD=60%; (i) initially at 25 °C before polymerization, (ii) directly after heating to 125 °C from 25 °C, and (iii) after 10 min once reaching 125 °C. Red arrows between scattering patterns indicate changes that occur during heating and polymerization. The blue traces indicate the presence of an ordered morphology, while red traces indicate disorder. (b) SAXS patterns showing the progression of the primary scattering peak,q*, over the first 90 min of polymerization until the first indication of higher-orderq-reflections. Scattering traces have been vertically shifted for clarity. (c) SAXS patterns showing the evolution of the structure from the first appearance of a second peak at 90-180 min. Scattering traces have been vertically shifted for clarity. (d) Isothermal time sweep for oscillatory shear DMS ofϕPS‑PBD=60% at 125 °C, strain amplitude of 0.5%, and frequency of 1 rad/s, showing a single disorder-to-order transition at ~10 min, as indicated by theG′ andG″ crossover (Reprinted with permission from Ref.[51]; Copyright (2020) American Chemical Society).
结合样品降温时的散射曲线以及振荡剪切动态力学谱的数据,最终得出聚苯乙烯-聚丁二烯嵌段共聚物/苯乙烯在整个过程中复杂的相结构转换,如图4所示. 材料最初为片层结构,升温后有序结构被破坏,通过反应变为六方和片层复合结构. 降温后材料会变为六方堆积结构,除去未反应的苯乙烯并将材料退火,这种六方堆积又可变为片层结构. 但若延长反应时间至21 h,即使降温和除去体系中未反应的苯乙烯,上述六方和片层的复合结构仍然可以得到保存.
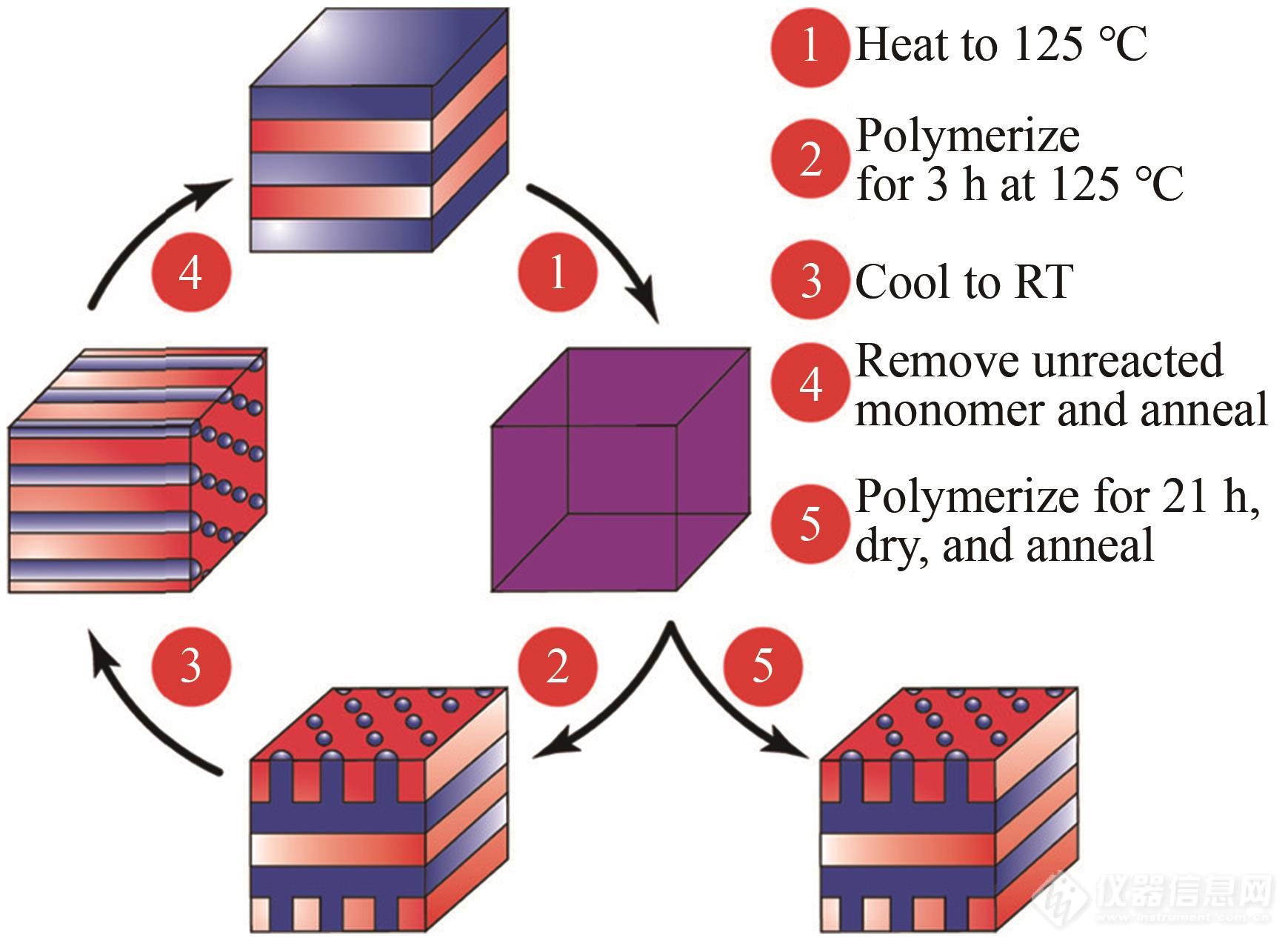
Fig. 4Morphology evolution during polymerization-induced nanostructural transitions for aϕPS‑PBD=60% PS-PBD/styrene mixture. At room temperature before polymerization, the LAM phase disorders at elevated temperatures (1), evolves from a disordered to a complex phase inferred to be a HPL phase over time (2), and then transitions to the HEX phase on cooling to room temperature if the polymerization is conducted for 3 h (3). The HEX phase will transition back to a LAM phase once the unreacted styrene is removed, and the sample is annealed (4). If the polymerization run for 21 h, then the high-temperature phase will persist at room temperature, and after unreacted styrene is removed (5) (Reprinted with permission from Ref.[51]; Copyright (2020) American Chemical Society).
3.3高分子片层结构
3.3.1一维相关函数
半结晶高分子通常由晶区非晶区交替组成,最简单的一种模型就是将体系简化为两相层状体系,其每层的面积无限大. 当这些片层的法向方向平行于z轴时,某特定位置的电子云密度只取决于其所处高度z. 此时相关函数可以简化为一维相关函数K(z):

Strobl团队考虑到样品内部结构,讨论了体系所对应的一维相关函数特征,如图5所示[8,52]. 对于晶区和非晶区界限明显,且不考虑各区域的多分散性,样品的电子云密度及其所对应的一维相关函数应如图5(a)所示. 图中L代表样品的长周期,d为晶区和非晶区中较小区域的厚度. 若体系中结晶度小于50%,则d对应晶区厚度,反之亦然.
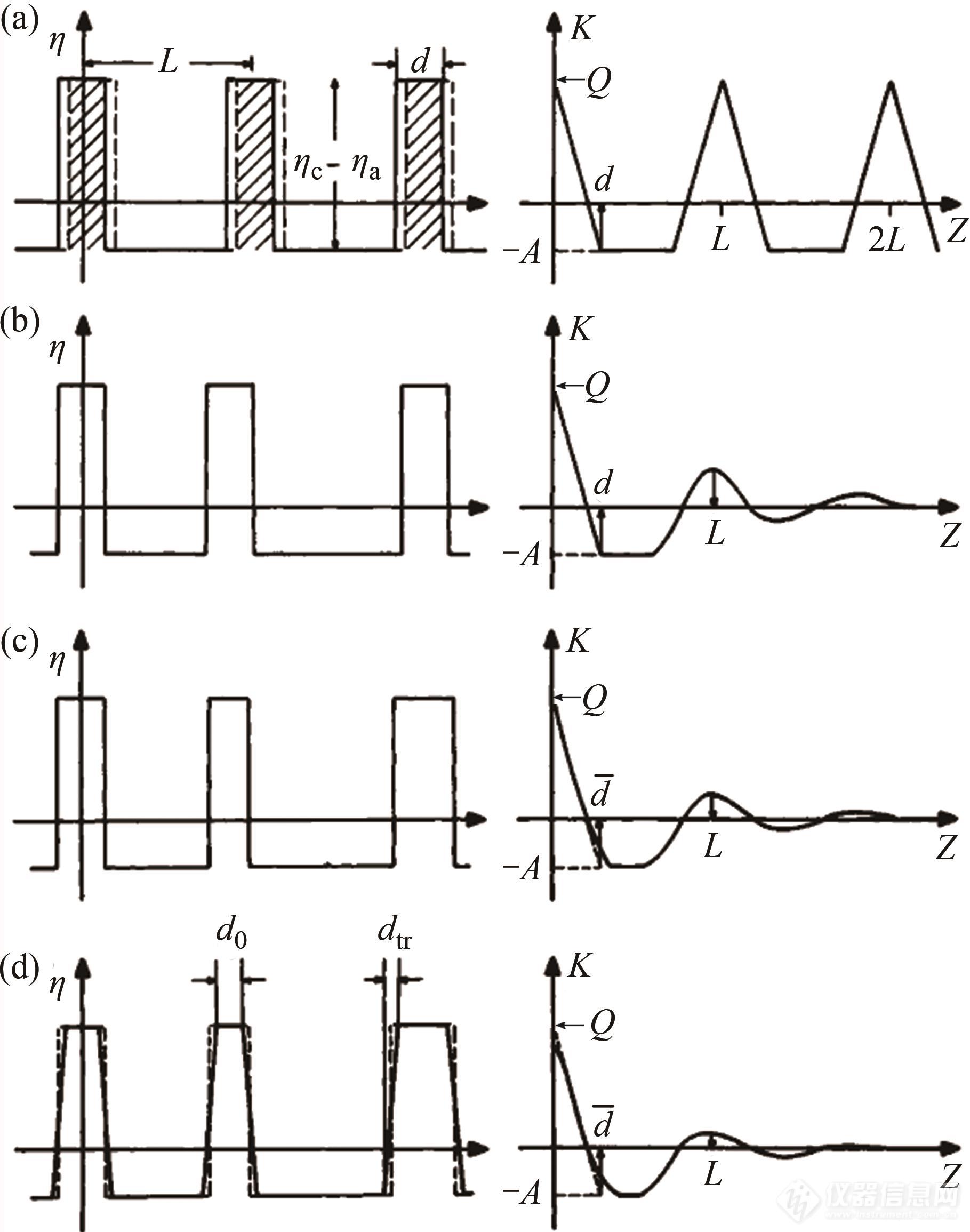
Fig. 5Electron density distributionρ(z)and the related correlation functionK(z)for lamellar systems of different regularities: (a) Periodic two-phase system; (b) Effect of long-spacing variations; (c) Effect of additional thickness fluctuations; (d) Effect of introduction of diffuse phase boundaries (Reprinted with permission from Ref.[52]; Copyright (1980) Wiley).
但实际体系和理想模型有一定差距,图5(b)~5(d)给出了理想模型向实际样品体系过渡时,体系内电子云密度分布特征及相关函数的演变.图5(b)考虑到了体系内片晶间距的多分散性,图5(c)在图5(b)的基础上考虑到了片晶厚度的多分散性,图5(d)则在图5(c)的基础上引入了晶区和非晶区间厚度为dtr的过渡层.图5(d)中长周期的值可由相关函数的第一个极大峰值位置确定,体系中较薄片区的厚度则可通过斜直线的延长线和基线的交点得出. 值得注意的是,这种方法最佳适用体系结晶度范围ϕ<0.3或ϕ>0.7,若结晶度不在此范围内,基线如图6所示,难以观察,此时就需要其他数据来辅助确定基线,从而判断晶区或非晶区的厚度.
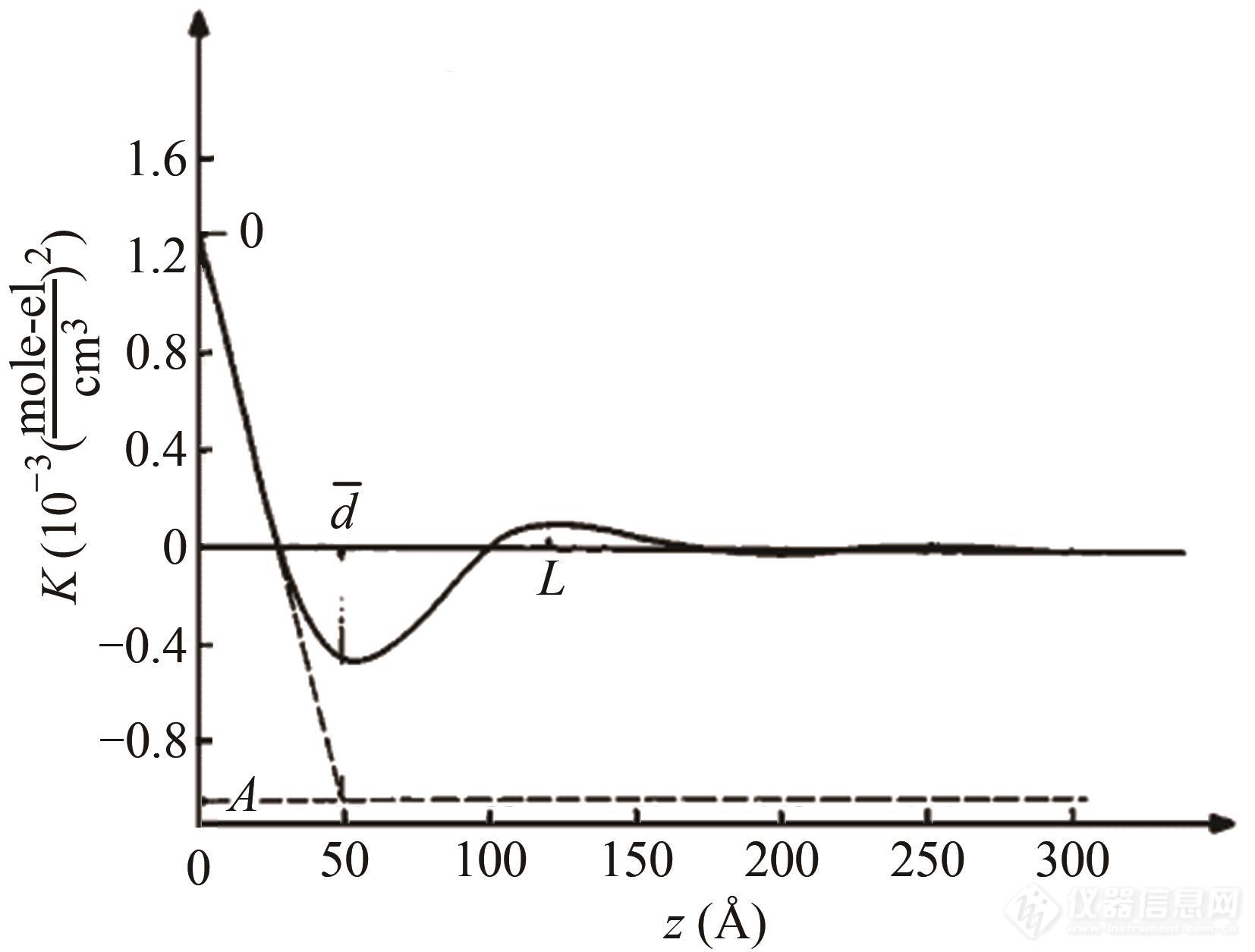
Fig. 6Experimental correlation function obtained for LDPE at room temperature (Reprinted with permission from Ref.[52]; Copyright (1980) Wiley).
利用相关函数算法计算片晶的长周期及片晶厚度十分简便,目前已得到广泛应用[53~61].
3.3.2弦分布函数算法和界面分布函数算法
相关函数算法最佳适用体系是理想两相片层体系,但实际上,半结晶高分子形成的片层结构并不十分规整. 对于晶区非晶区厚度分布较宽,且二者间分布差异明显的体系,相关函数及Bragg算法会由于散射峰的叠加偏移而引入较大误差[62],而使用界面分布函数(IDF)[63,64]则可以得到更加准确的数值.
门永锋团队与Thurn-Albrecht团队合作利用其发展的IDF曲线拟合的方法揭示了聚丁烯晶型II向晶型I转变时的结构变化[65]. 界面分布函数由一系列的距离分布构成:
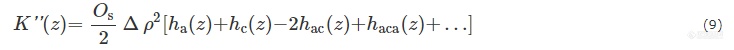
其中,ha(z)和hc(z)分别代表非晶区(da)和晶区厚度(dc)的分布,hac(z)是长周期dac=da+dc的厚度分布,后面的项以此类推.Os为特征内表面,Δρ为晶区非晶区间的电子云密度差. 则各向同性的样品的散射强度与K''(z)的关系:
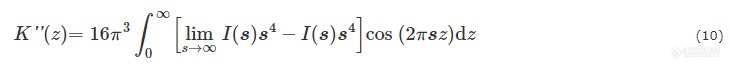
其中s为散射矢量且s=q2π. 对于有明确边界的一维两相体系,I(s)通常在s-4附近振荡,并在大s区域与s-4成正比[66].lims→∞I(s)s4也就是Porod常数P.
在利用上式计算前,首先要在较大s处进行Porod拟合I(s)≈Ps−4+c,确定P和体系热密度涨落带来的背景散射c,如图7上图所示. 之后对曲线进行平滑,拟合等操作,即可得出晶区非晶区厚度及厚度分布宽度等结构信息[67,68],图7给出了拟合过程中的一些曲线.
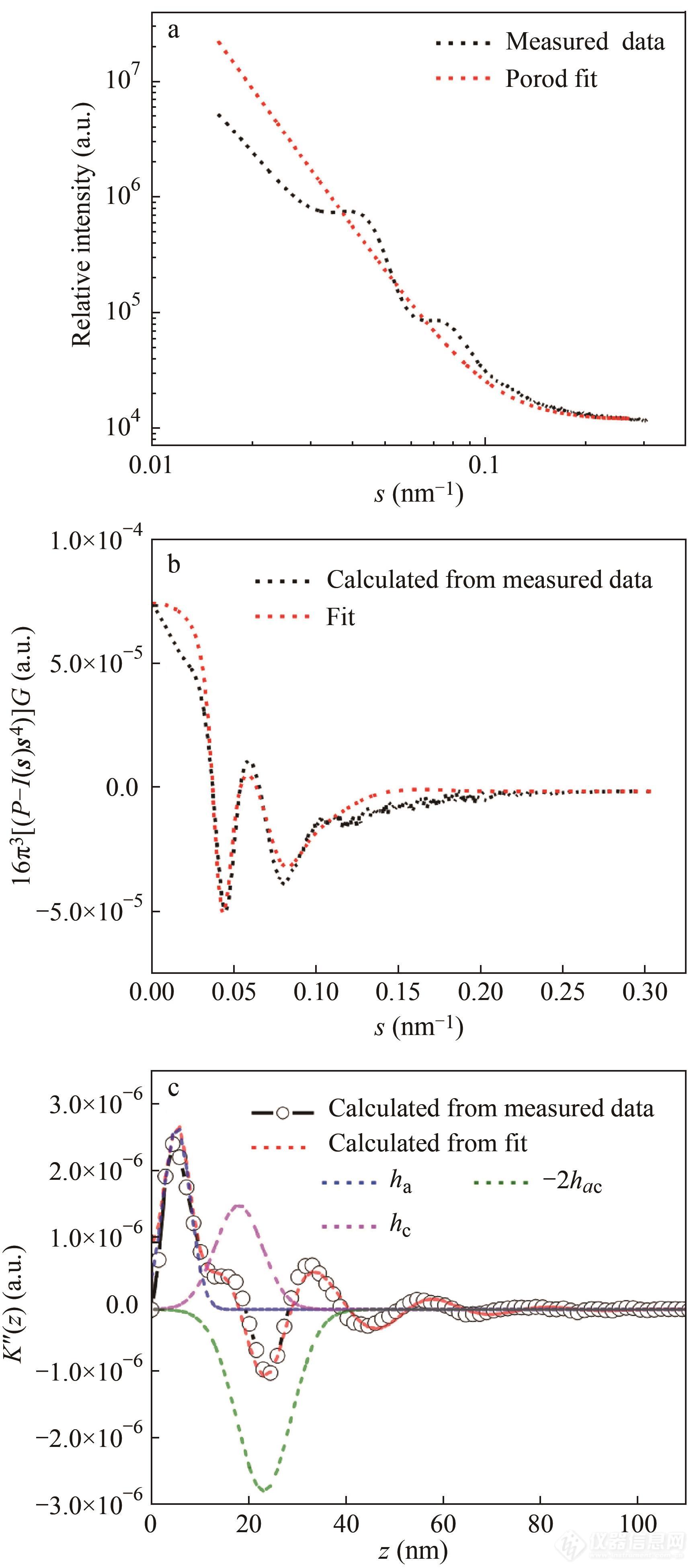
Fig. 7Exemplary analysis of SAXS data for the sample crystallized at 60 ℃ in form II. (a) Original scattering curve measured directly after crystallization and the fit describing Porod behavior and contribution due to density fluctuations; (b) The function 16π3[P-I(s)s4]Gas calculated fromI(s) after subtraction of the contribution due to density fluctuations; (c) IDF as calculated from the original scattering data and from the fit. The first three contributions to the IDF,ha,hc, and -2hac are shown separately in addition. (Reprinted with permission from Ref.[ 65]; Copyright (2020) Elsevier).
随着SAXS技术的发展以及计算机领域的进步,利用Mering等提出的适用于随机形状体系的弦分布函数(CDF)[69]能够计算得出更多的结构信息,从而更加精准地描述体系的微观结构. CDF和IDF实际都是相关函数的二阶导数,其中前者可认为由一系列后者构成[70,71]. 因此,在实际利用CDF算法描述体系时,也经常会涉及到IDF的拟合.
李许可等利用Stribeck发展的CDF算法衡量了不同硬段含量聚氨酯高温拉伸时的形变特征[71]. 在经线上CDF峰表明,沿拉伸方向体系中存在着由状态介于被破坏的硬段与被拉伸的软段之间的均质体组成的柱状微纤. 这些有较低硬段含量的聚氨酯微纤表现出侧向周期性排列模式. 基于Bonart纵向和横向投影,利用界面分布函数和弦长分布可以分别定量表征微区和微纤的排布.
在计算前,首先需要将信号投影到某一特定方向上[72]. 在本工作中,Bonart的横向投影被用来分析赤道区的信号.

从投影线{I}2(s12)可以得出两相体系的二维干涉函数G2(s12):
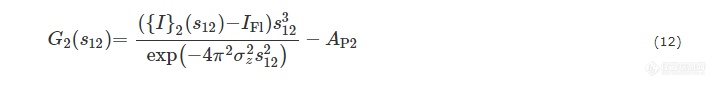
其中AP2是投影后SAXS强度的Porod渐进值.IFl和σz用于描述真实体系的非理想特性.IFl是由电子密度波动产生的散射强度.σz是微区边界过渡区的宽度. 由G2(s12)可以得到微纤的二维弦长分布g2(r12):
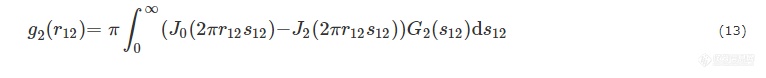
其中,J0和J2代表第一类Bessel函数的第零阶和第一阶.
将散射强度投影在纤维轴上可以得到Bonart纵向投影:
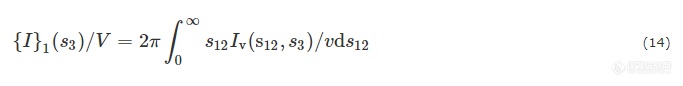
此时,弦会沿纤维轴交替穿过硬段和软段. 通过{I}1(S3)/V计算得来的g1(r12)可以定量描述硬弦和软弦的结果. IDF可以理解为是一系列硬弦和软弦的高度分布. 因此,可以通过拟合来得到参数信息[70,73].
在完成投影后,为拟合数据,还需根据体系的特征选择合适的模型[70,73,74]. Stribeck详细总结了应用于描述微区堆叠的模型及其物理意义[75]. 在此工作中,测得的IDF由一个大的微区峰和小的周期峰构成. 这意味着有多个独立的硬区,且硬区间联系较弱. 因此,利用独立硬段及软硬段一维阶梯排列模型,对g1(r3)进行进一步拟合[70,76],可以计算出平均硬段高度H¯¯¯H,平均软段高度H¯¯¯S,以及硬段软段的相对偏差,独立和非独立硬段的权重等参数,由此更好地描述体系形变过程中微观结构的变化,图8为数据拟合过程示意图.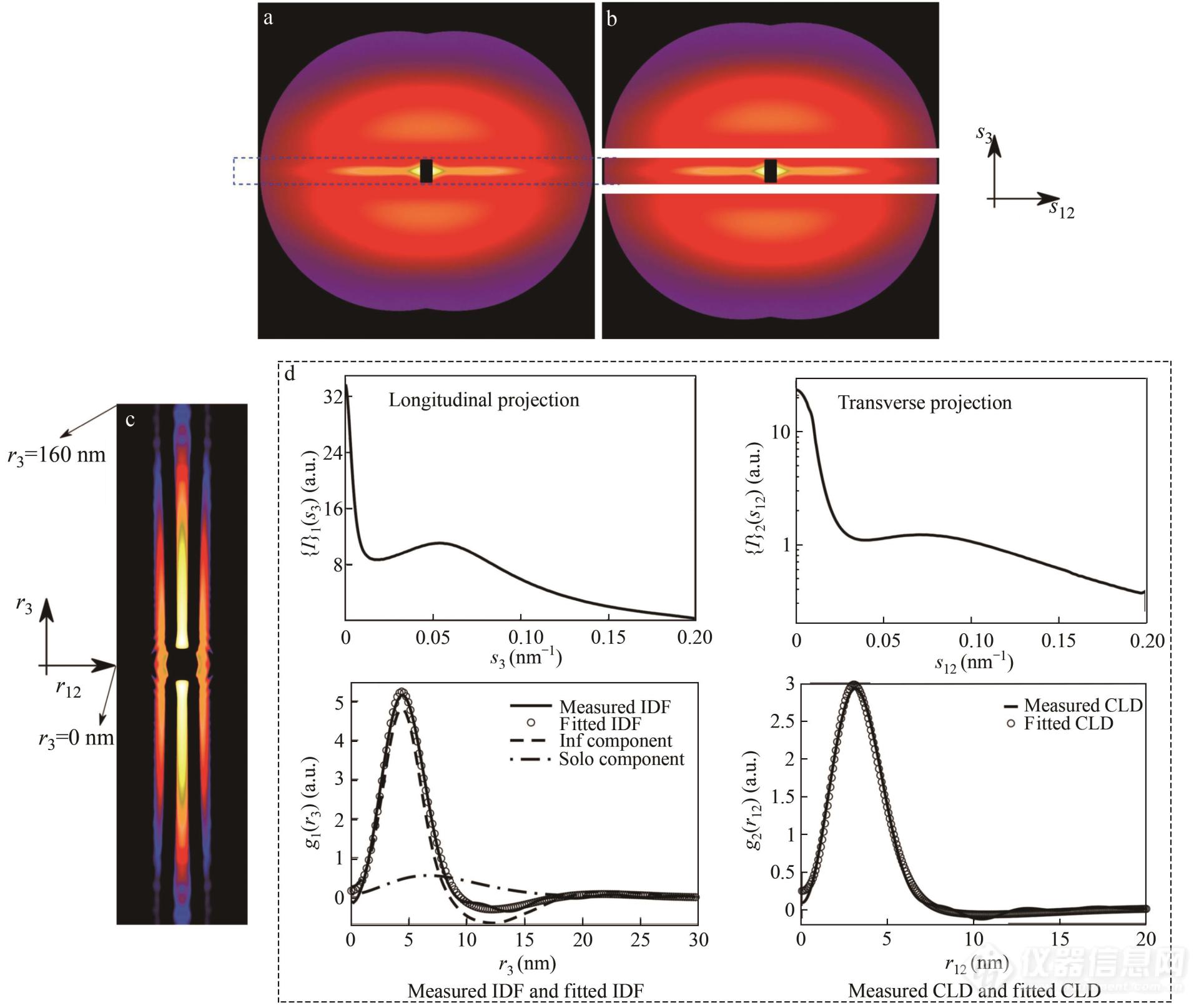
Fig. 8Schematic diagram of data evaluation. 2D SAXS patternI(s12,s3) with 0.01 nm-1≤|s12,s3|≤0.49 nm-1 processed in mirror symmetry (a) and separated into meridional two-spot scattering pattern and equatorial scattering streak (b) with a mask function. CDF (c) with -40 nm≤ r12≤40 nm and -160 nm≤r3≤160 nm computed from the extrapolated 2D SAXS pattern is employed to present the domain topology. The subfigure (d) indicates the Bonart's longitudinal projection from the separated meridional two-spot scattering pattern, transverse projection from the equatorial scattering streak, and their corresponding measured IDF/CLD and fitted IDF/CLD. (Reprinted with permission from Ref.[71]; Copyright (2017) Elsevier).
3.3.3Bragg公式
除以上2种方式外,另一种更加简便的方式是根据体系的散射峰位置,当体系为结晶高分子并呈现片晶叠层结构时,利用Bragg方程直接确定体系的长周期[77~79],即

除计算片晶厚度外,当体系为单轴取向体系时,可利用散射峰位置处的散射强度随方位角变化的分布计算片晶的侧向尺寸,根据倒易原理,该分布越窄则片晶侧向尺寸越大[80~82].
3.4球状及球壳粒子溶液体系
当体系中粒子浓度较高时,除了考虑单个粒子的散射,还需要考虑到粒子间的相互作用,此时探测器收集到的强度可表示为[10]:

其中,P(q)为形状因子,与单个粒子的散射强度有关,S(q)为结构因子,与粒子间的相互作用有关.
形状因子P(q)为散射振幅的平方. 半径为R的球体粒子,其散射振幅可表示为:
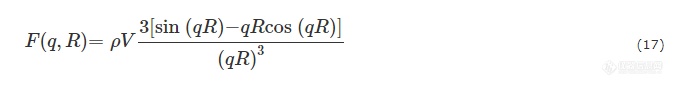
对于乳胶体系而言,粒子的尺寸分布通常可用高斯函数来描述,即:

其中,R0为粒子尺寸分布平均值,σR为标准差. 此时,体系的散射振幅可表示为:
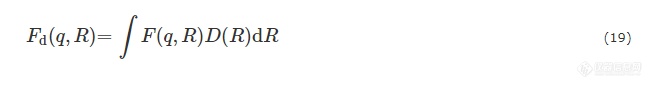
尺寸均一的核壳体系,散射振幅为:
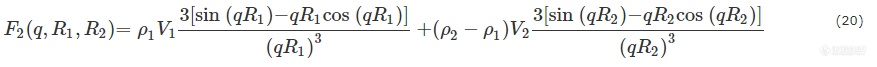
其中,R1为核壳粒子总半径,R2为核的半径,V1和V2分别为核壳粒子的总体积及核的体积,ρ1和ρ2分别为壳和核的电子密度. 考虑到尺寸的不均一性,核壳粒子体系散射振幅应为:
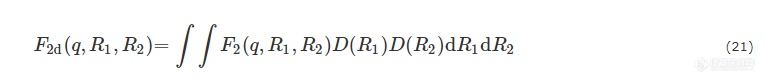
其中,D(R1)和D(R2)分别为核壳粒子和核的尺寸高斯分布.
以此类推,对于尺寸均一的多层核壳壳结构,其散射振幅为:
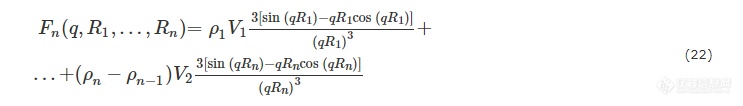
考虑到尺寸的不均一性,则多层核壳壳结构体系的散射振幅为:
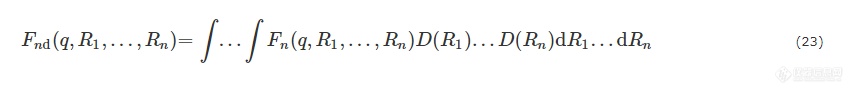
根据Percus-Yevick提出的适用于球形粒子的硬球模型[83~85],结构因子S(q)可表达为

其中
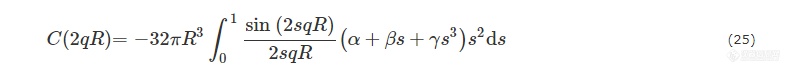
其中,体积分数ϕ可表示为
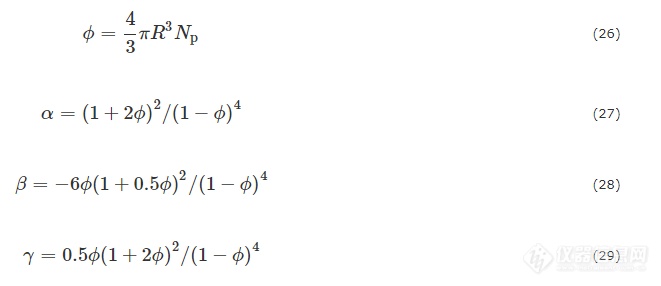
对于稀溶液体系而言,可认为S(q)=1,从而简化计算.
图9中给出了门永锋团队利用上述公式拟合核壳壳结构水溶液的实例[13]. 粒子的核为聚苯乙烯,中层壳为聚甲基丙烯酸甲酯,外层是聚丙烯酸乙酯和聚丙烯酸丁酯的共聚物. 如图9所示,拟合结果与实际曲线吻合很好,在各层电子密度已知的情况下,精确计算出了核的平均半径为85.9 nm,核壳平均尺寸为100.7 nm,核壳总体的平均半径为138.0 nm.
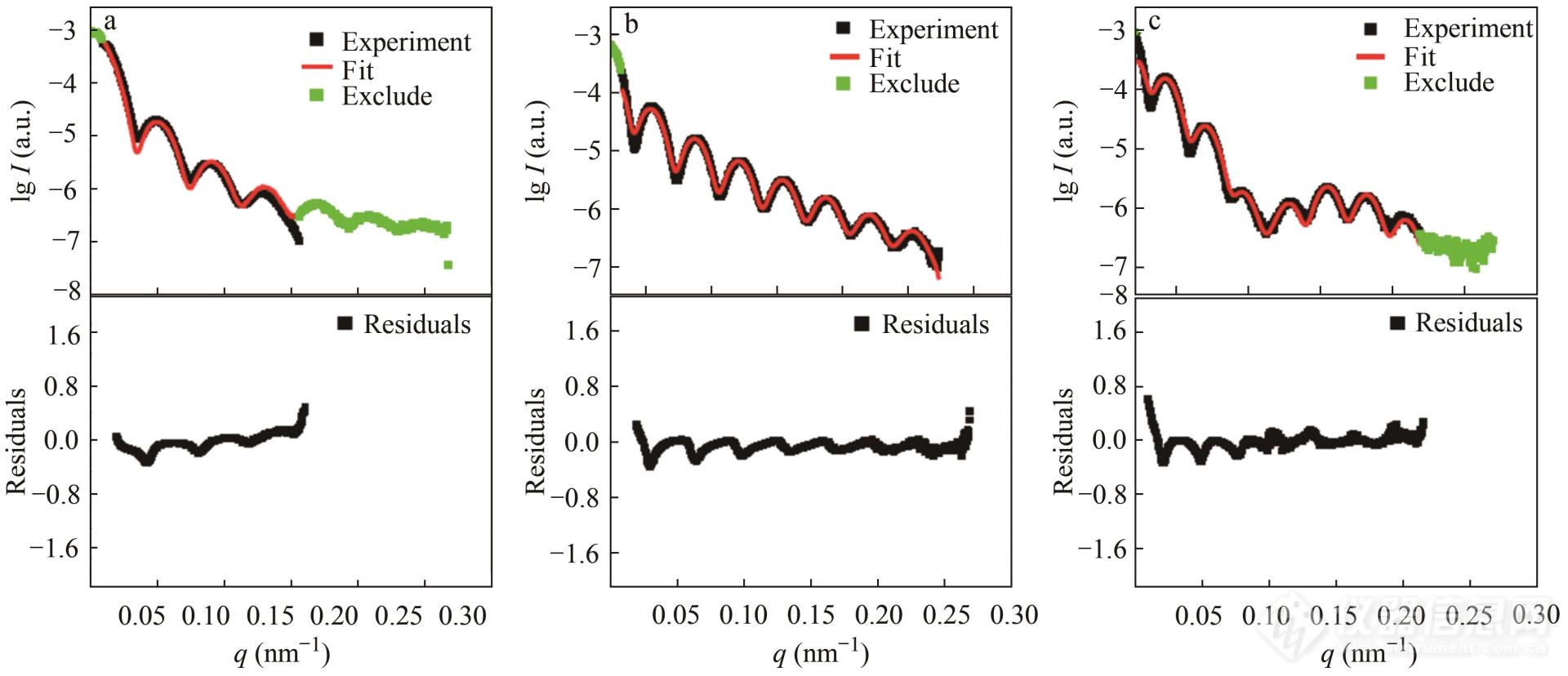
Fig. 9The fit results of (a) core, (b) core-shell and (c) core-shell-shell particles in colloidal dispersions and corresponding residuals[13].
郭旭虹团队以接枝了聚丙烯酸的聚苯乙烯球壳粒子为模板,构建了径向电子云分布十分复杂的聚苯乙烯-二氧化硅-聚丙烯酸球壳粒子,并用SAXS技术表征了其微观结构[86]. 在不考虑结构因子时,体系的散射主要由三部分构成:

其中I0(q)为单一粒子的散射强度,Ics(q)为整个核壳结构的贡献.Ishell(q)为壳的不均一性导致的散射,IPS(q)来源于PS核本身的电子云密度涨落(在本工作讨论中忽略).
对于球状粒子而言,Ics(q)可由散射振幅B(q)得到,B(q)可表达为:
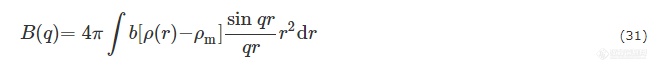
其中b为汤普森散射长度,ρm溶剂的电子云密度,ρ(r)为粒子的径向电子云密度.ρ(r)可根据粒子内部电子云分布特性分解为多个函数[86~88],在此工作中,二氧化硅构成的壳被分为i=1~5层,聚丙烯酸分子链被分为j=6~7层,如图10所示. 则核壳结构贡献的散射强度可表示为:
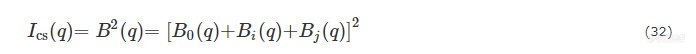
其中
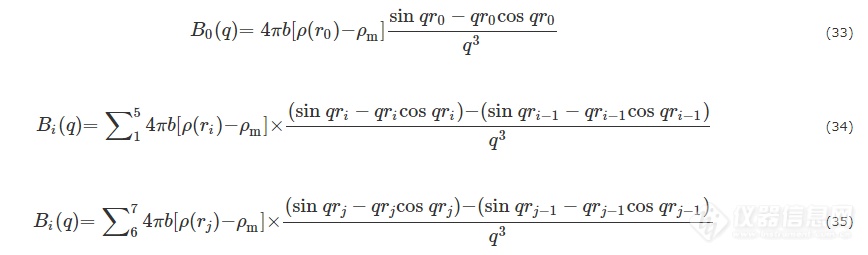
核不均一性导致的散射则可分解为二氧化硅壳的不均一性Iin(q)和聚丙烯酸分子链的热致密度涨落Ifluct(q),并且利用经验公式能够很好地描绘二者:
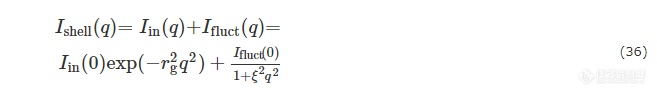
其中,rg为粒子的均方回转半径,量级在10 nm,ξ是分子链的相关长度,量级在几纳米. 在实际操作中,Iin(0)和Ifluct(0)作为可调节的变量.图11体系散射曲线和按上述方式拟合的曲线,数据吻合度高.
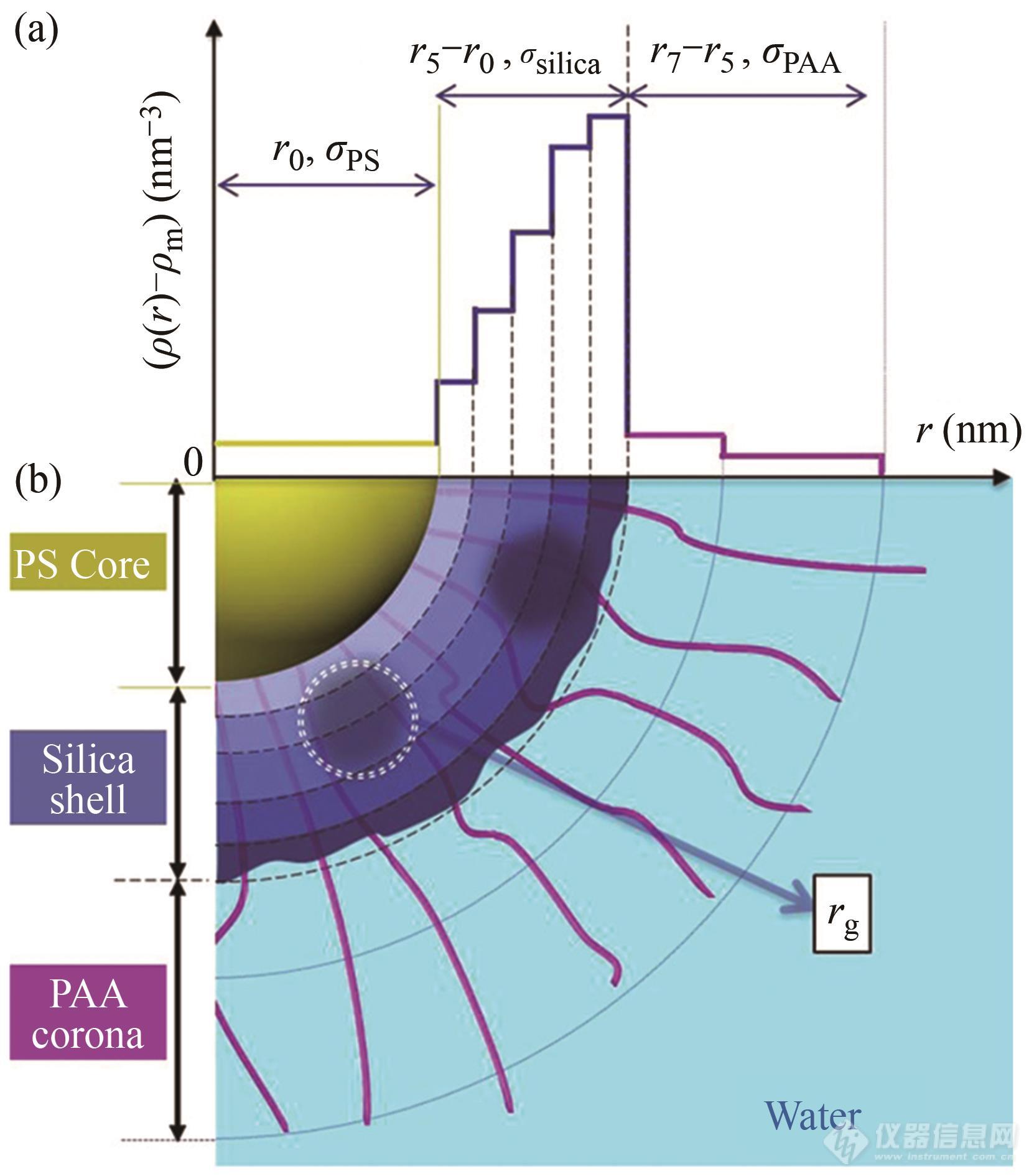
Fig. 10Double-shell fitting model of silica hybrid nano-particles. (a) Radial excess electron density [ρ(r)-ρm] distribution of the particle. Thex-axis is the radial distance r representing the distance from the center to local positions of silica hybrid nanoparticles. They-axis refers to the excess electron density of individual layers with respect to water. (b) Corresponding schematic illustration of the double-shell model. The colors yellow, dark blue, purple, and light blue refer to polystyrene core, silica shell, PAA shell, and water, respectively. (Reprinted with permission from Ref.[86]; Copyright (2017) American Chemical Society).
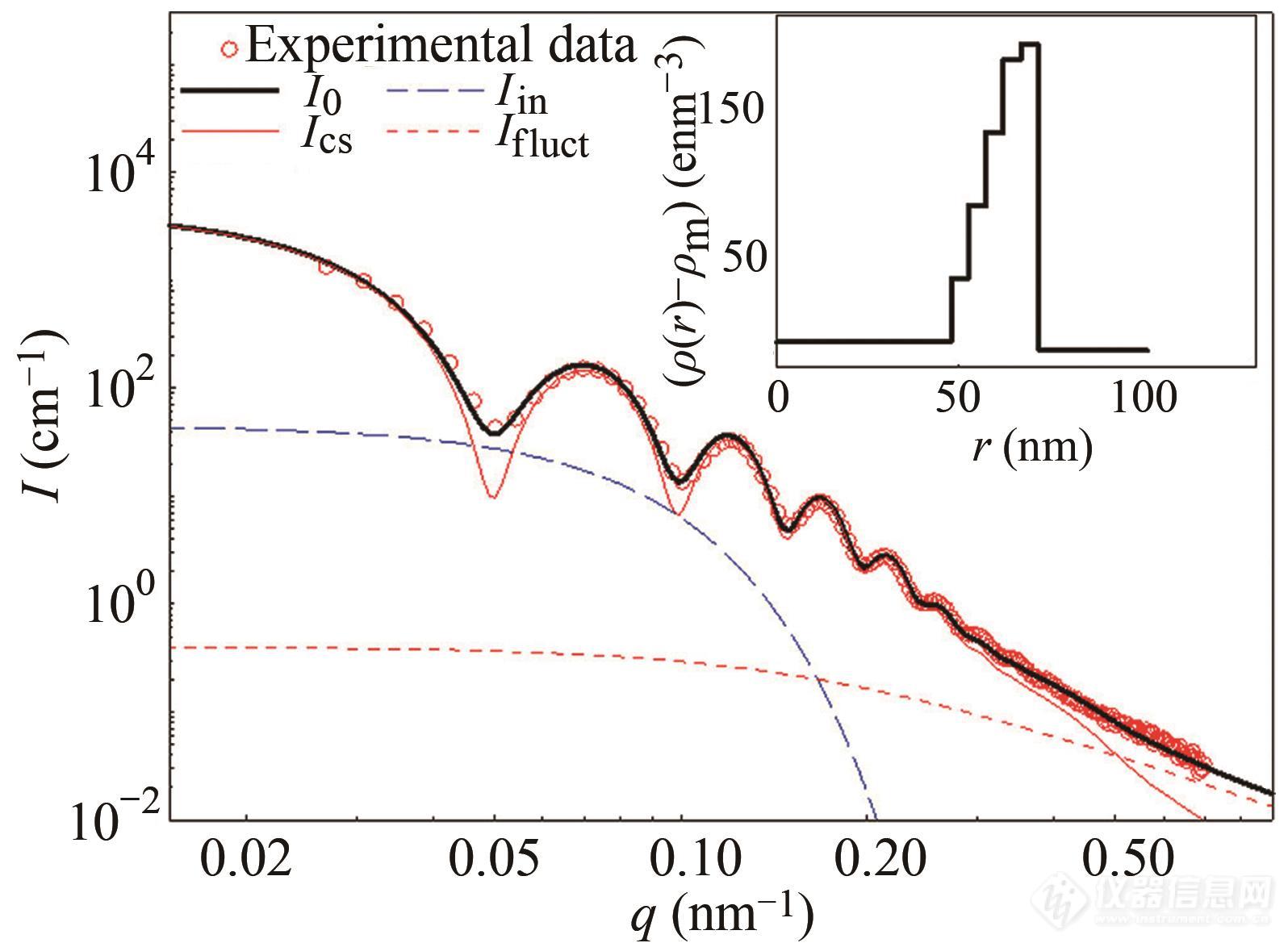
Fig. 11Decomposition of the scattering intensity of silica hybrid nanoparticles. The circles represent experimental data. Lines denote the fitting result (solid line), the spatial inhomogeneity of the silica shell (long dashed line), and the fluctuations of the PAA shell (short dashed line). The radial excess electron density profile is shown in the inset (Reprinted with permission from Ref.[86]; Copyright (2017) American Chemical Society).
3.5纤维状散射体的计算(Ruland方程)
最初Ruland使用此种算法来描述取向纤维[89,90],目前这种算法已经被很好地推广到串晶、微纤以及空洞领域[82,91~93]. 下面以计算空洞长轴为例,简要介绍这种算法[93].
首先,对取向明显的空洞散射信号进行如图12左侧所示的环形积分,并得到如图13右侧所示一系列强度曲线. 对于各个积分位置q都有与之对应的一维曲线且该曲线有着特定的积分面积A,可以得出积分宽度Bobs=A/q. 将一系列Bobs和q值代入
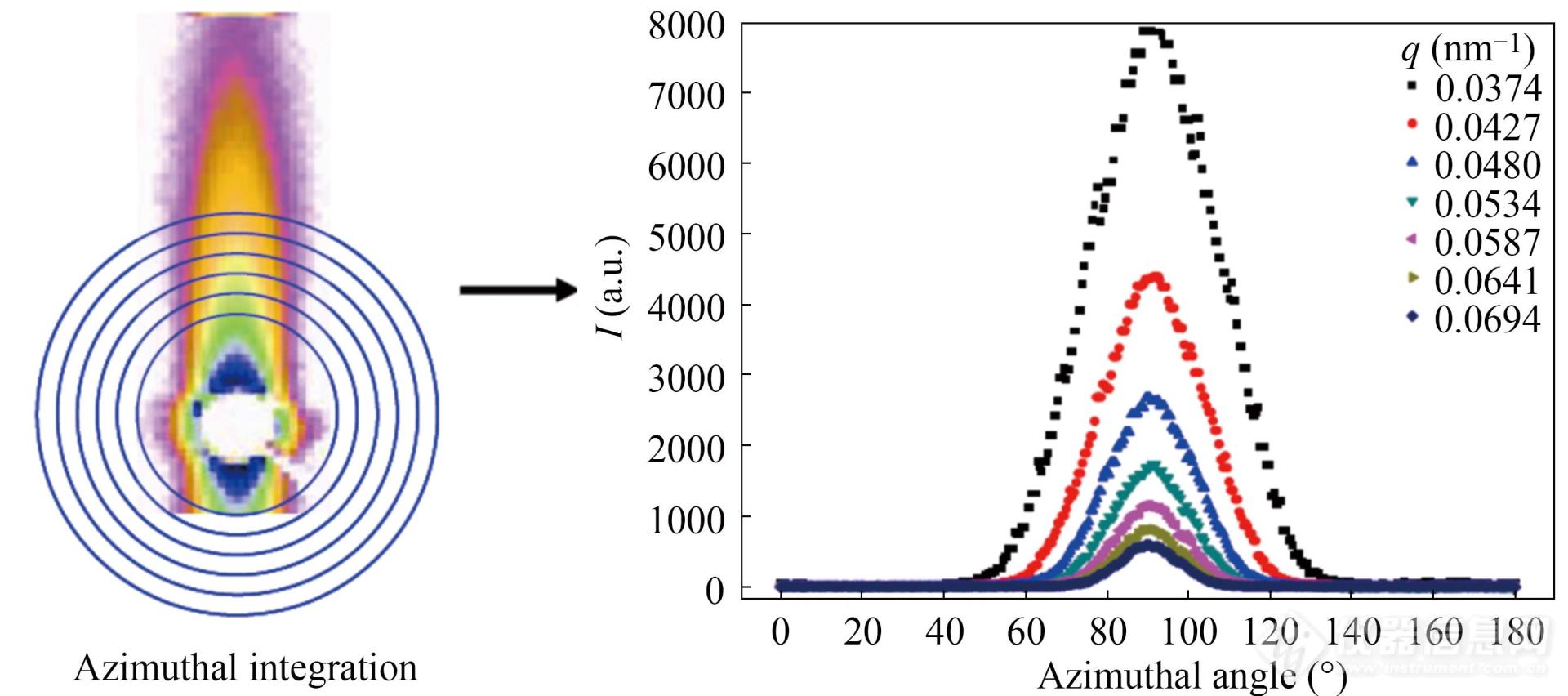
Fig. 12Azimuthal intensity distribution of highly orientediPP at different scattering vectors (Reprinted with permission from Ref.[93]; Copyright (2015) American Chemical Soc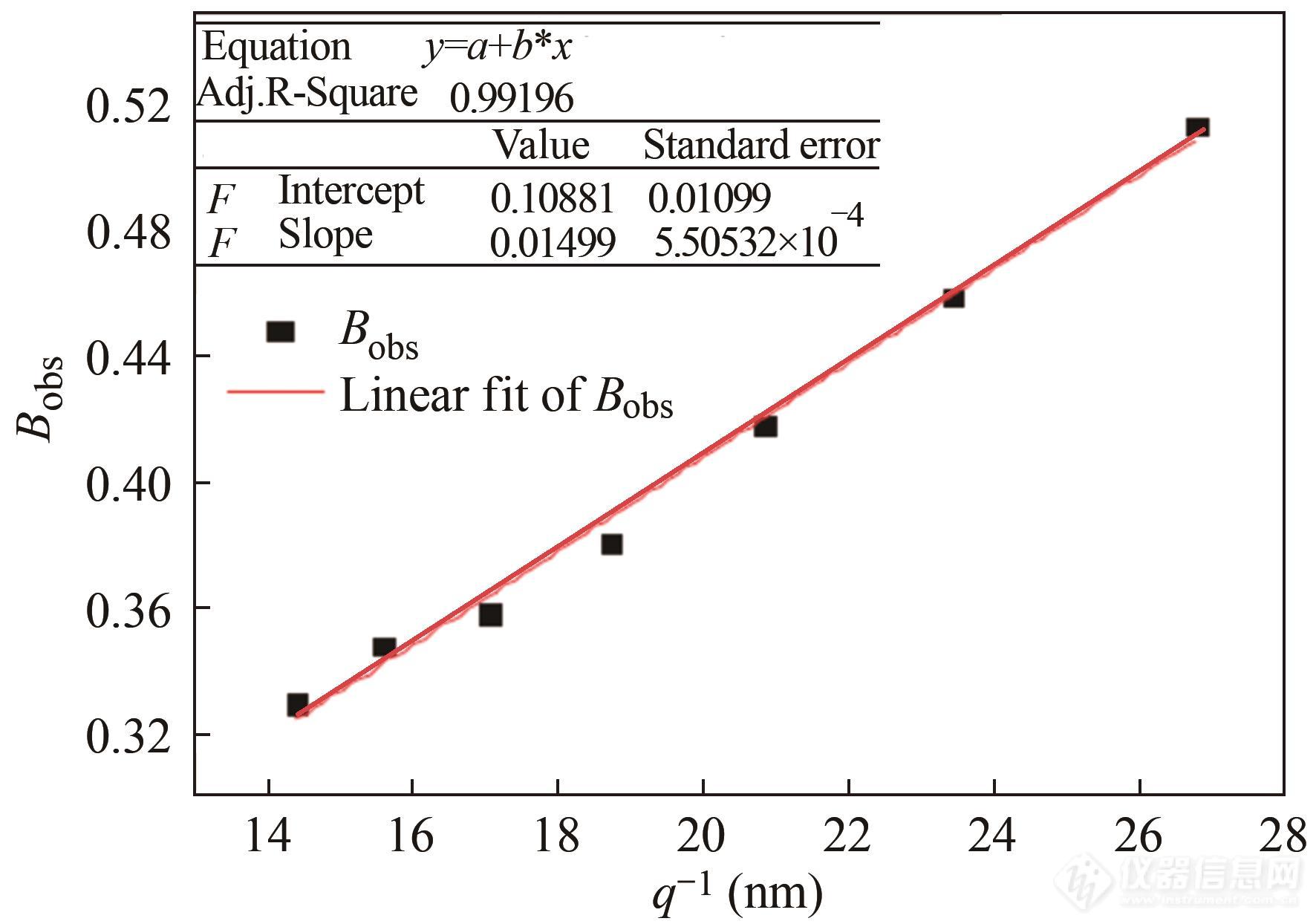 iety).
iety).
Fig. 13Relationship between the inverse scattering vectorq and the integral breadth Bobs (Reprinted with permission from Ref.[ 93]; Copyright (2015) American Chemical Society).

并对对应的数据点进行线性拟合,如图13所示,即可通过拟合曲线的斜率和截距得出散射体长度lc和散射体的取向误差BΦ.
门永锋团队利用上述算法对等规聚丙烯拉伸过程中的大形变空洞化进行了追踪,定量计算了空洞长轴尺寸[93],结果如图14所示.
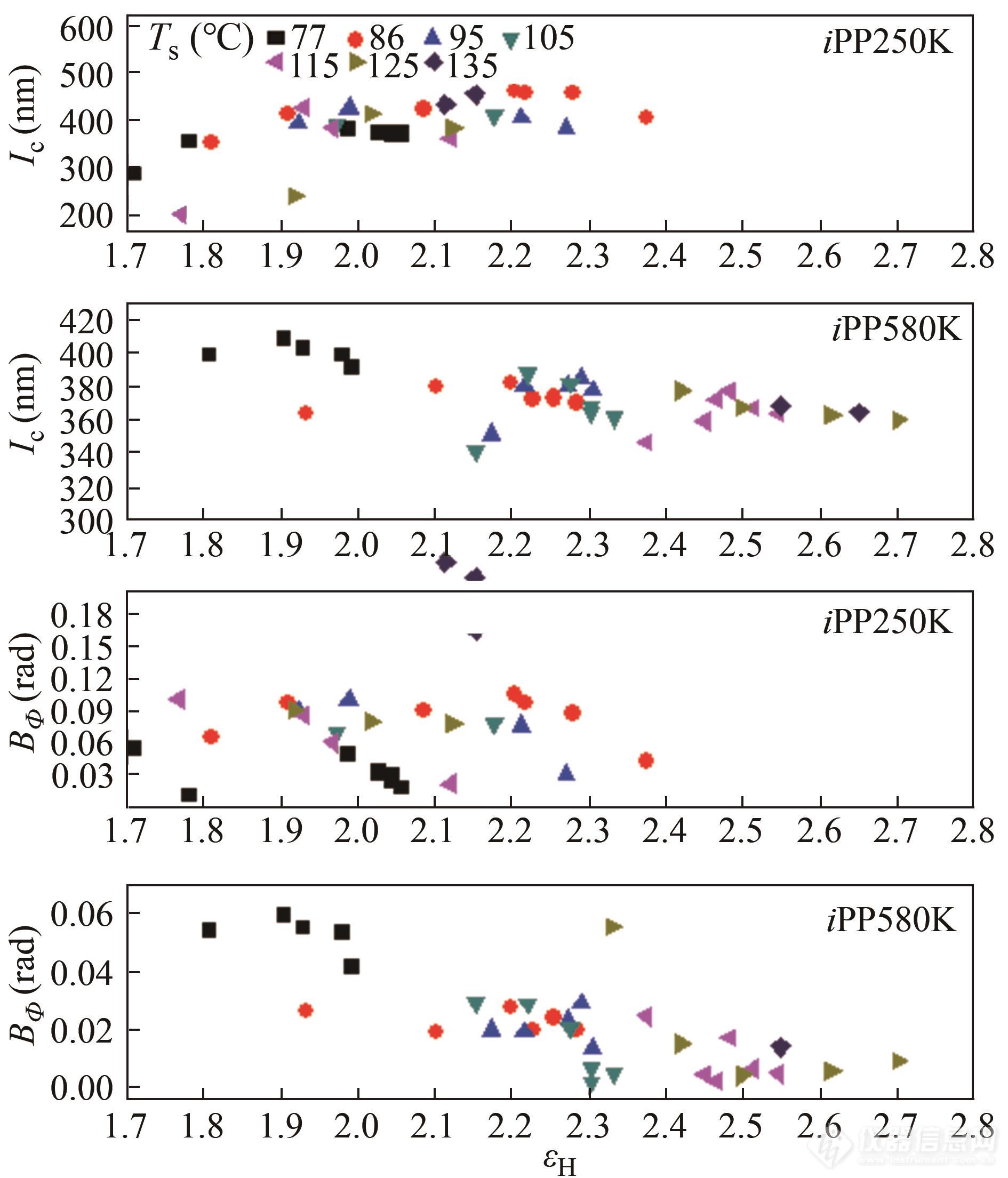
Fig. 14Length of long axis and misorientation of cavities obtainedvia Ruland method as a function of strain for iPP250K (top) andiPP580K (bottom) stretched at different temperatures (Reprinted with permission from Ref.[93]; Copyright (2015) American Chemical Society).
3.6圆柱状散射体的计算(Fischer算法)
为拟合聚丙烯样品中的空洞,Fischer等在前人的基础上提出了一种将空洞视为随机分布的且尺寸为对数正态分布圆柱体模型的算法[94]. 与Ruland算法拟合空洞相比,这种算法对空洞取向程度要求不高,更适合探究复杂空洞分布体系. 对于平行排列的圆柱体,其散射强度:
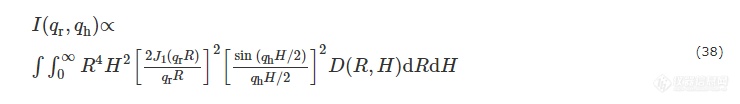
若其中的圆柱体底面半径R和高度H不相关,则有
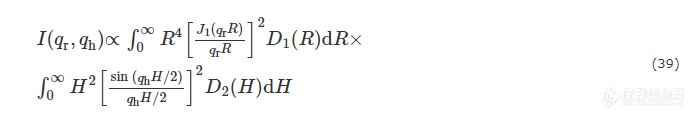
其中D1(R)和D2(H)分别为底面半径和高度的尺寸分布函数,其大小遵循对数正态分布.
门永锋团队在这个模型的基础上,引入了取向分布因子h(β)[29,95,96],散射强度变为:

其中,β为圆柱体法相方向和拉伸方向(x轴)的夹角,γ是圆柱体法相方向在yz平面(垂直于拉伸方向)的投影和y轴的夹角.ϕ是圆柱体的法相方向和散射矢量的夹角. 这个公式的引入,拓宽了Fischer模型的适用范围,使其能够描述空洞沿任意方向排列的体系.
3.7分形理论衡量自相似的随机结构
分形理论可以用来描述具有一定自相似性的任意结构[97],这种结构中一个非常重要的参数,分形维数D,可以用SAXS技术有效地测量[7,98,99]. 简单来说,就是利用指数函数对小q处散射数据进行拟合,所得指数即为分形维数D.D可用于描述聚集体的形态特征,在q溶液中的高斯链,D=2;有粗糙表面的团聚物对应的D为3~4;而具有平滑表面的紧密微区对应的D为4.
潘鹏举团队基于PNaAMPS网络,利用中性AM及阳离子单体构建了双网络结构,并利用分形理论探讨水凝胶的结构参数[99]. 阳离子为DAC, DBC, DMC,对应的名字为DN-A-x,DN-B-y,DN-M-z. 其中x,y,z代表第二网络内相应阳离子的摩尔浓度. 如图15,对于DN-A-x水凝胶的散射信号,没有明显的相关峰,这可能是由于第二网络的分子链穿插进入到了PNaAMPS的交联点,导致主网络中的相关关系模糊化. 这种DN-A-x水凝胶的信号可以用普适Ornstein-Zernike(GOZ)公式来描述:
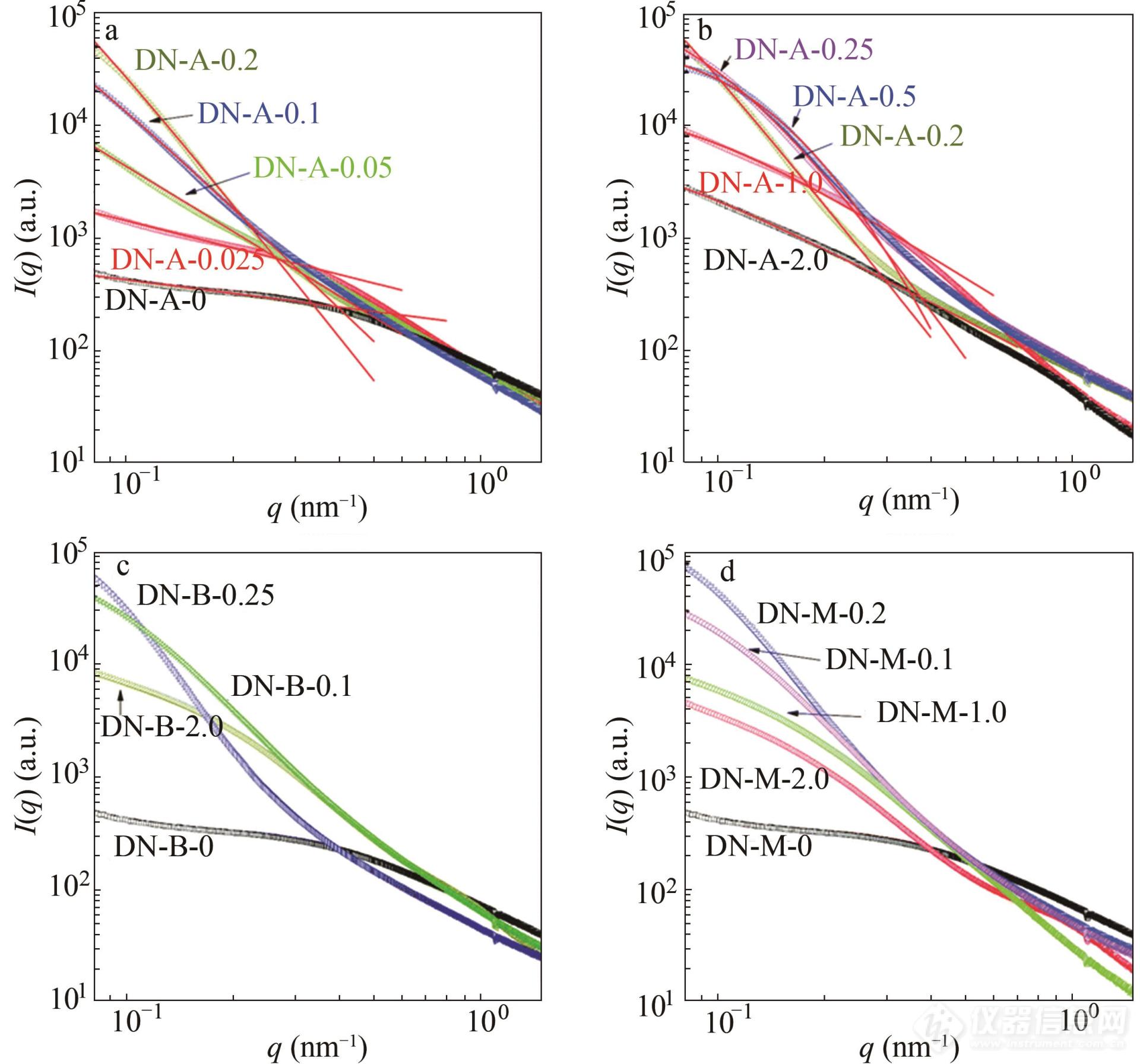
Fig. 15SAXS profiles of DN-A-x hydrogels (a) and (b), DN-B- y hydrogels (c), and DN-M- z hydrogels (d) in the swollen equilibrium state. The solid lines indicate the fits by generalized Ornstein-Zernike equation in the low- q region. Double-logarithmic coordinates are employed. (Reprinted with permission from Ref.[ 99]; Copyright (2019) The Royal Society of Chemistry).
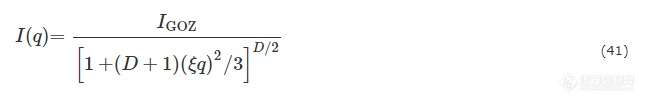
其中IGOZ为当q趋近于0时强度的渐近值,ξ是水凝胶中不均一聚集体的特征相关长度,在这里正比于交联微区的平均半径,D是分形维数.
通过拟合得到的ξ和D被收录到图16中. 在双网络中,第一个网络的紧密交联点作为不均一的聚集体和第二网络的松散交联链缠结. 这些聚集体的特征相关长度ξ可以用来衡量交联微区的尺寸.ξ随DAC的增加而增大,在超过离子平衡点后,随DAC的增加而减小. 当没有DAC时,第二网络的PAM链与PNaAMPS网络物理交联,使得ξ与基体网络的特征相关长度基本相等. 另一方面,添加DAC后,2个网络间形成了离子复合物,使得键合更加紧密. 除了物理缠结,离子的相互作用使得第二网络中更多的分子链穿插进入交联微区,增大了聚集体的特征相关长度. 随着DAC的增加,这种离子作用更强,ξ也逐渐到达峰值,超出平衡点后,ξ又变小. 这可能是由于第二网络中过剩的阳离子与交联微区随机作用,形成了有缺陷的离子复合物. 此外,P(DAC-co-AM)分子链和过剩的阳离子之间强大的静电互斥作用也能够使ξ变小. 分维数D的变化趋势与ξ一致.D先从2.06逐渐增加到4,超过离子平衡点后又逐渐减小,也就标志着在离子平衡点附近水凝胶形成了紧密的微区. 通过拟合样品的散射信号,可以很好地判断出水凝胶在各条件下的微区尺寸以及聚集体的形态特征.
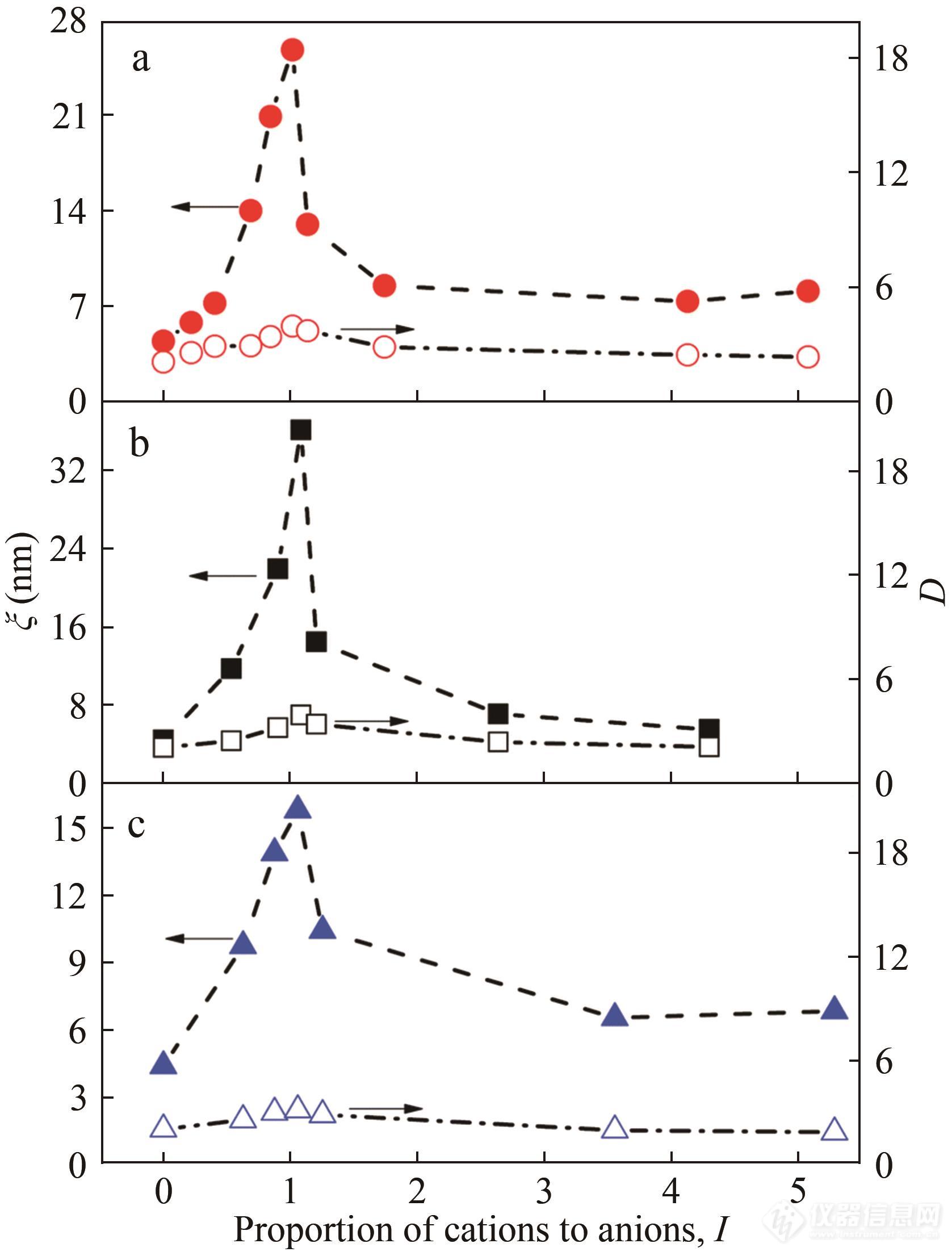
Fig. 16Effect of ion proportion on parameters,ξ (solid) and D (hollow), obtained by fitting the SAXS profiles of DN-A- x hydrogels (a), DN-B- y hydrogels (b), and DN-M- z hydrogels (c) with the GOZ equation in the low- q region (Reprinted with permission from Ref.[ 99]; Copyright (2019) The Royal Society of Chemistry).
4总结与展望
本文从小角X射线散射技术的基本原理出发,介绍了实验操作中的相关经验,并给出了几种经过实践的适用于不同微观结构的散射模型及结构参数的计算方法. 希望使初学者简单理解小角X射线散射基本理论的同时,还能快速判断这种技术是否对自己的研究有所帮助. 也希望文中介绍的实验技巧能够增添初学者的经验并在初期实验时少走弯路.
小角X射线散射技术操作简便,可与之搭配使用的小型装置多样化,适用于多种条件下材料的微观结构测试;此外,小角X射线散射技术属于无损测试,能得到样品体系内结构的统计平均信息,在高分子材料的表征中有着广泛的应用. 但是,由于分析散射图像需要结合相应理论模型,相对于其他结果直观的测试手段,这种技术常常令初学者望而却步. 随着近年来理论模型的丰富,以及批量数据处理软件的飞速发展,小角X射线散射技术的数据分析不再需要初学者具备深厚的数学功底,其处理程序变得简单明了,对用户越来越友好. 同时,随着同步辐射光源的升级,对材料在各种过程中的微观结构演变进行实时跟踪成为可能,因而小角X射线技术备受学术界和工业界的青睐.
我们认为小角X射线技术的进一步发展主要依赖于三部分,一是仪器,尤其是探测器的进一步优化;二是理论模型的再次精炼;三是数据分析软件的优化. 在目前的实验中,为保护探测器或/及去除窗口散射信号,常常需要利用直通光挡板(beamstop)遮挡直通光,在此过程中可能会有些结构信息被一并掩盖. 虽然现在已有算法能够还原出beamstop处散射强度,但这种方法在一定程度上受到主观干扰,并不是很理想. 现有的理论模型还没有覆盖到全部高分子材料的微观结构. 此外,在部分模型中,理想化的参数较多,分析结构时同样可能会受到主观因素的干扰. 第三,也是决定小角X射线散射技术能否实现应用大飞跃的一步,即数据处理软件的发展. 现阶段虽然已经出现了部分数据批处理软件,但仍处于小众化阶段,而且操作不够简便. 随着人工智能大数据的发展,是否能出现更智能的一键数据分析软件呢?我们拭目以待.
希望通过本文的介绍,能够激发初学者的学习兴趣,消除畏难心理,使得有需要的科研工作者可以更好地理解与应用小角X射线散射技术.
参考文献
1
Glatter O,Kratky O.Small-Angle Scattering of X-Rays.New York:Acadenic Press,1982
2
Guinier A.X-Ray Diffraction in Crystals, Imperfect Crystals, and Amorphous Bodies.San Francisco: W. H.Freeman and Company,1963
3
Guinier A,Fournet G.Small-Angle Scattering of X-Rays.New York:Wiley,1955
4
Stribeck N.X-Ray Scattering of Soft Matter.Berlin:Springer,2007
5
Lu Y.Molecular Weight and Chains Configuration Dependencies of Crystallization and Deformation in Polypropylene.Doctoral Dissertation of the University of Chinese Academy of Sciences,2015
6
Vonk C G,Kortleve G.Kolloid Z,1967,220:19-24.doi:10.1007/bf02086052
7
Zhu Yuping(朱育平).Small Angle X-ray Scattering-Theory, Measurment, Calculation and Application (小角X射线散射-理论、测试、计算及应用).Beijing(北京):Chemical Industry Press(化学工业出版社),2008
8
Strobl G.The Physics of Polymers.Berlin:Springer,2007
9
Lindner P, Zemb T. Neutrons, X-rays and Light: Scattering Methods Applied to Soft Condensed Matter.Amsterdam:Elsevier,2002.doi:10.12173/j.issn.1004-5511.2020.02.06
10
Roe R J.Methods of X-Ray and Neutron Scattering in Polymer Science.New York:Oxford University Press,2000
11
Stein R S.Scattering and Birefringence Methods Applied to Polymer Texture(散射和双折射方法在高聚物织构研究中的应用).Beijing(北京):Science Press(科学出版社),1983.doi:10.1002/app.1983.070280414
12
Chu B,Hsiao B S.Chem Rev,2001,101:1727-1762.doi:10.1021/cr9900376
13
Chen R.Application of Data Processing for Small-Angle X-ray Scattering in Polymer System.Doctoral Dissertation of the University of Chinese Academy of Sciences,2016
14
Pauw B R.J Phys:Condens Matter,2013,25:383201.doi:10.1088/0953-8984/25/38/383201
15
Hammersley A.J Appl Crystallogr,2016,49:646-652.doi:10.1107/s1600576716000455
16
Semenyuk A V,Svergun D I.J Appl Crystallogr,1991,24:537-540.doi:10.1107/s002188989100081x
17
Bressler I,Kohlbrecher J,Thunemann A F.J Appl Crystallogr,2015,48:1587-1598.doi:10.1107/s1600576715007347
18
Boon N,Schurtenberger P.Phys Chem Chem Phys,2017,19:23740-23746.doi:10.1039/c7cp02434g
19
Forster S,Apostol L,Bras W.J Appl Crystallogr,2010,43:639-646
20
Konarev P V,Petoukhov M V,Volkov V V,Svergun D I.J Appl Crystallogr,2006,39:277-286.doi:10.1107/s0021889806004699
21
Bressler I,Pauw B R,Thunemann A F.J Appl Crystallogr,2015,48:962-969.doi:10.1107/s1600576715007347
22
Hopkins J B,Gillilan R E,Skou S.J Appl Crystallogr,2017,50:1545-1553.doi:10.1107/s1600576717011438
23
Lyu D,Sun Y Y,Lu Y,Liu L Z,Chen R,Thompson G,Caton-Rose F,Coates P,Wang Y,Men Y F.Macromolecules,2020,53:4863-4873.doi:10.1021/acs.macromol.0c00005
24
Wang B H,He K Z,Lu Y G,Zhou Y F,Chen J L,Shen C Y,Chen J B,Men Y F,Zhang B.Macromolecules,2020,53:6476-6485.doi:10.1021/acs.macromol.0c00885
25
Zhou J,Zheng Y,Shan G R,Bao Y Z,Wang W J,Pan P J.Polymer,2020,188:122121.doi:10.1016/j.polymer.2019.122121
26
Lu Y,Lyu D,Tang Y J,Qian L,Qin Y N,Xiang M Y,Men Y F.Polymer,2020,210:123049.doi:10.1016/j.polymer.2020.123049
27
Lyu D,Tang Y J,Qian L,Chen R,Lu Y,Men Y F.Polymer,2019,167:146-153.doi:10.1016/j.polymer.2019.01.081
28
Jiang Z Y,Liao T,Chen R,Men Y F.Polymer,2019,185:121984.doi:10.1016/j.polymer.2019.121984
29
Lyu D,Chen R,Lu Y,Men Y F.Ind Eng Chem Res,2018,57:8927-8937.doi:10.1021/acs.iecr.8b01650
30
Lu Y,Men Y F.Chinese J Polym Sci,2018,36:1195-1199.doi:10.1007/s10118-018-2123-x
31
Lu Y,Men Y F.Macromol Mater Eng,2018,303:1800203.doi:10.1002/mame.201800203
32
Hu S S,Rieger J,Lai Y Q,Roth S V,Gehrke R,Men Y F.Macromolecules,2008,41:5073-5076.doi:10.1021/ma800451n
33
Shen J F,Zhou Y F,Lu Y G,Wang B H,Shen C Y,Chen J B,Zhang B.Macromolecules,2020,53:2136-2144.doi:10.1021/acs.macromol.9b01880
34
Lu Y G,Li H,Wei H X,Wang B H,Shen C Y,Zhang B,Chen J B.Polymer,2020,199:122562.doi:10.1016/j.polymer.2020.122562
35
Konko I,Guriyanova S,Boyko V,Sun L C,Liu D,Reck B,Men Y F.Langmuir,2019,35:6075-6088.doi:10.1021/acs.langmuir.8b04327
36
Lin Y F,Li X Y,Chen X W,An M F,Zhang Q L,Wang D L,Chen W,Yin P C,Meng L P,Li L B.Polymer,2019,178:121579.doi:10.1016/j.polymer.2019.121579
37
Lv C,Wang R Y,Gao J,Ding N,Dong S N,Nie J J,Xu J T,Du B Y.Polymer,2019,185:121982.doi:10.1016/j.polymer.2019.121982
38
Lu Y P,Chen T Q,Mei A X,Chen T Y,Ding Y W,Zhang X H,Xu J T,Fan Z Q,Du B Y.Phys Chem Chem Phys,2013,15:8276-8286.doi:10.1039/c3cp50376c
39
Yan J J,Tang R P,Zhang B,Zhu X Q,Xi F,Li Z C,Chen E Q.Macromolecules,2009,42:8451-8459.doi:10.1021/ma901494z
40
Liu X B,Zhao Y F,Chen E Q,Ye C,Shen Z H,Fan X H,Cheng S Z D,Zhou Q F.Macromolecules,2008,41:5223-5229.doi:10.1021/ma800517k
41
Hsiao B S,Chu B,Burger C.Synchrotron Radiat News,2002,15:20-34.doi:10.1080/08940880208602974
42
Xiang M Y,Lyu D,Qin Y N,Chen R,Liu L Z,Men Y F.Polymer,2020,210:123034.doi:10.1016/j.polymer.2020.123034
43
Ding S S,Fang C,Wang X H,Wang Z G.Polymer,2020,186:121993.doi:10.1016/j.polymer.2019.121993
44
Wang W,Wang X,Jiang F,Wang Z.Polym Chem,2018,9:3067-3079.doi:10.1039/c8py00375k
45
Wu Q W,Lv C,Zhang Z J,Li Y Q,Nie J J,Xu J T,Du B Y.Langmuir,2018,34:9203-9214.doi:10.1021/acs.langmuir.8b01575
46
Jiang H,Ye L,Wang Y H,Ma L,Cui D M,Tang T.Macromolecules,2020,53:3349-3357.doi:10.1021/acs.macromol.0c00159
47
Liu K,Yang C M,Yang B M,Zhang L,Huang W C,Ouyang X P,Qi F G,Zhao N,Bian F G.Chinese J Polym Sci,2020,38:92-99.doi:10.1007/s10118-019-2315-z
48
Bhaumik S,Ntetsikas K,Hadjichristidis N.Macromolecules,2020,53:6682-6689.doi:10.1021/acs.macromol.9b02326
49
Tap T D,Nguyen L,Hasegawa S,Sawada S,Luan L,Maekawa Y.J Appl Polym Sci,2020,137:e49029.doi:10.1002/app.49029
50
Zhang Q L,Li L F,Su F M,Ji Y X,Ali S,Zhao H Y,Meng L P,Li L B.Macromolecules,2018,51:4350-4362. 10.doi:10.1021/acs.macromol.8b00346
51
Zofchak E S,LaNasa J A,Torres V M,Hickey R J.Macromolecules,2020,53:835-843.doi:10.1021/acs.macromol.9b01695
52
Strobl G R,Schneider M.J Polym Sci,Part B:Polym Phys,1980,18:1343-1359.doi:10.1002/pol.1980.180180614
53
Yang S,Wei Q Y,Gao X R,Zhou L,Xu L,Tang J H,Zhong G J,Ji X,Li Z M.Polymer,2020,187:122099.doi:10.1016/j.polymer.2019.122099
54
Hu T,Hua W Q,Zhong G J,Wang Y D,Gao Y T,Hong C X,Li Z M,Bian F G,Xiao T Q.Macromolecules,2020,53:6498-6509.doi:10.1021/acs.macromol.0c01177
55
Zhu H,Lv Y,Shi D,Li Y G,Miao W J,Wang Z B.Chinese J Polym Sci,2020,38:1015-1024.doi:10.1007/s10118-020-2427-5
56
Huang S Y,Li H F,Jiang S C.Polymer,2019,175:81-86.doi:10.1016/j.polymer.2019.05.020
57
Zhang W Y,Li J Q,Li H F,Jiang S C,An L J.Polymer,2018,143:309-315.doi:10.1016/j.polymer.2018.04.030
58
Li W Z,Gong P J,Huang Y J,Niu Y H,Li G X.Appl Surf Sci,2020,501:144251.doi:10.1016/j.apsusc.2019.144251
59
Li W Z,Niu Y H,Zhou C T,Luo H,Li G X.Chinese J Polym Sci,2017,35:1402-1414.doi:10.1007/s10118-017-1997-3
60
Wang H,Wu C J,Cui D M,Men Y F.Macromolecules,2018,51:497-503.doi:10.1021/acs.macromol.7b01943
61
Chen R,Lu Y,Jiang Z Y,Men Y F.J Polym Sci,Part B:Polym Phys,2018,56:219-224.doi:10.1002/polb.24536
62
Cruz C S,Stribeck N,Zachmann H G,Calleja F J B.Macromolecules,1991,24:5980-5990.doi:10.1021/ma00022a013
63
Ruland W.Colloid Polym Sci,1977,255:417-427.doi:10.1007/bf01536457
64
Ruland W.Colloid Polym Sci,1978,256:932-936.doi:10.1007/bf01383589
65
Qiao Y N,Schulz M,Wang H,Chen R,Schafer M,Thurn-Albrecht T,Men Y F.Polymer,2020,195:122425.doi:10.1016/j.polymer.2020.122425
66
Porod G.Kolloid Z,1951,124:83-114.doi:10.1007/bf01512792
67
Albrecht T,Strobl G.Macromolecules,1995,28:5827-5833.doi:10.1021/ma00121a020
68
Albrecht T,Strobl G.Macromolecules,1995,28:5267-5273.doi:10.1021/ma00121a020
69
Tchoubar D,Mering J.J Appl Crystallogr,1969,2:128-138.doi:10.1107/s0021889869006716
70
Stribeck N.Colloid Polym Sci,2002,280:254-259.doi:10.1007/s00396-001-0601-z
71
Li X K,Lu Y,Wang H,Poselt E,Eling B,Men Y F.Eur Polym J,2017,97:423-436.doi:10.1016/j.eurpolymj.2017.10.014
72
Bonart R.Kolloid Z,1966,211:14-33.doi:10.1007/bf01500205
73
Stribeck A,Li X,Zeinolebadi A,Pöselt E,Eling B,Funari S.Macromol Chem Phys,2015,216:2318-2330.doi:10.1002/macp.201500255
74
Li X K,Lu Y,Sun Y Y,Che Y H,Men Y F.Ind Eng Chem Res,2017,56:8535-8542.doi:10.1021/acs.iecr.7b01757
75
Stribeck A,Pöselt E,Eling B,Jokari-Sheshdeh F,Hoell A.Eur Polym J,2017,94:340-353.doi:10.1016/j.eurpolymj.2017.07.020
76
Stribeck N.Colloid Polym Sci,1993,271:1007-1023.doi:10.1007/bf00659290
77
Hu J,Wang J P,Gowd E B,Yuan Y,Zhang T P,Duan Y X,Hu W B,Zhang J M.Polymer,2019,167:122-129.doi:10.1016/j.polymer.2019.01.088
78
Wang Y,Zhao J,Qu M J,Guo J,Yang S G,Lei J,Xu J Z,Chen Y H,Li Z M,Hsiao B S.Polymer,2018,134:196-203.doi:10.1016/j.polymer.2017.11.040
79
Zhao H Y,Li L F,Zhang Q L,Xia Z J,Yang E J,Wang Y S,Chen W,Meng L P,Wang D L,Li L B.Biomacromolecules,2019,20:3895-3907.doi:10.1021/acs.biomac.9b00975
80
Lu Y,Thompson G,Lyu D,Caton-Rose P,Coates P,Men Y F.Soft Matter,2018,14:4413-4650.doi:10.1039/c7sm02446k
81
Tang Y J,Jiang Z Y,Men Y F,An L J,Enderle H F,Lilge D,Roth S V,Gehrke R,Rieger J.Polymer,2007,48:5125-5132.doi:10.1016/j.polymer.2007.06.056
82
Lei C H,Xu R J,Tian Z Q,Huang H H,Xie J Y,Zhu X Q.Macromolecules,2018,51:3433-3442.doi:10.1021/acs.macromol.7b02335
83
Percus J K,Yevick G J.Phys Rev,1958,110:1-13.doi:10.1103/physrev.110.1
84
Ashcroft N W,Lekner J.Phys Rev,1966,145:83-90.doi:10.1103/physrev.145.83
85
Kinning D J,Thomas E L.Macromolecules,1984,17:1712-1718.doi:10.1021/ma00139a013
86
Han H Y,Li L,Wang W H,Tian Y C,Wang Y W,Wang J Y,von Klitzing R,Guo X H.Langmuir,2017,33:9857-9865.doi:10.1021/acs.langmuir.7b02239
87
Ye Z S,Li L,Zhao F,Tian Y C,Wang Y W,Yang Q S,Dai L H,Guo X H.J Polym Sci,Part B:Polym Phys,2019,57:738-747.doi:10.1002/polb.24828
88
Yang Q S,Li L,Zhao F,Han H Y,Wang W H,Tian Y C,Wang Y W,Ye Z S,Guo X H.J Mater Sci,2019,54:2552-2565.doi:10.1007/s10853-018-2996-7
89
Ruland W.J Appl Phys,1967,38:3585-3589.doi:10.1063/1.1710176
90
Ruland W.J Polym Sci Polym Symp,1969,28:143-151.doi:10.1002/polc.5070280113
91
Liao T,Zhao X T,Yang X,Coates P,Whiteside B,Jiang Z Y,Men Y F.Polymer,2019,180:121698.doi:10.1016/j.polymer.2019.121698
92
Zhao J Y,Yang X,Sun Y Y,Men Y F.Ind Eng Chem Res,2018,57:4967-4977.doi:10.1021/acs.iecr.8b00194
93
Lu Y,Wang Y T,Chen R,Zhao J Y,Jiang Z Y,Men Y F.Macromolecules,2015,48:5799-5806.doi:10.1021/acs.macromol.5b00818
94
Fischer S,Diesner T,Rieger B,Marti O.J Appl Crystallogr,2010,43:603-610.doi:10.1107/s0021889810006163
95
Chen R,Lu Y,Jiang Z Y,Men Y F.J Phys Chem B,2018,122:4159-4168.doi:10.1021/acs.jpcb.8b00060
96
Chen R,Lu Y,Zhao J Y,Jiang Z Y,Men Y F.J Polym Sci,Part B:Polym Phys,2016,54:2007-2014.doi:10.1002/polb.24108
97
Mildner D F R, Hall P L.J Phys D:Appl Phys,1986,19:1535-1545.doi:10.1088/0022-3727/19/8/021
98
Pizzorusso G,Fratini E,Eiblmeier J,Giorgi R,Chelazzi D,Chevalier A,Baglioni P.Langmuir,2012,28:3952-3961.doi:10.1021/la2044619
99
Zhao X Y,Liang J,Shan G R,Pan P J.J Mater Chem B,2019,7:324-333.doi:10.1039/c8tb02803f
原文链接:
http://www.gfzxb.org/thesisDetails#10.11777/j.issn1000-3304.2020.20249&lang=zh《高分子学报》高分子表征技术专题链接:
http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304
DOI:10.11777/j.issn1000-3304.2020.20249
[来源:高分子学报]
高分子表征技术专题——透射电子显微镜在聚合物不同层次结构研究中的应用
2021.12.28

2024.07.26

首届江西南昌高校联合检测机构大型仪器操作技能大赛在南昌成功举办
2024.07.25

第3届测量仪器国际会议暨第13届精密工程测量与仪器国际会议通知
2024.07.24

2024.07.24

AI时代,质谱软件也被卷到了——访科迈恩(北京)科技有限公司总经理/创始人田润涛
2024.07.24
品牌合作伙伴






























版权与免责声明:
① 凡本网注明"来源:仪器信息网"的所有作品,版权均属于仪器信息网,未经本网授权不得转载、摘编或利用其它方式使用。已获本网授权的作品,应在授权范围内使用,并注明"来源:仪器信息网"。违者本网将追究相关法律责任。
② 本网凡注明"来源:xxx(非本网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责,且不承担此类作品侵权行为的直接责任及连带责任。如其他媒体、网站或个人从本网下载使用,必须保留本网注明的"稿件来源",并自负版权等法律责任。
③ 如涉及作品内容、版权等问题,请在作品发表之日起两周内与本网联系,否则视为默认仪器信息网有权转载。
![]() 谢谢您的赞赏,您的鼓励是我前进的动力~
谢谢您的赞赏,您的鼓励是我前进的动力~
打赏失败了~
评论成功+4积分
评论成功,积分获取达到限制
![]() 投票成功~
投票成功~
投票失败了~











