视频号

抖音号

哔哩哔哩号

前沿资讯手机看

热分析&电镜&表面分析,分享最新国内外仪器技术成果进展



分享到微信朋友圈
打开微信,点击底部的“发现”,
使用“扫一扫”即可将网页分享到朋友圈。
2021年,《高分子学报》邀请了国内擅长各种现代表征方法的一流高分子学者领衔撰写从基本原理出发的高分子现代表征方法综述并上线了虚拟专辑。仪器信息网在获《高分子学报》副主编胡文兵老师授权后,也将上线同名专题并转载专题文章,帮助广大研究生和年轻学者了解、学习并提升高分子表征技术。在此,向胡文兵老师和组织及参与撰写的各位专家学者表示感谢。
更多专题内容详见:高分子表征技术专题

高分子表征技术专题前言
孔子曰:“工欲善其事,必先利其器”。 我们要做好高分子的科学研究工作,掌握基本的表征方法必不可少。每一位学者在自己的学术成长历程中,都或多或少地有幸获得过学术界前辈在实验表征方法方面的宝贵指导!随着科学技术的高速发展,传统的高分子实验表征方法及其应用也取得了长足的进步。目前,中国的高分子学术论文数已经位居世界领先地位,但国内关于高分子现代表征方法方面的系统知识介绍较为缺乏。为此,《高分子学报》主编张希教授委托副主编王笃金研究员和胡文兵教授,组织系列从基本原理出发的高分子现代表征方法综述,邀请国内擅长各种现代表征方法的一流高分子学者领衔撰写。每篇综述涵盖基本原理、实验技巧和典型应用三个方面,旨在给广大研究生和年轻学者提供做好高分子表征工作所必须掌握的基础知识训练。我们的邀请获得了本领域专家学者的热情反馈和大力支持,借此机会特表感谢!
从2021年第3期开始,以上文章将陆续在《高分子学报》发表,并在网站上发布虚拟专辑,以方便大家浏览阅读. 期待这一系列的现代表征方法综述能成为高分子科学知识大厦的奠基石,支撑年轻高分子学者的茁壮成长!也期待未来有更多的学术界同行一起加入到这一工作中来.
高分子表征技术的发展推动了我国高分子学科的持续进步,为提升我国高分子研究的国际地位作出了贡献. 借此虚拟专辑出版之际,让我们表达对高分子物理和表征学界的老一辈科学家的崇高敬意!


光散射技术在高分子表征研究中的应用
Laser Light Scattering and Its Applications in Polymer Characterization
作者机构:
中国石化北京化工研究院,北京,100013;北京大学化学与分子工程学院,北京,100871
作者简介:
梁德海,男,1971年生. 1994年获南开大学环境科学系理学学士,同年进入南开大学化学系攻读硕士. 2001年在美国纽约州立大学石溪分校获得理学博士学位,并留任博士后. 2006年加入北京大学化学与分子工程学院高分子科学与工程系,任副教授;2012年任教授. 2011年得到教育部新世纪优秀人才计划的支持,2015获得Elsevier第九届冯新德高分子奖最佳文章奖. 研究方向为高分子溶液物理,主要项目包括:基于生物大分子的非平衡态原始细胞模型的构筑及动态行为研究;多肽诱导脂质体膜内吞及外吐机理研究;大分子拥挤及限制作用的定量化研究.
摘要
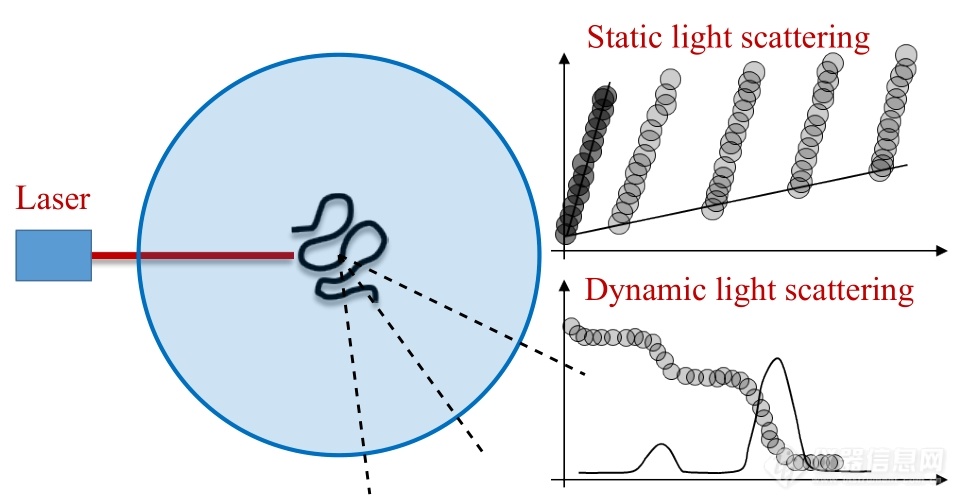
光散射技术是高分子领域中重要的表征手段之一. 静态光散射和动态光散射的结合能够获得丰富的关于高分子的信息,如重均分子量、回转半径、第二维里系数、流体力学半径、尺寸分布、分子链构象等. 除合成高分子外,光散射技术同样适用于研究生物大分子、微生物、胶体、纳米粒子、病毒、囊泡等在溶液或悬浮液中的行为. 本综述重点介绍稀溶液中静态光散射和动态光散射的历史、基本理论和实验技巧. 对于浓溶液适用的交叉相关技术和扩散波谱技术以及固体光散射也做简要介绍. 为了帮助初学者更好地理解并掌握光散射技术,综述的最后介绍了4个应用实例:动、静态光散射相结合跟踪研究线团到密实球的转变过程,光散射确定超支化分子的标度关系,时间可分辨的光散射来剖析聚合诱导胶束化的机理,以及去偏振动态光散射研究纳米粒子在生物介质中的聚集行为.
Abstract
Laser light scattering (LLS), which includes static light scattering (SLS) and dynamic light scattering (DLS), has been widely applied in characterization of polymer samples in dilute solutions. SLS measures the angular dependence of the excess scattered intensity, from which the weight average molecular weight, radius of gyration, and second viral coefficient are obtained. DLS measures the intensity-intensity time correlation functions, from which the hydrodynamic radius and size distribution are obtained. The combination of SLS and DLS enables information on chain conformation. Beside synthetic polymers, LLS is also suitable for the solutions and suspensions of biopolymers, microbial, colloids, nanoparticles, virus, and vesicles. The history, theory, and experimental techniques of SLS and DLS specific for dilute solutions are summarized. In recent years, the cross-correlation techniques, diffusing wave spectroscopy, and other related techniques have been developed to expand LLS to study samples in semi-dilute and even concentrated solutions. These techniques, as well as solid light scattering, are also briefly introduced in this review. In the last, we provide four typical examples of light scattering experiments: the coil-to-globule transition as studied by the combination of SLS and DLS, the scaling of hyperbranched polymers as determined by LLS, the polymerization-induced micellization process as monitored by time-resolved LLS, and the aggregation of nanoparticles in biological media as investigated by depolarized DLS.
关键词
Keywords
Laser light scattering; Polymer characterization; Molecular weight; Radius of gyration; Correlation function
1光散射技术的发展简史
人们对光散射的认识最早可以追溯到1869年著名的丁达尔(Tyndall)凝胶散射实验. 1871年,瑞利对空气中的光散射现象进行了理论研究[1],推导出了球形粒子的散射公式,解释了晴空蓝和夕阳红的成因[2]. 之后,德拜(Debye)和甘(Gans)分别把瑞利的散射理论拓展到了非球形粒子[3] 和大尺寸的粒子[4],完善了气体中粒子的光散射理论.
在液体等凝聚相(condensed phase)中,散射强度的实测值通常比瑞利理论的预测值小一个数量级以上,这是由散射波的相消干涉造成的. 针对这种现象,斯莫鲁霍夫斯基(Smoluchowski)和爱因斯坦(Einstein)[5]从密度涨落的角度出发,提出了光散射的涨落理论(fluctuation theory of light scattering),极大地拓展了光散射的应用范围. 1940年前后,德拜和齐姆(Zimm)将涨落理论与溶液中的高分子表征相结合,实现了光散射对高分子的分子量、分子尺寸、分子形状和分子间相互作用的测量[6].
静态光散射(static light scattering, SLS)也称为弹性光散射,是指不考虑散射波长(或能量)变化的光散射. 1914年,布里渊(Brillouin)预测固体中热声波的散射光频率会出现双峰分布,后被实验所证实,从而开启了人们对准弹性光散射,即动态光散射(dynamic light scattering, DLS)的研究. 由于对光源单色性的苛求,动态光散射技术直到1960年前后激光光源趋于成熟之后,才得到了较好的发展. 1964年,佩科拉(Pecora)[7]利用高分子溶液中散射光的频率变化,计算出了高分子的扩散系数,并得到了高分子的流体力学半径、链柔顺性等信息.
当溶液中粒子的浓度增加到一定程度时,就会发生多重散射,即散射光再次或多次与粒子发生作用. 这种浓度下溶液的光散射理论较为复杂. 近年来,科学家们针对这类体系设计了许多特殊的方法或仪器,如折射率匹配法(1991年)[8],微样品池法(1998年)[9,10]、光纤准弹性散射法(fiber optical quasi elastic light scattering, FOQELS,1991年)[11,12]、时间交叉相关法(1981年)[13]、3D交叉相关法(1999年)[14]、互相关法(1997年)[15]等. 2006年,得益于电荷耦合器件(charge coupled device,CCD)以及计算机的发展,基于光斑(speckles)的互相关法得到了实质性发展[16],得以对亚浓溶液或浓溶液进行较为深入的研究. 当溶液体系达到浑浊状态时,极其严重的多重散射使得光在体系中的行进可以按扩散过程来处理,扩散波谱(diffusing wave spectroscopy, DWS)理论应运而生[17],基于该理论的技术可适用于多种不同的浑浊体系.
固体介质中也存在光散射现象,但在原理和应用等方面与溶液中的光散射都有很大差别. 固体中很容易产生严重的多重散射,且固体表界面的强烈散射常会对内部的散射造成严重干扰,这些都使得固体的光散射结果难以解读. 早在1922年,布里渊[18]就用光散射对固体振动进行了研究,但这不是严格意义的弹性光散射. 1960年斯坦因(Stein)[19]优化了垂直偏振光散射方法,极大地简化了散射结果,使得固体光散射在测定聚合物的链取向和晶体结构的研究中得到广泛应用[20,21].
2光散射原理
2.1气体光散射
光的本质是电磁波,含有周期变化的电场E. 原子或分子在电场作用下会发生极化,强度与极化率α相关. 原子在周期性变化的电场中会被周期性地极化,从而转变为一个次级光源,向周围发射同频率的电磁波,即散射光(图1).
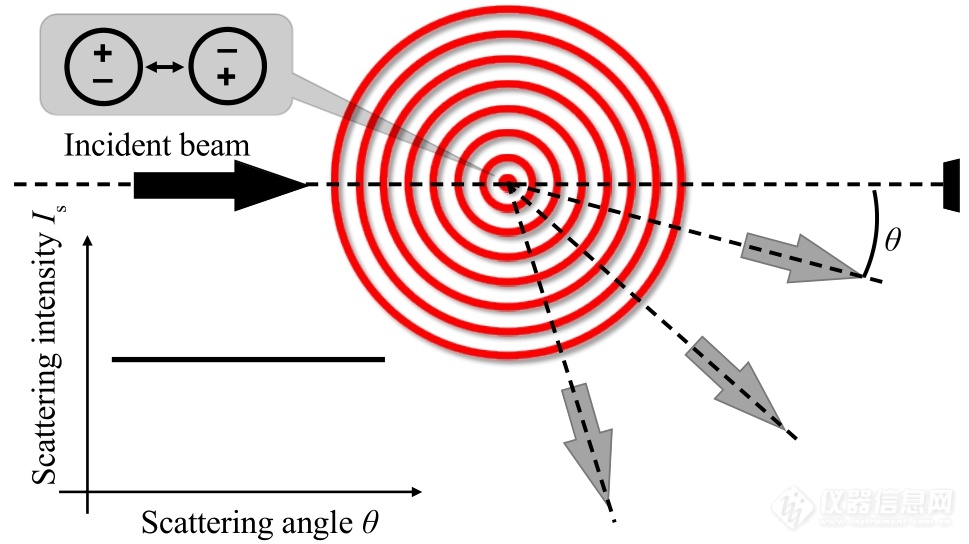
Fig. 1Scattered light generated by a scatterer as it is induced to be an oscillating dipole in the incident beam. θ is the scattering angle, and the inset shows the angular dependence of the scattered light from small particles, such as atoms or molecules. The polarization of incident beam is not considered.
单原子产生的散射光强Is由原子的极化率α和入射光波长λ决定. 另外,在空间某点测定的散射光强还与观测点到散射点的距离r有关. 1871年,瑞利推导出如下的散射公式:

其中I0为入射光强度. 单个原子、分子和粒子在空气中的散射光强都可以用公式(1)描述. 对于多粒子体系,可表示为体积V中存在N个散射粒子,如果粒子尺寸小(半径小于入射光波长的1/20),且数目较少,粒子之间的散射光不发生干涉,散射光强可表示为:

公式(2)表明,散射光强度与波长的4次方成反比,波长短的蓝色光的散射明显强于波长更长的红色光,因此天空在阳光的照耀下显示为蓝色.
2.2溶液光散射
光散射技术在溶液体系中具有非常广泛的应用. 在稀溶液中,利用静态光散射技术能够测定散射粒子的绝对分子量M、回转半径Rg、第二维里(Virial)系数A2等信息;利用动态光散射技术能够测定散射粒子的流体力学半径Rh及其分布等信息. 光散射技术在亚浓溶液或浓溶液中也发挥了重要作用,但该类体系中的多重散射使得散射理论变得十分复杂. 本文重点介绍稀溶液中的光散射理论,对非稀溶液体系的散射理论只做简要介绍.
2.2.1稀溶液中的静态光散射
在稀溶液中,根据Clausius-Mossoti公式,可将难以测量的极化率α转化容易测量的折光指数n:

其中n0是纯溶剂的折光指数,M为粒子的绝对分子量,NA为阿伏伽德罗(Avogadro)常数,c(=MN/VNA)为质量浓度. 值得一提的是dn/dc, 即溶液折光指数n对溶液质量浓度c的导数,称为折光指数增量,可以用专有仪器测定,或是从相关手册[22]中查到. 当dn/dc= 0时,预示体系中测不到反映溶质结构信息的光散射信号.
对于dn/dc≠0的单组分体系,将公式(3)代入(2)中,可得到瑞利散射公式:
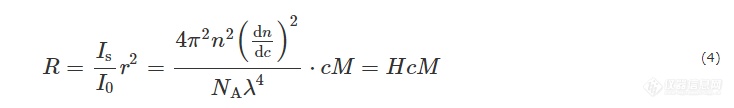
其中H称为光学常数,R为瑞利比.
忽略由溶剂自身密度涨落引起的散射. 根据涨落理论,散射光强I仅与光学常数H、质量浓度c和渗透压π相关,并遵循如下的关系式:

根据van’t Hoff关系式:
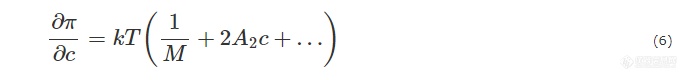
其中,M为溶液中粒子的绝对分子质量,A2为第二维里系数,用来定量描述溶剂-溶质之间的相互作用. 将公式(6)代入(5)中,可以得到:

式(7)中只有2个未知数M和A2. 理论上只要测量2个不同浓度溶液的散射光强I,就可以计算得到粒子的绝对分子量M和第二维里系数A2. 但是,由于每一台光散射仪的探测器面积和探测器到样品的距离都可能不同,激光束的粗细和样品池的大小也可能存在差异,因此对于同一个样品,每台光散射仪得到的信号都可能是不同的. 仪器测得的光强,必须要转化为绝对散射光强,才可以进行下一步的计算. 在实际操作中,常用瑞利比R代替I,并考虑以下这些影响因素:
第一步,偏振校正. 取决于样品的性质,散射光的偏振方向会发生变化,且会影响散射光强的大小. 偏振的校正较复杂[23]. 目前绝大多数光散射仪均使用了VV偏振散射设计,即入射光与观测的散射光都是垂直(vertical)偏振的,相应的散射光强标记为Rvv.
第二步,散射体积校正. 常见的散射仪器一般用小孔和狭缝来限制检测器接收的散射光. 激光束中被小孔或狭缝截留的光路在空间中所占的体积称为散射体积(图2). 对于同一个体系,散射体积越大,测得的散射光越强. 在激光光束和小孔或狭缝固定的情况下,散射体积与散射角θ(入射光矢量与散射光矢量的夹角)存在sinθ的定量关系. 因此在静态光散射实验中,在θ角测定的散射光强需要进行sinθ的校正.
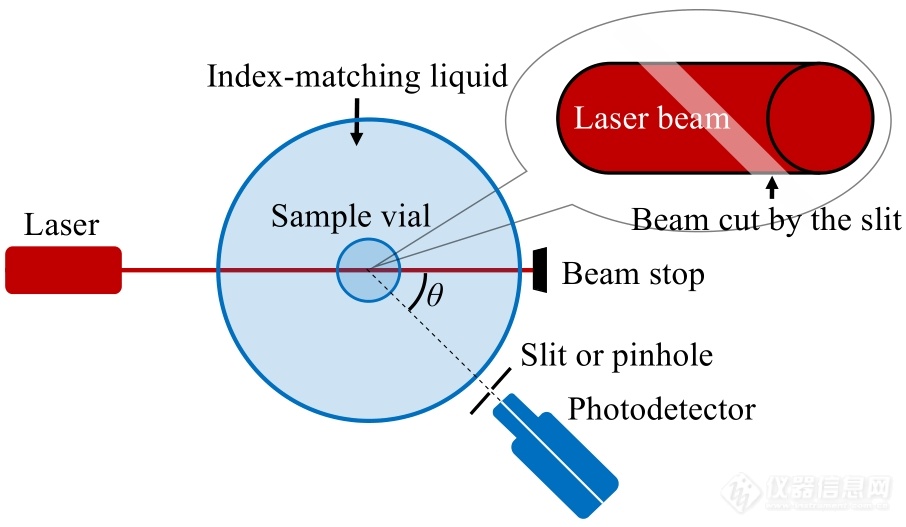
Fig. 2Geometry of a typical laser light scattering setup (top view).
第三步,净剩光强校正. 公式(7)中的光强是散射粒子自身的光强,在溶液中又称净剩光强,即溶液的散射光强Isolution减去溶剂的散射光强Isolvent.
在实验中,以瑞利比Rvv已知的标准溶剂为参照,在同一台散射仪器上进行样品的测量是最常用的做法. 例如温度为T时,样品在θ角的瑞利比RTθ通过以下公式得到:
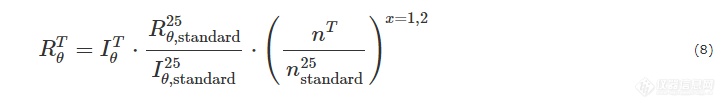
其中ITθ、RTθ、nT为样品在温度T下的净剩光强、瑞利比和折光指数,I25θ,standard、R25θ,standard和n25standard分别为标准溶剂在25 oC的散射光强、瑞利比和折光指数,也可以选用其他温度的配套数值. 当样品溶液和标准试剂的折光指数不同时,也需要进行校正. 狭缝和小孔所对应的指数分别为1和2. 甲苯是目前最常用的标准试剂,25 °C和632.8 nm波长下的瑞利比为8.70×10-6 cm-1. 甲苯与苯在不同波长和温度下的瑞利比可以从参考文献中查阅[24,25].
将散射光强用瑞利比表示后,公式(7)可改写为:

公式(9)适用于描述小粒子(尺寸小于波长的1/20)在溶液中的散射行为. 通常测量多个浓度下的Rvv值,将Hc/Rvv对c作图,从拟合直线的截距和斜率中分别求得M和A2值.
当高分子的尺寸较大时,同一高分子内部不同重复单元的散射光会发生干涉现象,从而导致散射光强出现了散射角度的依赖性(图3). 从光强角度依赖性数据可以反推粒子的尺寸和形状. 具体做法是在公式(9)的基础上,引入与散射角度相关的形状因子(form factor)P,其中包含了粒子的尺寸和结构信息.
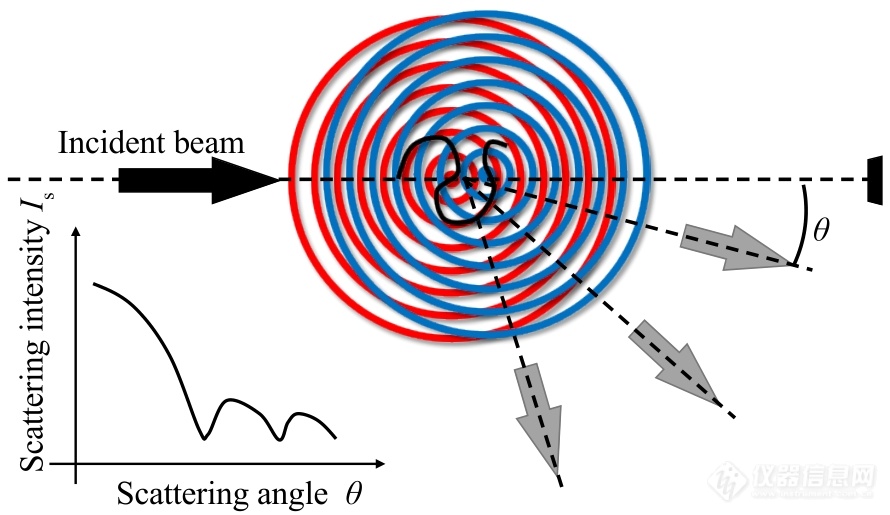
Fig. 3Interference pattern of light scattered from two segments in a large particle or polymer chain. The inset shows the angular dependence of the scattered light.
在光散射中,习惯上使用散射矢量q表示散射角. 散射矢量q定义为散射光波矢量与入射光波矢量的差.q与散射角度θ之间的数值关系为[24]:

由式(10)可知,散射矢量q的单位为长度的倒数. 在波长和溶液体系固定的前提下,q是由散射角θ决定的变量,此时形状因子可相应地记为P(q). 经P(q)修正后的散射光强公式为[23]:

对于小粒子而言,P(q) = 1,与散射角度无关.
用回转半径Rg来描述高分子的尺寸,当qRg < 1时:

将公式(12)代入公式(11)中,并做近似处理,可得到:
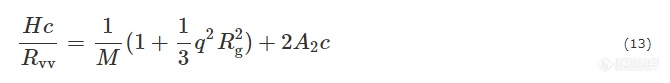
公式(13)是经典的静态光散射方程. 通过配置若干不同浓度的样品,测定每个样品的散射光强随角度的变化,利用公式(13)就可以得到样品的分子量M,回转半径Rg以及第二维里系数A2. 需要强调的是,对于具有一定多分散度的高分子样品,静态光散射测定的是绝对“重均”分子量和“z均”回转半径. 因此对于关联分子量和回转半径的研究,如确定二者的标度关系,必须采用分布尽可能窄的样品,测得的光散射数据才有分析处理的意义.
对于浓度较高或分子量较大的样品,公式(13)有时并不能给出令人满意的结果. 在这种情况下,可以尝试利用改进的公式来进行数据处理:
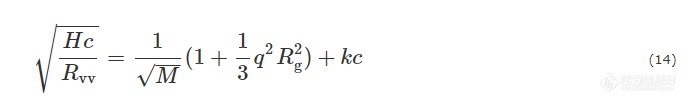
其中k为和第二维里系数相关的常数. 根据公式(14)绘制的图称为Berry Plot,同样可以得到重均分子量和回转半径.
当qRg > 1时,不同形状粒子的P(q)存在较大差别[23,26].
回转半径为Rg的无规高分子线团:
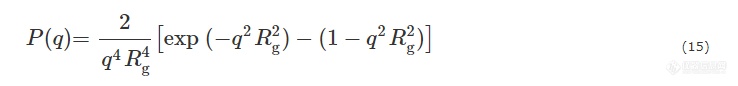
半径为R的均匀实心球:
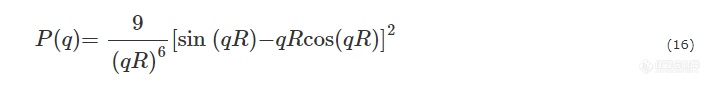
半径为R的空心薄球壳:

半径为R的薄圆盘:
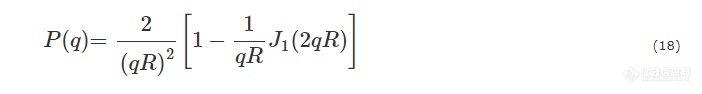
其中J1为一阶贝塞尔函数.
长度为L的细圆柱:
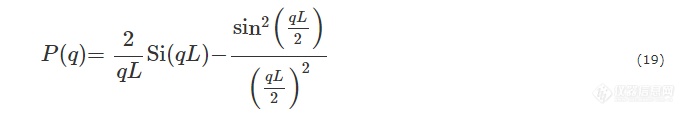
其中Si(x)为sinus积分函数:

通过测定待研究体系的形状因子P(q),并与标准体系进行对比,就能够判断粒子的构象并确定其特征尺寸参数. 当体系浓度足够小,2A2c一项相对于1/MP(q)可以忽略时,公式(11)可转化为:

即:

在公式(22)中,M/Hc是与散射角θ或散射矢量q无关的量. 因此,测定各个散射角度下的Rvv,用零角度的数值归一化,再对q作图就得到了P(q)曲线. 为了提高用P(q)确定体系构象的准确性,尽量选用窄分布的样品,并在测定时覆盖尽可能宽的散射角度.
利用静态光散射来测定共聚物比均聚物要复杂很多. 由公式(4)可知,决定体系散射性能及强度的内在因素是dn/dc. 共聚物等体系包含有2种或2种以上的组分. 当这些组分的(dn/dc)不同时,散射方程将急剧地复杂化. 以AB两嵌段共聚物为例,体系总的(dn/dc)AB =wA(dn/dc)A +wB(dn/dc)B,wA和wB分别为A和B嵌段的质量分数. 按照均聚物的测定方式,利用公式(13)能够得到共聚物的表观分子量Mapp[27]:
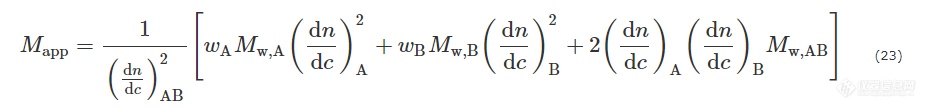
其中:
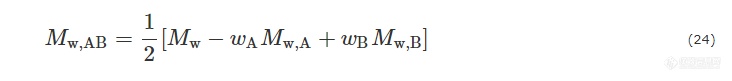
由公式(23)和(24)可以得到如下结论:
(1)Mapp由两嵌段的(dn/dc)决定. 当所选溶剂的(dn/dc)AB接近0时,Mapp趋于无穷大.
(2) 公式中有3个独立的未知数Mw,A,Mw,B和wA,因此需要在3种不同折光指数的溶剂中测定样品的Mapp,然后解方程得到两嵌段共聚物的真实分子量Mw [27]. 对大多数嵌段共聚物体系,找到3种可单分散溶解共聚物的溶剂并不容易. 吴奇等人在1994年报道了只用2种溶剂就可利用静态光散射测定共聚物分子量的方法[28],但数据处理稍显繁琐.
(3) 当在选用的溶剂中A嵌段的(dn/dc)A= 0时,直接测定的是B嵌段的分子量,反之亦然. 利用这种掩盖法,只需要2种溶剂就能精确测定A嵌段、B嵌段以及共聚物总的分子量.
公式(23)还可以改写为:[28]
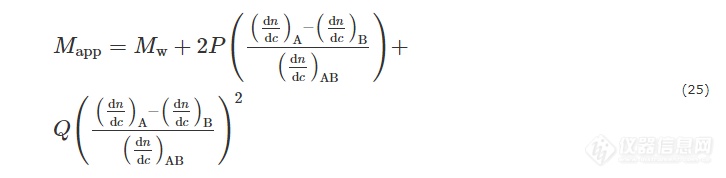
其中P和Q是与嵌段共聚物组分非均匀分布相关的常数.
由上式可知,当A和B两嵌段的dn/dc相等或接近时,所测定的表观分子量与真实值一致. 同理,也只有在这种情况下,才能够利用公式(13)来测定共聚物的回转半径Rg. 如果A和B两嵌段的dn/dc相差较大,特别是当(dn/dc)AB接近0时,Hc/Rvv在小角度会出现负斜率,导致外推得到的Rg为负值.
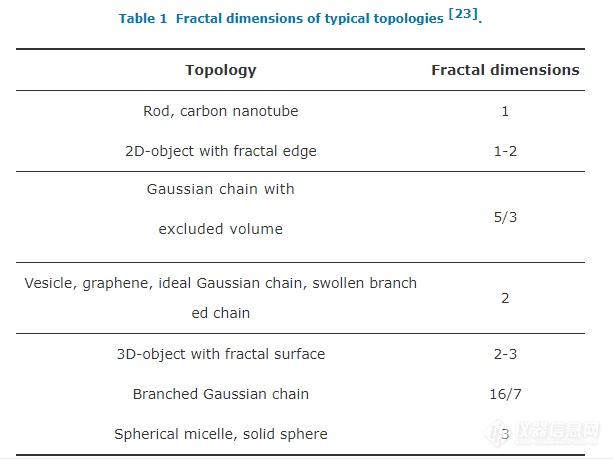
利用静态光散射还可以测定粒子的分形维数. 一般来讲,若物体的维数是d,则其质量M和尺寸R应满足如下的标度关系:

例如:三维的实心物体,质量M与R3成正比,而二维的实心物体,M与R2成正比. 维数d在一定程度上反应了粒子的结构和形状. 而高分子线团、空心粒子或具有不规则形状的物体,其维数通常不是整数. 静态光散射是测定粒子分形维数的有效工具. 对于尺寸为R的粒子,当满足qR>>1 (一般大于3)时,绝对散射光强Rvv和散射矢量q之间的标度将满足[23]:

Rvv和q的双对数图是一条直线,直线斜率的相反数就是该粒子的分形维数d. 该方法的准确度与q有效的数据范围有关,一般需要跨越数量级. 因此,不是所有体系都适用这种方法.表1列出了常见拓扑结构的分形维数.
2.2.2稀溶液中的动态光散射
散射体积一般是固定的,其中往往包含有多个散射粒子. 由于布朗运动,散射体积内粒子的数目和位置都在发生变化,这导致在固定检测位置测定的散射光强会随时间发生涨落.图4所示是2个高分子相对位置发生改变引起的光强涨落. 看似无规的涨落信号中埋藏了粒子扩散的信息. 挖掘扩散信息的途径是从随时间变化的I~t曲线得到光强-光强的时间相关函数.
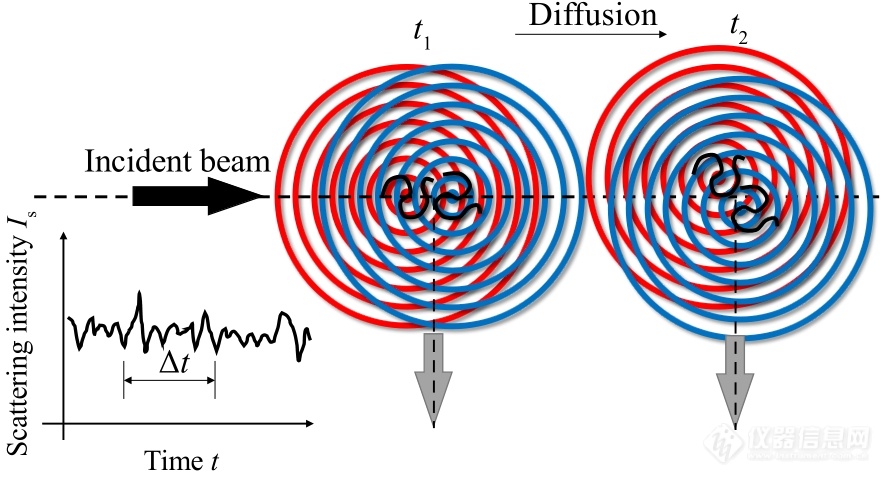
Fig. 4Time dependence of the interference pattern. The inset shows the change of scattered intensity with time at fixed scattering angle.
首先介绍相关函数的概念. 在I-t曲线中,t和t+τ时刻分别对应着光强It和It+τ,τ称为延迟时间. 当τ→0时,总有It=It+τ,而当τ→∞时,It和It+τ则是围绕平均光强
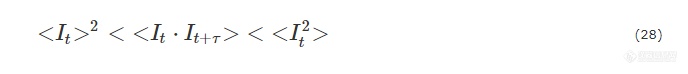
令:

g2(τ)称为归一化的光强-光强时间相关函数[29].
将动态光散射中的g2(τ)对τ作图,得到如图5中所示的曲线. 如果体系中只包含一种散射体A,则g2(τ)随τ呈现单一的快速衰减,衰减最快处对应的时间τA反映了体系的特征性质.
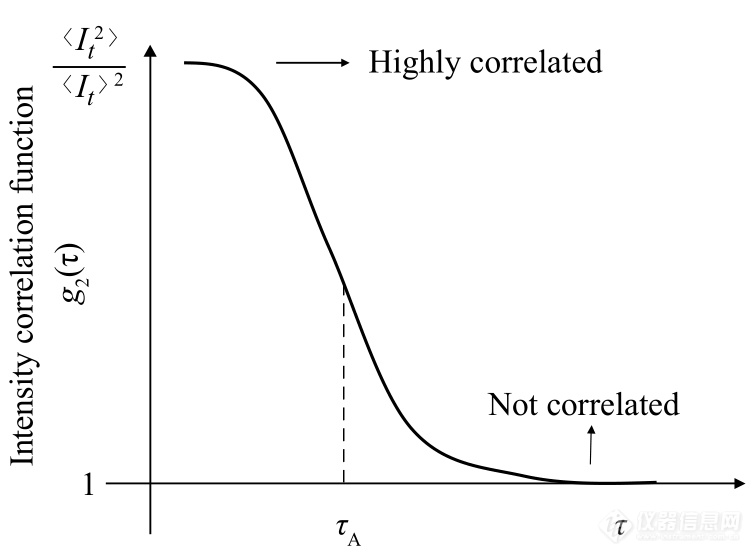
Fig. 5Intensity-intensity correlation function.
在现代的光散射仪中,光强的测定和g2(τ)的计算都是由硬件直接完成. 测定光强常用的仪器是雪崩光电二极管探测器(avalanche photodiode detector, APD);从光强到g2(τ)是由相关器来完成的[24].
从g2(τ)到粒子扩散的信息,还需要经过以下步骤:
第一步,求解电场-电场时间相关函数g1(τ).g2(τ)是光强的相关函数,需要将其转换为电场的相关函数g1(τ),才能和扩散过程直接相关联. 在光的波动理论中,光强是电场的平方. 而g2(τ)和g1(τ)的关系比简单的平方关系要复杂,称为西格特关系式(Siegert relation)[30]:

其中β是和测量光路相关的系数. 当检测器前的狭缝或小孔合适,只测到单光斑(speckle)时,β=1.
第二步,求解粒子自扩散系数Ds. 这个求解的过程是动态光散射理论的核心. 这里只简单介绍基于van Hove自相关函数Gs(r,τ) 的推导过程. 假定某个粒子在时间t的位置为0,Gs(r,τ)就是在时间t+τ时在位置r处发现该粒子的概率. 由于g1(τ)是随散射矢量q而变化的,可写成g1(q,τ).g1(q,τ)和Gs(r,τ)符合傅里叶变换(Fourier trans-formation)的关系:
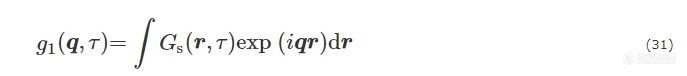
对于单分散、各向同性粒子的扩散运动(布朗运动或无规行走),Gs(r,τ)仅依赖于距离r= |r|,且符合高斯方程:
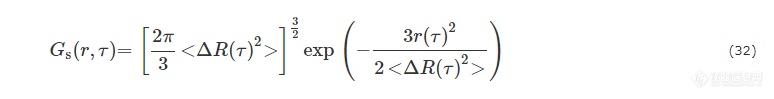
从Gs(r,τ)的半峰宽可以解出散射粒子的均方位移<ΔR(τ)2>. 在布朗运动中,<ΔR(τ)2>与粒子的自扩散系数D0的关系为:

求解方程(31)可得:
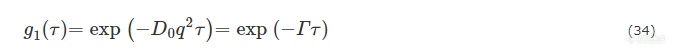
其中Γ=q2D0,称为线宽. 据公式(34),将ln(g1(τ))对τ作图,从直线的斜率直接得到D0.
第三步,求解流体力学半径Rh. 利用Stokes-Einstein方程:

其中k为玻尔兹曼(Boltzmann)常数(1.38×10-23 J/K),T为绝对温度,η为溶剂黏度,可从扩散系数直接得到流体力学半径. 对于有一定分散度的样品而言,DLS测定的流体力学半径和扩散系数都是z均值.
由于粒子各向异性等因素的影响,在不同散射角度测定的扩散系数存在差异,因此在固定角度测定的是表观扩散系数Ds,app. 另外,光散射直接测定的是粒子的互扩散系数(mutual diffusion coefficient),只有在零浓度时才与自扩散系数一致[23,31,32]. 因此,利用动态光散射求算扩散系数的公式包含了散射角度和浓度的依赖性:
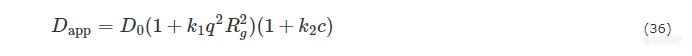
其中k1和k2是2个常数.k1和样品的分散度以及拓扑形状有关,k2和样品与溶剂的相互作用有关. 公式(36)与静态光散射公式(13)在形式上是类似的. 在实验中,同样需要对不同浓度的样品在不同的散射角进行测量,然后按照公式(36),通过角度和浓度的外推,得到粒子扩散系数D0.
以上介绍的是单分散粒子的动态光散射理论. 当样品呈多分散时,扩散系数D0或线宽Γ会出现相应的分布,一般用G(Γ)表示. 由公式(34)可得:
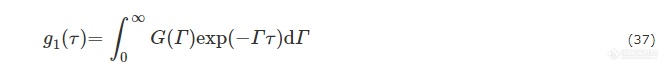
g1(τ)是由G(Γ)经拉普拉斯变换得到的,而实际过程中是通过测定g1(τ)来反推样品的分布G(Γ),因此是反拉普拉斯变换. 针对动态光散射实验开发的反拉普拉斯变换的方法有许多,如累积矩(cumulant)法、双指数(double-exponential, DE)法、直方图(histogram)法,离散变换(discrete inversion)法、熵最大化(maximum entropy method, MEM)法、非负值最小二乘法(nonnegatively least squares, NNLS)法、指数抽样(exponential sampling, ES)法和CONTIN法等. 关于各算法的优劣,可参考具体文献[33~36]. 在这些方法中,CONTIN是使用较为广泛且适用大多数多分散体系的算法.
2.2.3稀溶液中静态光散射和动态光散射的结合应用
不难看出,静态光散射和动态光散射是对同一个样品的浓度系列进行了2种不同方式的测量. 2种测量方式的有机结合,能够得到关于样品更多或更深入的信息.
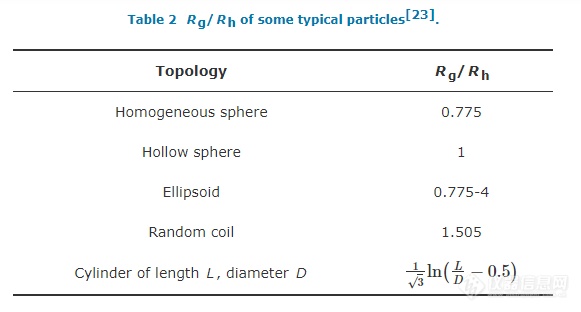
首先,对于单分散样品,比值Rg/Rh反应了粒子的拓扑结构.表2列出了一些常见粒子的Rg/Rh的理论值.
其次,对于双分布或多分布样品,静态光散射只能得到样品Rg和Mw的平均值. 而如果动态光散射能够在不同的散射角对多分布,特别是双分布,进行明确区分,就可以把在该角度的散射总光强按照峰的比例进行分配,从而得到各个组分的光强角度依赖性,再利用静态光散射理论,得到不同组分的Rg和Mw[37~39].
最后,结合静态散射理论,能够把动态光散射测到的线宽分布G(Γ)转换为分子量的分布G(M),前提是需要知道样品分子量和扩散系数的标度关系[40~42].
2.2.4非稀溶液中的动态光散射
非稀溶液体系中的动态光散射研究近年来取得了较多进展,已有不少成功应用的例子,并可以预期它在未来的科研中将发挥更重要的作用. 非稀溶液动态光散射主要面临2个共性问题:多重散射和非遍历(non-ergodicity). 扩散波谱也是一种特别且重要的非稀溶液动态光散射技术. 下面将分别进行介绍.
非稀溶液中的多重散射可以通过设计特殊的仪器设备来进行削弱或抑制. 例如:扁平池光散射仪[43]就是采用光程非常小的扁平样品池(厚度可小至10 μm),并辅以相应的散射体积校正,从而大幅减少多重散射,使得测量体系浓度可以比通用光散射仪大1000倍左右.
光纤准弹性光散射仪(FOQELS)[11,12]是利用背散射来消除多重散射的影响. 入射光通过光纤导入到待测溶液中,该光纤同时也是信号接收器,接收(180±3)°范围内的散射光,背散射光和主光束用单模光纤定向耦合器进行区分. 浓度高达40 wt%的浑浊乳胶样品中也能利用该仪器进行DLS研究,且无需除尘.
利用2束激光进行交叉相关是抑制多重散射的有效方式[14,44]. 双色交叉相关仪采用2束不同波长的激光同时照射样品;3D交叉相关仪则采用2束同波长但分别略高和略低于散射平面的激光同时照射样品. 这2种仪器大致上是利用非相干光的相关性为0,来消除有限次多重散射对相关函数的影响,从而得以对高浓体系进行光散射的测量. 这类仪器的测量角度也是大幅度可变的,在这一点上比FOQELS具有明显的优势. 双色交叉相关仪对光路准直的要求非常高,甚至0.01 oC的温度涨落所导致的光路波动都有可能破坏仪器的准直性. 相对而言,3D交叉相关仪对此的要求低得多.
在非稀溶液中,由于粒子运动过慢或粒子过大等因素,导致实际的测量结果不是对样品所有可能状态的综合,这就是非遍历问题. 非遍历测量的直接后果就是数据不具有统计性,导致测得的g2(τ)数据无法解出样品真实的g1(τ).
解决非遍历问题的首要思路是如何尽可能多地得到g2(τ)的信息. 可采用的方法包括对同一个体系用不同的方法测得g2(τ),如用CCD面探测器测得多个光斑的变化然后进行互相关,对不同位置的测量结果取平均,或是用串联的双样品池进行目标样品和参考溶液的相关等.
如何从g2(τ)中解出接近真实的g1(τ)也是解决非遍历问题的必经步骤. 目前常用的方法是对西格特关系式(公式(30))进行变换,如

其中f(g1(τ))是与实验装置相关的函数,具体的装置设计和对应的算法可参考文献[45]. 根据公式(37)可在非遍历条件下求得较准确的g1(τ).
扩散波谱是针对极浓溶液的一种特殊的动态光散射方法,基本思路和常规的动态光散射法相同:仪器测定g2(τ),算出g1(τ),通过变换得到扩散系数Ds,从而算出Rh. 所不同的是,从g1(τ)到Ds涉及了特殊的理论,具体的推导过程可参考文献[17,45,46]. 对于单分散样品,g1(τ)和Ds的关系式可表示为:
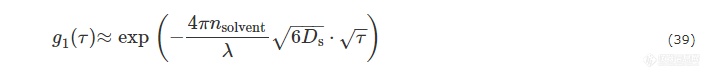
将ln(g1(τ))对τ−−√作图,数据将呈现一条直线,从斜率即可求出Ds. 可以看出,对于极浓溶液,g1(τ)和q或散射角无关,这也是合理的.
更重要的是,扩散波谱能够测定介质的储能模量G'和损耗模量G''的频率依赖性,也就是介质的黏弹性[47~49],这类似于流变仪扫频实验得到的数据. 由Stokes-Einstein方程(公式(35))可知,扩散系数D与ηR的乘积呈反比关系,这3个参数可以知二求一. 对于常规的动态光散射而言,溶剂黏度η已知,可求出Rh. 在极浓溶液中放入给定尺寸Rh的小球,根据小球的D(τ)能够得到η*(ω),即溶液复合黏度随频率的变化曲线. 由该曲线可计算求得G'(ω)和G''(ω).
2.3固体光散射
固体光散射在高分子球晶的研究中发挥了重要作用,可得到球晶分布、取向和尺寸信息. 虽然球晶也可用偏光显微镜(POM)和原子力显微镜(AFM)进行观测,但偏光显微镜有光学分辨极限,对尺寸小于5 μm的球晶几乎无法观测,而原子力显微镜对样品制备有着较为严格的要求,也无法观测固体内部的球晶形态. 因此,在球晶研究方面,固体光散射有着不可替代的优势. 球晶固体光散射的理论比较复杂[19~21], 本节仅简单介绍球晶呈现的四叶草瓣形状的散射图样和球晶尺寸的求算.
2.3.1球晶的VH散射四叶草瓣图样
光穿过具有取向的结构后,沿非取向方向偏振的光将被抑制或滤去(图6(a)),这也是许多偏振片的工作原理. 常用的VH固体光散射的光路设计是在样品的前后分别放置偏振片,偏振方向相互垂直(图6(b)). 这样的实验设计滤去了偏振不变的散射光,只有改变了偏振方向的那部分散射光才能被检测到. 对于许多结晶高分子而言,球晶的散射信号是唯一偏振有变化的散射信号.
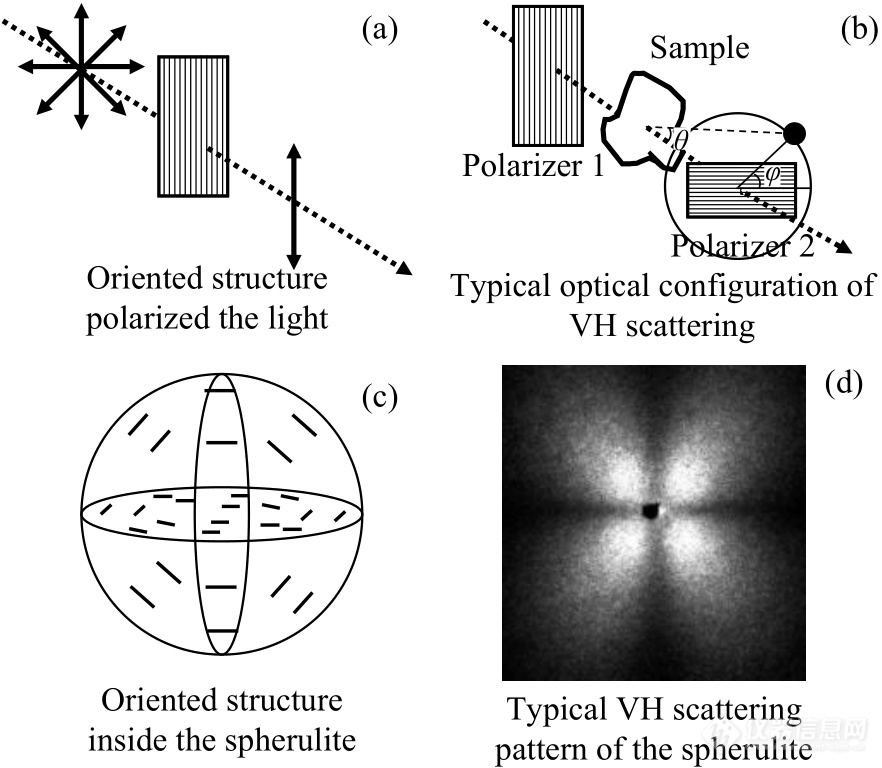
Fig. 6Spherulite studied by solid light scattering.
球晶内部的取向结构是中心对称的(图6(c)). 经过第一个V偏振片的入射光,在球晶的V方向和H方向上遇到的球晶内部的取向结构均是垂直或平行于V方向的,光将直接通过或是被完全滤去,方向不发生偏转. 因此,在这2个方向上的散射光在第二块H偏振片后面,完全不会被检测到. 而除了V方向和H方向,散射光均和球晶内部的取向结构有一定夹角,光将偏转方向,得以被最终检测到. 因此,散射图样常出现黑十字消光现象(图6(d)),呈现四叶草瓣形状. 消光十字的方向分别平行于2个偏振片的取向方向. 图6(d)还表明散射图样不是连续的,而是由多个分散的斑点所构成,其中每一个亮斑都是之前动态光散射理论中所说的斑点(speckle). 这不是因为检测器的精度不够造成的.
2.3.2球晶的尺寸计算
球晶属于大粒子,其固体散射也存在形状因子P(θ). 在VH光路下[19],

其中:

R'为球晶半径.
对于无取向的球晶时,理论和实验均表明,在花瓣散射光强最亮点处,近似有U=4.0[19]. 因此:

其中θm即最亮点处的散射角. 公式(42)即广泛使用的无取向球晶的尺寸计算公式. 对于有取向的球晶,最亮点处的U值有时会发生变化. [21]
3实验技巧
在上面介绍的光散射技术中,稀溶液体系的光散射应用目前最为广泛,所得到的信息也最丰富,但相应的样品制备和实验过程也比较复杂. 本节将简要介绍稀溶液光散射的实验技巧和数据处理方式.
3.1样品溶解
首先是要选择合适的溶剂来溶解样品. 重点考虑光散射衬度,即(dn/dc)的大小. 若(dn/dc) = 0,将得不到任何散射信号. 在保证溶解性能的前提下,通常选择折射率和溶质差别较大的溶剂. 对于共聚物体系而言,需要根据体系的性质和实验的需求来选择溶剂. 例如:在测定有机共聚物的精确分子量时,则应当选择多种良溶剂或共溶剂进行实验.
其次是要选择合适的样品浓度来进行测量. 一方面浓度要足够稀,使得分子间的相互作用可以忽略. 高分子的临界交叠浓度(overlap concen-tration)c*是浓度上限的参考点. 另一方面,浓度越稀,散射信号也越弱,测量将变得困难. 对于未知且不易估算c*的高分子体系,0.1 mg/mL可以作为初始的浓度进行尝试.
最后需要溶解样品,形成均一体系. 高分子的溶解过程耗时较长,通常需要2~24 h. 搅拌仅能有限地加速溶解过程. 升温会使得高分子体系氧化,应尽量避免. 超声也是不推荐的.
3.2除尘
由于散射光强与粒子尺寸的4~6次方成正比,直径在微米级的灰尘粒子会对高分子样品的散射实验造成毁灭性的破坏,因此要尽量避免样品溶液中掺杂有灰尘粒子. 灰尘是极性的. 水溶液体系的除尘往往比有机溶液体系要困难. 除尘操作包括样品瓶除尘和溶液样品除尘.
光散射样品瓶的除尘通常采用类似于索式提取的装置,利用蒸发后再冷凝的丙酮间歇性地对倒置样品瓶的内部进行多次冲刷. 除尽灰尘的样品瓶要封口并倒置保存.
样品的除尘通常有过滤法和离心法. 过滤法更易操作,需要在空气尽量净化的环境中,使用孔径在样品尺寸之上,且在灰尘粒径之下的滤膜,用注射器将待测样品过滤后注入到除尘后的样品瓶中. 可供选择的商业化滤膜有很多,可选用的孔径在200~600 nm之间. 过滤时滤膜上的压力不宜过大,因此过滤需缓慢进行. 如果所测体系较为复杂,没有合适的滤膜可选,则可考虑离心法.
3.3仪器准直
仪器的准直性是光散射实验的前提. 溶剂分子(一般选甲苯)的散射光强在校正散射体积后是没有角度依赖性的(图1),可用来验证仪器的准直程度. 对除尘后的甲苯样品进行角度扫描,角度范围一般在20°~150°. 如果每个角度的散射光强都围绕某一平均值波动,且波动不超过2%,则可认为仪器的准直是良好的. 若该条件不满足,则需要对仪器的准直进行校准.
3.4实验过程
静态光散射实验中散射角度的选择很重要. 原则上,只有在qRg < 1的情况下才能用公式(13)准确测定粒子的回转半径. 对于尺寸较大的样品,需要在小的散射角度或q范围内测量多个数据点(减小角度间隔),以保障角度外推的可靠性. 另外,在小角度时,灰尘的影响会变得更加明显,这对样品特别是水溶液中的样品的除尘提出了更高的要求. 大尺寸样品的光强角度依赖性很强,小角度的光强比大角度会高出有4~5个数量级,因此要注意检测器的线性响应范围,必要时可用非偏振类滤光片调节入射光的强度.
动态光散射数据的根源是g2(τ). 在样品除尘合格的前提下,选择合适的延迟时间τ范围,并累积足够长的时间是获得可靠g2(τ)的前提.
检测器前端的小孔(pinhole)或狭缝是可调的. 对于静态光散射,通常需要选择较大尺寸(如1 μm)以测得具有统计性的散射光强. 对于动态光散射,通常需要选择较小的尺寸(如200 nm),以保证只测到单一光斑,从而使得西格特关系式中的β值趋近于1.
对于碳纳米管、石墨烯、金纳米颗粒、荧光分子等具有光吸收能力的样品,静态光散射和动态光散射的校准方式也是不同的. 静态光散射需要通过测定光吸收系数,通过朗伯比尔定律来校正不同角度的净剩散射光强;而动态光散射则需要测定在不同入射光强下的样品扩散系数,通过外推到零入射光强的方式来消除光吸收对扩散的影响. 如果样品的吸光性太强,引入的误差增加,不提倡用光散射进行测量.
3.5数据处理
绘制Zimm图是静态光散射最常用的数据处理方法. 这是一个初学者经常会出错的处理过程,其中最关键的是各物理量单位的转化. 简单的处理方式是采用非国际单位:q以nm作为长度单位,其他所有物理量的长度单位均转化为cm. 光学常数H和质量浓度的单位则分别为cm2⋅g-2⋅mol和g⋅cm-3. 在绘制Zimm图时,如果数据点偏离了线性,可以从样品是否多分散、是否聚集以及是否满足qRg < 1等方面进行分析.
尺寸小于激光波长1/20的粒子通常不会出现散射角度的光强依赖性,不需要做角度扫描. 为了尽量降低灰尘对散射实验的影响,一般选择90°进行各浓度溶液的测量,然后直接运用公式(9)计算M和A2.
如果实验中只关注回转半径,且要求的准确度不高,可选择一个较低的样品浓度进行角度扫描,不需要dn/dc的测量. 具体处理如下:
取x列为散射角度θ,y列为光强值I原始数据,将x列转换为q2,单位为nm-2,将y值转换为(I - Isolvent)⋅sinθ(即只做净剩光强校正与散射体积校正),单位任意;(2)将x和1/y作图,线性拟合,取3倍截取/斜率,并开平方,即得到回转半径Rg,单位为nm.
对于多组分体系的动态光散射,尺寸相差2倍以上的粒子才有可能被分辨为2个组分. 如果体系中组分的数量大于3,或得到的Rh分布图的峰数量大于3,则需要对结果的准确性持较谨慎的态度,需要从原理上判断结果是否合理,或通过其他手段适当进行辅证.
3.6(dn/dc)测量
(dn/dc)通常需要专用的仪器进行测量. 折光指数和原子极化率相关,极大地受原子序数的影响. 相对于C和H元素而言,Na和K等元素的原子序数要大得多,因此溶剂中的微量溶盐将极大地影响(dn/dc)的测量准确性. 为了确保对未知体系的准确测量,最好使用同一批溶剂,分别进行(dn/dc)的测量以及所有的光散射实验.
4典型应用
光散射技术在高分子表征中的应用非常广泛. 感兴趣的人士可以查阅相关书籍、专著和文献. 从掌握光散射基本理论和实验技巧、了解光散射技术发展趋势的角度出发,结合实验体系的代表性, 我们选取了4个经典的应用案例,来具体说明动、静态光散射的使用技巧,二者相结合的必要性,时间可分辨光散射技术的优势,以及如何开发光散射技术在复杂溶液体系中的应用.
4.1动、静态光散射相结合表征溶液中高分子行为
动、静态光散射技术相结合能够对溶液中的高分子进行深入、系统的表征. 跟踪高分子链从线团到球的转变(coil-to-globule transition)过程是该技术最典型的应用之一. 在不良溶剂中,高分子链会发生塌陷,同时会伴随着高分子链之间的聚集. 如果观测单个高分子链在不良溶剂中的构象转变,要考虑多方面的因素[50,51],一般采用尽可能高的分子量、尽量窄的分布、并在尽可能稀的溶液中来进行. 一方面可以避免分子链之间的聚集,另外也可以保持较高的净剩散射光强. 吴奇课题组结合分级和过滤得到了分子量极高、多分散度窄的水溶性聚N-异丙基丙烯酰胺(PNIPAM)样品(Mw=1.3×107 g/mol,Mw/Mn < 1.05),并配制了10-7 g/mL级别的极稀水溶液,用光散射首次观测到了高分子单链塌缩的构象转变.
PNIPAM的低临界溶液温度(lower critical solution temperature,LCST)约为32 °C.图7对比了6.7×10-7 g/mL PNIPAM在相变前后的动、静态光散射结果. 在35.9 °C时,水是PNIPAM的不良溶剂,Rg从30.1 oC的127 nm减小到17.9 nm,Rh也发生了类似变化.Rg/Rh在2个温度的数值分别为1.5和0.72,表明PNIPAM在30.1 oC时为线团构象,而升温到35.9 °C时则转变为密实球的构象.
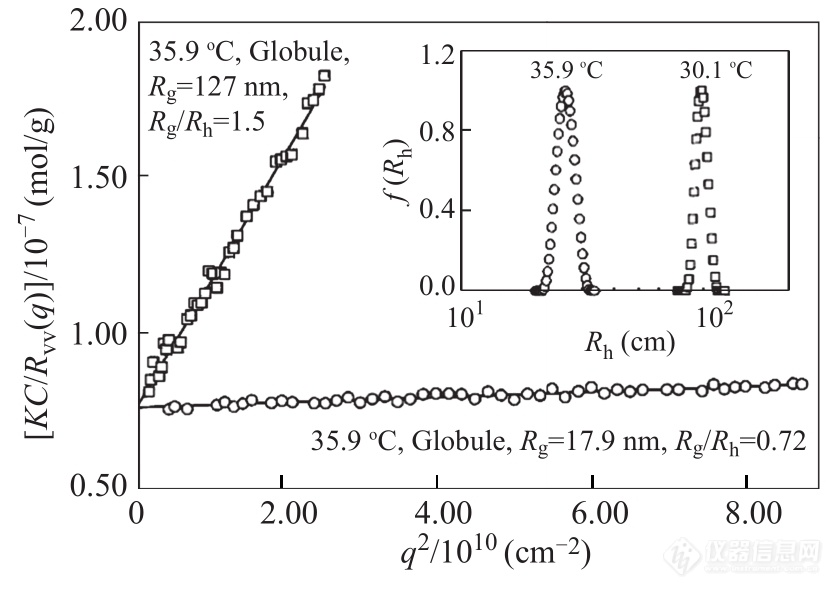
Fig. 7Typical angular dependence of Hc/Rvv of PNIPAM in water at two different temperatures, where the polymer concentration is 6.7×10-7 g/mL. The inset shows the corresponding hydrodynamic radius distributions f(Rh) of the PNIPAM chains respectively in the coil and the globule states. (Reprinted with permission from Ref.[50]; Copyright (1998) American Chemistry Society).
在连续的升温和随后的降温过程中,Rg/Rh随温度并不是单调变化的. 如图8(a)所示,在升温过程至30.6 °C之前,Rg/Rh基本保持在1.5左右,表明PNIPAM为无规线团构象;在30.6~31.6 °C 温度区间,Rg/Rh 从1.5快速降低到1.0,此时的链构象可归结为褶皱的线团(crumpled coil);继续升温到32.4 °C时,Rg/Rh骤降到0.56,所对应的是熔融球构象(molten globule),即表面密度低、内部密度高的球体;在随后的升温过程中,Rg/Rh逐渐增加到0.775, 所对应的是常规的球体. 图8(b)对比了不同温度时PNIPAM的链构象示意图及相应的链密度分布. 在随后的降温过程中,Rg/Rh的变化过程出现了明显的滞后,这可能是在球体状态下形成了某种链内结构所造成的.
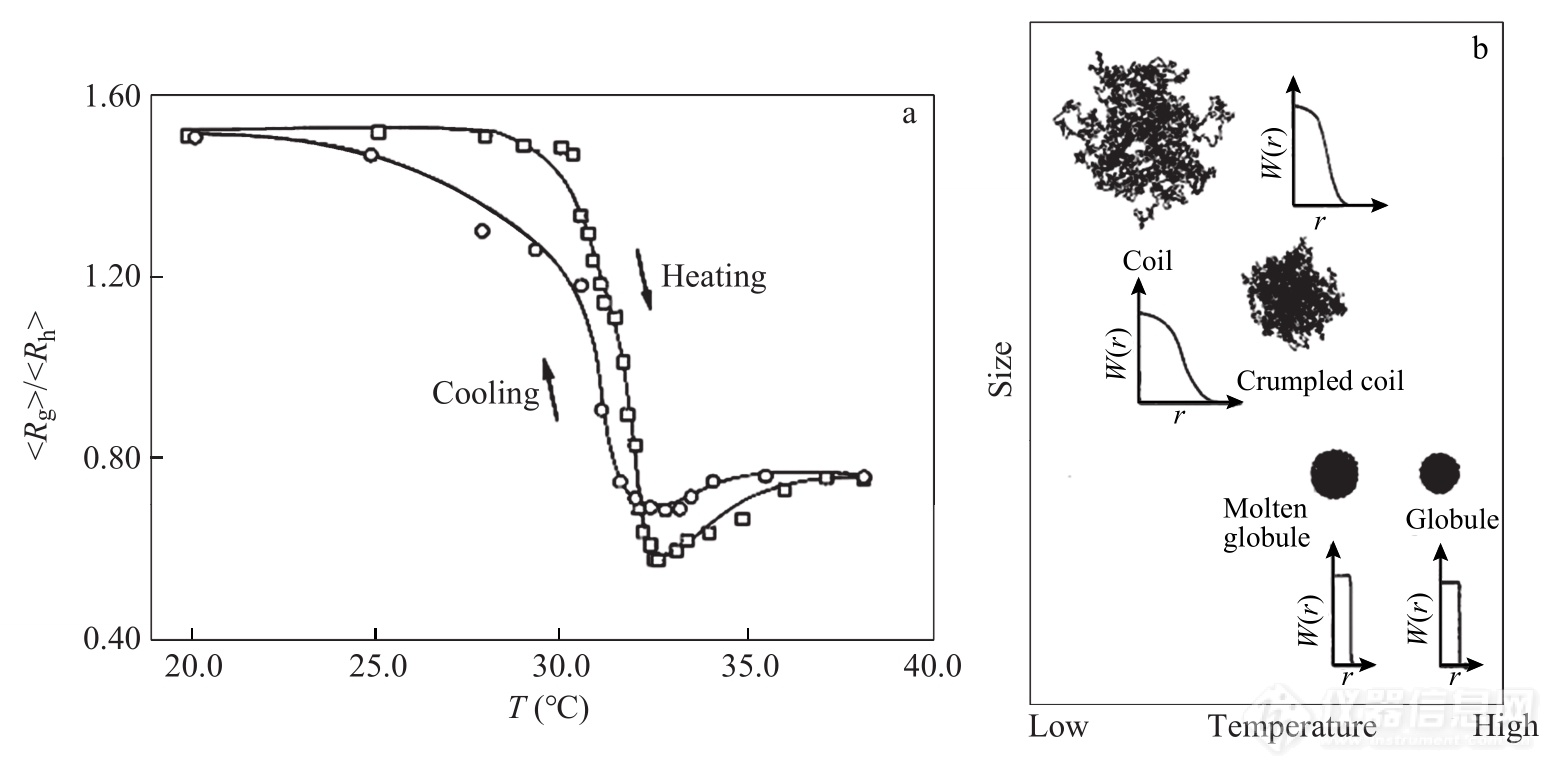
Fig. 8(a) Temperature dependence of
4.2光散射测定超支化分子的标度关系
除线性高分子外,光散射在测定具有复杂构型的高分子样品中也具有独到的优势. 以支化高分子为例,李连伟课题组制备得到了支化点间长度等同的“完美”支化高分子,并利用光散射技术确定了支化高分子尺寸和聚合度之前的标度关系[52].
对于满足支化随机、支化点间长度等同的单分散高分子样品,其回转半径Rg与支化分子总的单体数Nt以及临界支化点间的单体数Ns之间存在如下的标度关系:

其中b是库恩长度. 对于在θ溶剂中ν值的大小,不同理论有着不同的认识. 平均场理论认为ν=0.25,而Flory理论则预测ν=0.44. 由于理想的支化高分子难以得到,在此之前尚无实验数据进行验证.
李连伟课题组合成了不同分子量的支化聚苯乙烯(h-PS),并用静态光散射测定了重均分子量. 对于高分子量样品,qRg > 1,采用Berry plot(参见公式(14))进行数据处理. 低温下,环戊烷是h-PS的不良溶剂,而高温下是良溶剂. 通过测量多个温度下体系的第二维里系数A2,找到其由正值转变为负值的临界点,即可得到θ温度,其值为304~307 K.
通过对静态光散射数据进行处理得到了形状因子Rvv(q)/Rvv(0) (图9(a)). 线性拟合qRg > 3的数据,利用公式(27)得到支化分子的分形维数为2.4,并进一步求得ν约为0.42. 另外,ν值还可以从支化样品的Rg~Mw 的双对数关系中直接得到. 如图9(b)所示,h-PS在环戊烷溶剂中302.1 K的ν约为0.47. 2种方法得到的结果是吻合的,均支持Flory理论的预测.
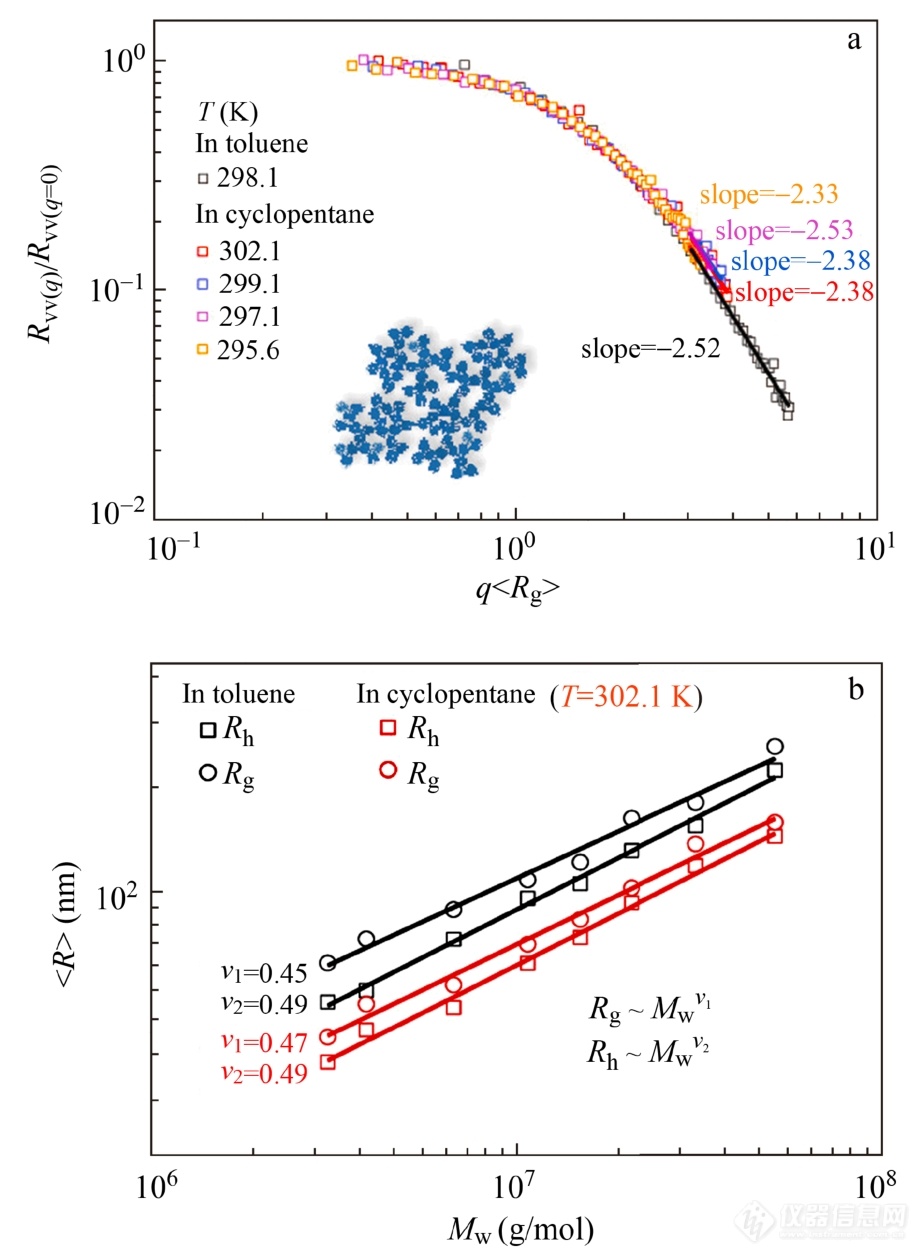
Fig. 9(a) qRg dependence of the normalized excess Rayleigh ratio [RVV(q)/RVV (q=0)] for h-PS and (b) weight-average molar mass (Mw) dependence of chain size (R) for different h-PS in cyclopentane at 302.1 K (Reprinted with permission from Ref.[52]; Copyright (2020) American Chemistry Society).
4.3用时间分辨光散射表征体系的动态变化
前文中介绍的光散射理论都是针对平衡态体系的. 如果体系发生变化所需的时间远超过光散射的采样时间,就能够在保证准确度的情况下,利用光散射技术原位、在线跟踪聚合、组装、解离、降解等过程,获得分子量、尺寸等随时间变化的信息,并以此来剖析机理,也就是常说的时间分辨的光散射技术. 这里以聚合诱导的胶束化过程为例来说明该技术的特点和优势[53]. 类似的经典案例还有利用GPC-LLS联用技术监测高分子的降解过程[54],监测支化高分子的聚集与解散[55],以及监测噬菌体喷射DNA的过程[56]等.
氯仿是聚氧乙烯(PEO)的良溶剂, 苯乙烯(S)和马来酸酐(MAn)交替共聚物的不良溶剂. 运用可逆加成断裂转移(RAFT)活性聚合技术,让含有PEO(聚合度114)的大分子链转移剂在氯仿中进行苯乙烯和马来酸酐的交替共聚,生成PEO-b-P(S-alt-MAn). 当P(S-alt-MAn)的聚合度达到某临界值时,就会发生胶束化. 取决于浓度、温度、链长等因素,该过程的时间跨度可达10 h,因此适合用时间可分辨的光散射技术进行表征.
聚合反应的各种试剂和溶剂经滤除尘后,收集于无尘的光散射样品瓶中,并用高纯氮吹扫5 min以除去体系中的氧气. 把样品瓶放入恒温(55±0.01) °C散射仪中,计时开始,交替进行SLS和DLS测量. 取决于散射光强,DLS的采样时间从10 s到2 min不等.图10 是PEO引发剂为1.38 mg/mL时,Rh分布随时间的变化情况. 在229 min时,体系中除了聚合物单分子外(Rh为2~3 nm),还出现Rh约100 nm聚集体(图10(A)),但散射光强弱,证明此类聚集体比较松散. 随时间推移,单分子含量减少,聚集体含量增加,尺寸分布也变窄(图10(B)). 在373 min时,体系中出现了Rh约20 nm的另外一种聚集体(图10(C)),并伴随着大分子单体和100 nm聚集体含量的减少(图10(D)),此时散射光强开始急剧增加,说明新聚集体的链密度较高. 最终体系中仅存在尺寸为20 nm的聚集体,即大分子胶束.
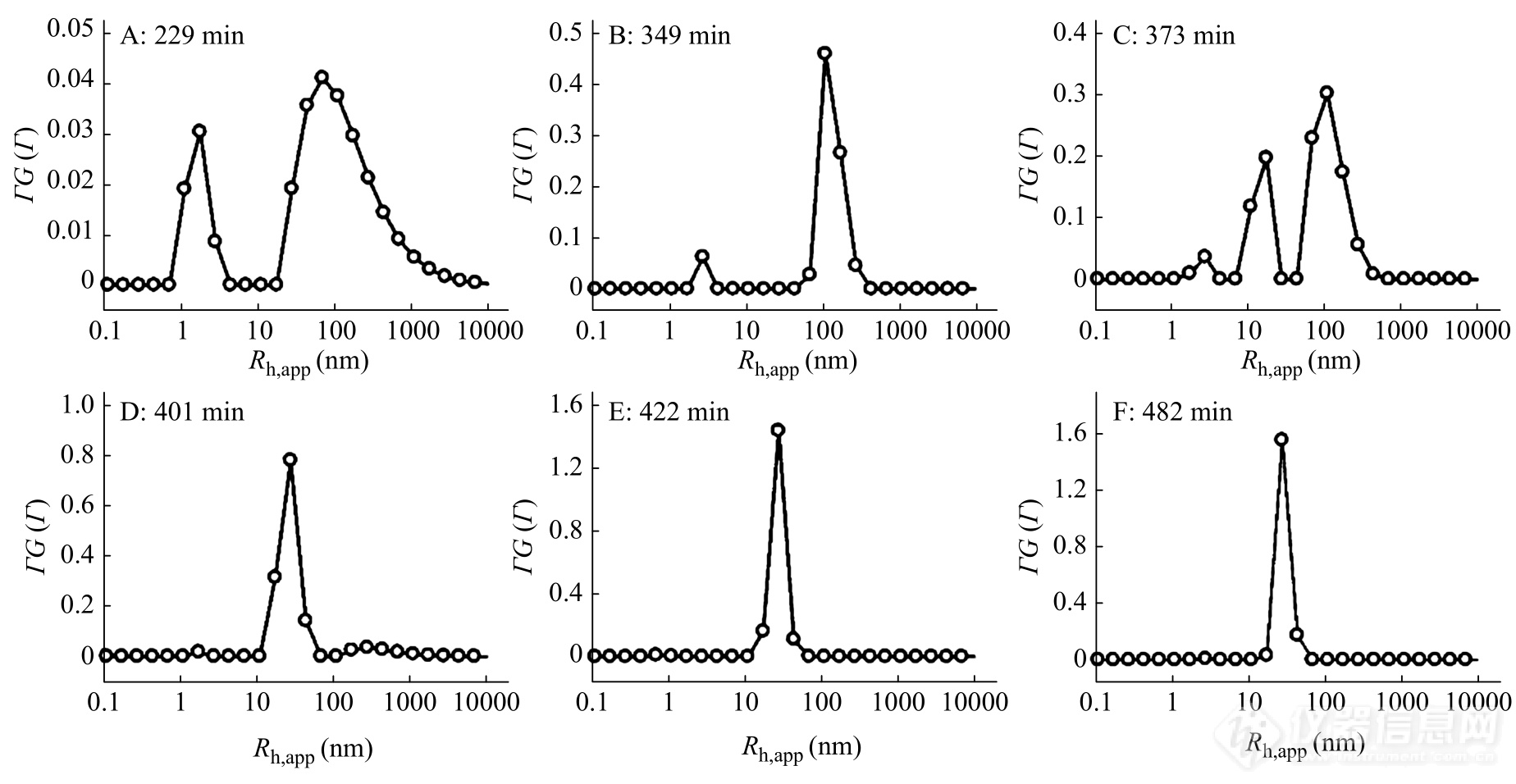
Fig. 10Distribution of hydrodynamic radius during polymerization at different time at 30°. The concentration of PEO macro-CTA is 1.38 mg/mL. (Reprinted with permission from Ref.[53]; Copyright (2008) American Chemistry Society).
由于在373 min之前体系中存在多分布,用静态光散射测定分子量和Rg没有实际意义. 当体系中只存在20 nm的聚集体时,就可以用静态光散射测定Rg,并结合动态光散射的结果,对粒子构象进行分析. 由于光强随时间在发生变化,而Rg的测定需要同一时间的光强角度依赖性数据. 可行的做法是依次测量30°、45°、60°、75°、90°这5个角度下光强数据,并记录时间,直至反应结束. 这样就得到了5条不同角度的散射光强随时间的变化曲线. 使用MATLAB中的cubic spline平滑拟合并插值,可得到任意时间下的光强角度依赖性数据,从而分析得到Rg和分子量. 尽管胶束化过程与浓度相关,无法进行浓度外推,但从严格意义上来讲,这种单一浓度测定的胶束尺寸仍然是表观数据. 如图11所示,随着聚合反应的进行,Rh,app从380 min的23 nm单调增加至840 min的40 nm;而Rg,app在500 min之前快速减小,从53 nm减至20 nm,后基本保持不变.Rg,app/Rh,app则从~1.8降低至~0.5,说明了该聚集体的构象从松散的聚集体向密实球转变. 由于最终聚集体的核是P(S-alt-Man)形成的密实球,而外围的PEO链仍然处在良溶剂中,为线团构象,因此Rg,app/Rh,app可低至0.5左右,类似熔融球构象. 这些结果表明,当P(S-alt-MAn)的聚合度到达临界聚集值时,嵌段共聚物并不是一步组装成胶束结构,而是首先形成具有松散结构的聚集体,继而发育成胶束结构.
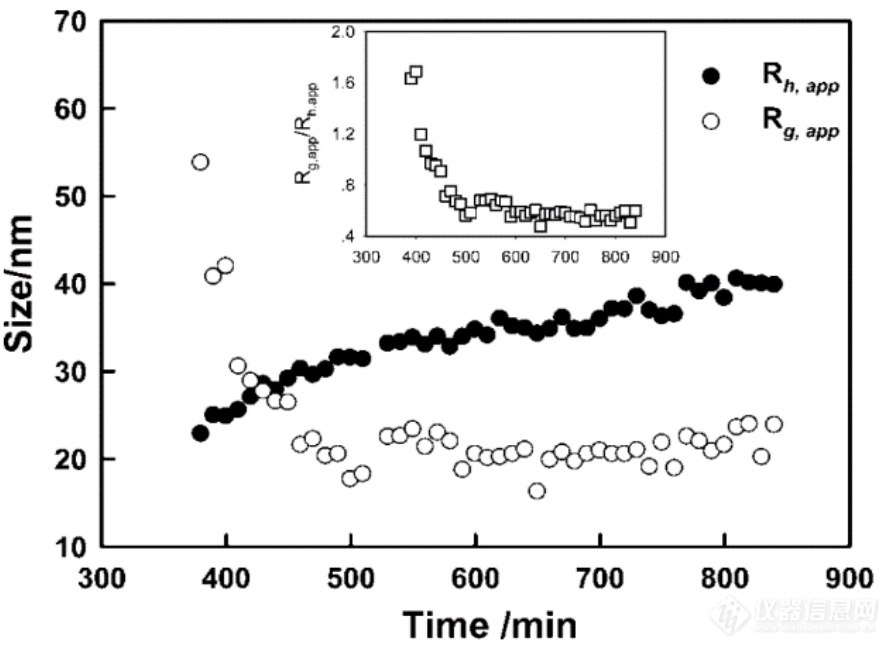
Fig. 11Time dependence of Rg,app and Rh,app in the polymerization-induced self-assembly process. The inset shows the changes in Rg,app/Rh,app. The concentration of PEO macro-CTA is 1.38 mg/mL. (Reprinted with permission from Ref.[53]; Copyright (2008) American Chemistry Society).
4.4去偏振光散射表征生理介质中的纳米粒子
随着现代生物医学技术的发展,纳米粒子在药物缓释、基因传递、生物传感和成像等领域得到了长足发展. 纳米粒子与生物介质的相互作用决定了纳米粒子的细胞中的归宿,包括吸附、分布、代谢和清除,因此原位、无扰跟踪纳米粒子在生物介质中的动态过程就显得尤为重要. 荧光标记是目前最常用的方法,但荧光基团毫无疑问会改变纳米粒子的表面性质.
原位、无扰对体系进行检测是光散射技术的优势. 由于生物介质中高含量的蛋白质等物质会严重干扰纳米粒子的散射光,这使得常规的偏振光散射(VV)并不适于复杂生物体系的研究(图12(a)). 但由于多晶结构的存在,无机纳米粒子不会是完美的球形,总会存在非均质的内部结构,从而能够改变偏振光的方向. 因此采用去偏振动态光散射(depolarized DLS,DDLS),即入射光为V方向偏振,但收集H方向偏振的散射光,就能够有效滤除生物介质产生的背景散射光(图12(b))[57].
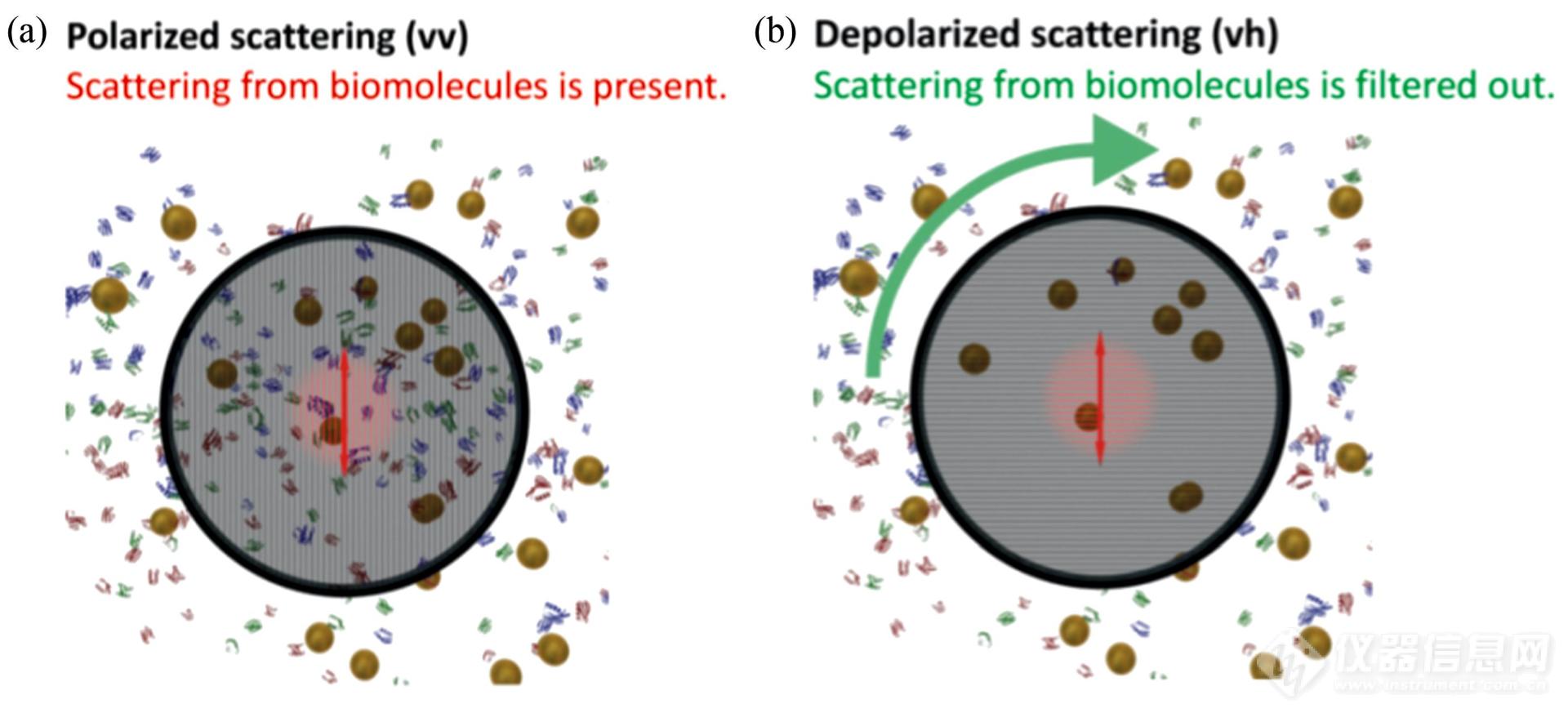
Fig. 12Depiction of nanoparticles and the bio-matrix background as seen in standard polarized (a) and depolarized (b) dynamic light scattering experiments, respectively. (Reprinted with permission from Ref.[57]; Copyright (2015) The Royal Society of Chemistry).
Balog团队利用DDLS技术对比研究了柠檬酸稳定的金纳米颗粒以及不同端基聚乙二醇链包覆的金纳米颗粒在四种不同的生物介质(磷酸盐缓冲液PBS、牛血清白蛋白的PBS溶液、培养基DMEM以及添加了牛血清蛋白的DMEM)中的动态行为. 所使用的仪器是商业化的3D光散射仪. 激光光源为21 mW,632.8 nm的氦氖激光器,散射光信号由装有集成准直光学元件的单模光纤收集,并传递至2个高灵敏度的APD探测器进行分析. 结果表明,DDLS有效地屏蔽了背景散射光,从而能够跟踪金纳米颗粒在不同介质中的聚集过程. 如图13所示,金纳米颗粒形成的聚集体尺寸及其分布既与颗粒表面的涂层有关,更受介质组分的影响. 所得结果得与扫描电镜的结果一致,证明了DDLS原位、无扰跟踪研究复杂体系动力学过程的可靠性.
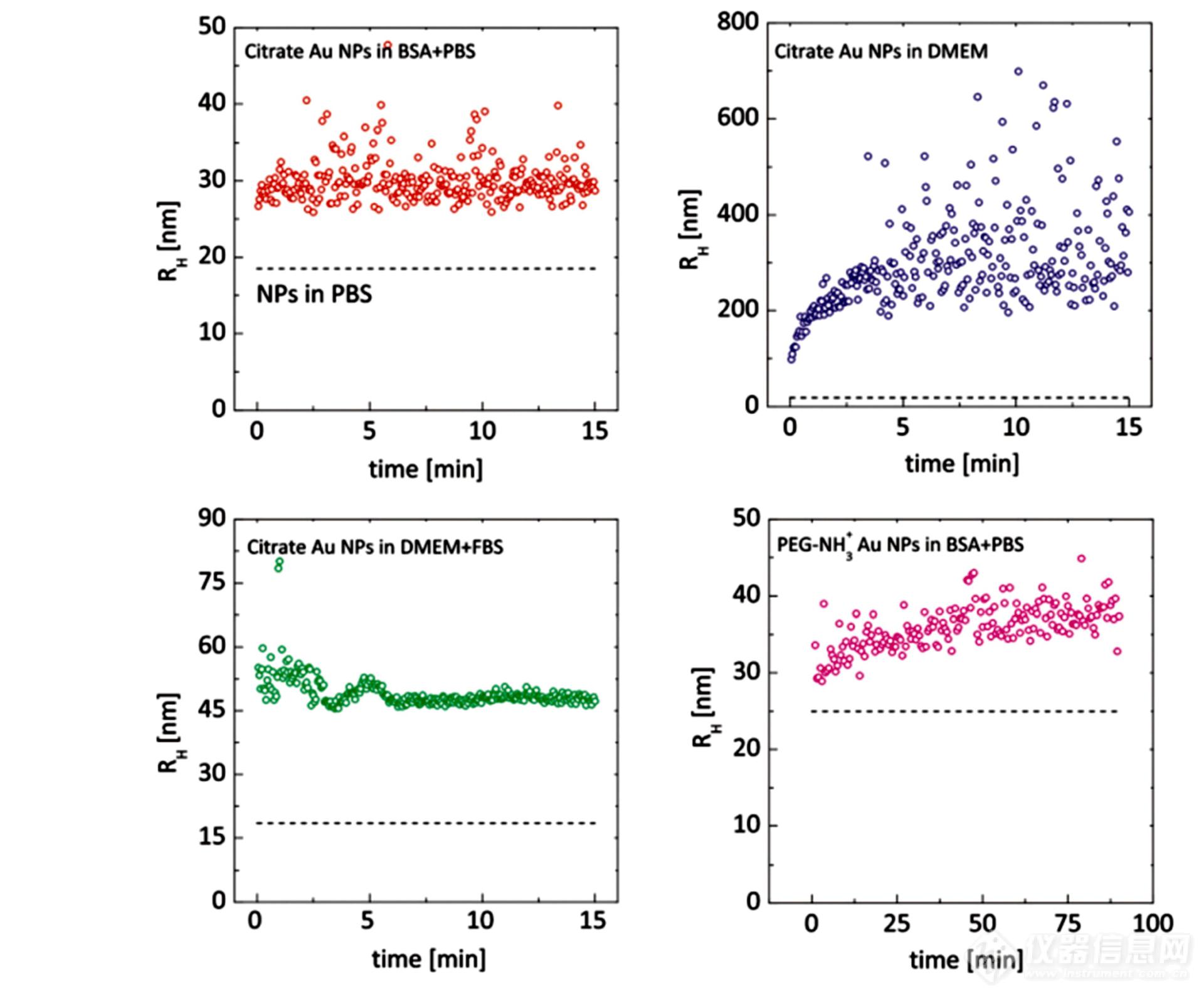
Fig. 13Time-resolved DDLS study started promptly after incubating the Au NPs in the biological media. The dashed lines correspond to the Au NPs in PBS buffer. (Reprinted with permission from Ref.[57]; Copyright (2015) The Royal Society of Chemistry).
5结语与展望
本文介绍了分别对应高分子稀溶液、浓溶液和固体的光散射技术. 其中针对高分子稀溶液的动、静态光散射技术和针对高分子球晶的固体散射技术都是比较成熟的手段,在高分子体系的研究中发挥着不可替代的作用. 光散射技术最显著的优势是能够对体系实现原位、无扰的表征. 伴随着生物医学、活性软物质等领域的发展,针对复杂体系的光散射技术将具有更广阔的应用前景.
致谢
感谢赛普瑞生的牛爱珍博士和布鲁克海文的王继军工程师提供商业化仪器的相关资料.
参考文献
1
Rayleigh L. Phil Mag, 1871, 41: 107-120
2
Rayleigh L. Phil Mag, 1899, 47:566-572. doi:10.1080/14786449908621298
3
Debye P. Ann Phys, 1915, 351: 809-823. doi:10.1002/andp.19153510606
4
Gans R. Ann Phys, 1925, 381: 29-38. doi:10.1002/andp.19253810103
5
Einstein A. Ann Phys, 1910, 338: 1275-1298. doi:10.1002/andp.19103381612
6
Berne B J, Pecora R. Dynamic Light Scattering. With Applications to Chemistrys, Biology, and Physics. New York: Dover Publications, Inc., 2000. 5
7
Pecora R. J Chem Phys, 1964, 40: 1604-1614. doi:10.1063/1.1725368
8
MegenVan, Pusey P N. Phys Rev A, 1991, 43: 5429-5441. doi:10.1103/physreva.43.5429
9
Urban C, Schurtenberger P. J Colloid Interface Sci, 1998, 207: 150-158. doi:10.1006/jcis.1998.5769
10
Lehner D, Kellner G, Schnablegger H, Glatter O. J Colloid Interface Sci, 1998, 201: 34-47. doi:10.1006/jcis.1997.5327
11
Lilge D, Horn D. Colloid Polym Sci, 1991, 269: 704-712. doi:10.1007/bf00657408
12
Wiese H, Horn D. J Chem Phys, 1991, 84: 6429-6443. doi:10.1063/1.460272
13
Phillies G D J. J Chem Phys, 1981, 74: 260-262. doi:10.1063/1.440884
14
Pusey P N. Curr Opin Colloid Interface Sci, 1999, 4: 177-185. doi:10.1016/s1359-0294(99)00036-9
15
Meyer W, Cannell D, Smart A, Taylor T, Tin P. Appl Opt, 1997, 36: 7551-7558. doi:10.1364/ao.36.007551
16
Zakharov P, Bhat S, Schurtenberger P, Scheffold F. Appl Opt, 2006, 45: 1756-1764. doi:10.1364/ao.45.001756
17
Maret G, Wolf P E Z. Phys B, 1987, 65: 409-413. doi:10.1007/bf01303762
18
Brillouin L. Ann Phys, 1922, 17: 88-122. doi:10.1051/anphys/192209170088
19
Stein R S, Rhodes M B. J Appl Phys, 1960, 31: 1873-1884. doi:10.1063/1.1735468
20
Stein R S, Chu W. J Polym Sci, Part A: Polym Chem, 1970, 8: 1137-1157. doi:10.1002/pol.1970.160080709
21
Van Aartsen J J, Stein R S. J Polym Sci, Part B: Polym Phys, 1971, 9: 295-311. doi:10.1002/pol.1971.160090206
22
Huglin M B. Light Scattering from Polymer Solutions. London: Academic Press, 1972. 204-289
23
Wolfgang S. Light Scattering from Polymer Solutions and Nanoparticle Dispersions Series. Translated by Zheng Cui, Liang Dehai. Beijing: China Machine Press, 2012. 1-25
24
Chu B. Laser Light Scattering: Basic Principles and Practice. 2nd ed. New York: Academic Press Inc, 1991. 19. doi:10.1016/b978-0-12-174551-6.50005-7
25
Hua W. Chem Phys, 2010, 367: 44-47. doi:10.1016/j.chemphys.2009.10.019
26
Zhao Zeqing(赵择卿), Lu Danian(陆大年), Yang Dingchao(杨定超). Light Scattering Technology(光散射技术). Beijing(北京): China Textile&Appare lPress(纺织工业出版社), 1989. 28-30
27
Bushuk W, Benoit H. Can J Chem, 1958, 36: 1616-1626. doi:10.1139/v58-235
28
Wu C, Fai K, Luo W, Zhu X, Ma D. Macromolecules, 1994, 27: 6055-6060. doi:10.1021/ma00099a018
29
Teraoka I. Polymer Solutions: An Indroduction to Physical Properties. New York: John Wiley&Sons, Inc. 2002. 168-171. doi:10.1002/0471440264
30
Chu B. Laser Light Scattering: Basic Principles and Practice. 2nd ed. New York: Academic Press Inc, 1991. 84. doi:10.1016/b978-0-12-174551-6.50005-7
31
Kanematsu T, Sato T, Imai Y, Ute K, Kitayama T. Polym J, 2005, 37: 65-73. doi:10.1295/polymj.37.65
32
Delaye M, Gromi Ec A. Biopolymers, 1983, 22: 1203-1221. doi:10.1002/bip.360220413
33
Vanhoudt J, Clauwaert J. Langmuir, 1999, 15: 44-57. doi:10.1021/la980747r
34
Gulari Esin, Gulari Erdogan, Tsunashima Y, Chu B. J Chem Phys, 1979, 70: 3965-3965. doi:10.1063/1.437950
35
Kim S H, Ramsay D J, Patterson G D, Selser J C. J Polym Sci, Part B: Polym Phys, 1990, 28: 2023-2056. doi:10.1002/polb.1990.090281111
36
Benmouna M, Vilgis T A, Hakem F. Macromolecules, 1992, 25: 1144-1152. doi:10.1021/ma00029a022
37
Buhler E, Rinaudo M. Macromolecules, 2000, 33: 2098-2106. doi:10.1021/ma991309+
38
Litmanovich E A, Ivleva E. M Polym Sci, 2010, 52: 671-678. doi:10.1134/s0965545x10060143
39
Corrotto J, Ortega F, Vázquez M, Freire J J. Macromolecules, 1996, 29: 5948-5954. doi:10.1021/ma9507397
40
Murphy R M, Yarmush M L, Colton C K. Biopolymers, 2010, 31: 1289-1295
41
Casassa Edward F. Polym J, 1972, 3: 517-525. doi:10.1295/polymj.3.517
42
Chi W. Polym Adv Technol, 2015, 8: 177-183
43
Lehner D, Kellner G, Schnablegger H, Glatter O J. Colloid Interface Sci, 1998, 201: 34-47. doi:10.1006/jcis.1997.5327
44
Stieber F, Richtering W. Langmuir, 1995, 11: 4724-4727. doi:10.1021/la00012a024
45
Zakharov P, Scheffold F. Light Scattering Reviews 4. Bremen: Berlin Heidelberg: Springer-Verlag, 2009. 433-467. doi:10.1007/978-3-540-74276-0_8
46
Pine D J, Weitz D A, Chaikin P M, Herbolzheimer E. Phys Rev Lett, 1988, 60: 1134-1137. doi:10.1103/physrevlett.60.1134
47
Mason T G, Gang H, Weitz D A. J Opt Soc Am A, 1997, 14: 139-149. doi:10.1364/josaa.14.000139
48
Oelschlaeger C, Schopferer M, Scheffold F, Willenbacher N. Am Inst Phys, 2008,1027: 1150-1152. doi:10.1021/la802323x
49
Morse D C. Macromolecules, 1998, 31: 7044-7067. doi:10.1021/ma980304u
50
Wang X, Qiu A X, Wu C. Macromolecules, 1998, 31: 2972-2976. doi:10.1021/ma971873p
51
Wu C, Zhou S. Macromolecules, 1995, 28: 8381-8387. doi:10.1021/ma00128a056
52
Zhu M, Yang J, Li L, Duan X, Li L. Macromolecules, 2020, 53: 7980-7987. doi:10.1021/acs.macromol.0c01407
53
Ji W, Yan J, Chen E, Li Z, Liang D. Macromolecules, 2008, 41: 4914-4919. doi:10.1021/ma8005312
54
Yang J, Li Y, Hao N, Umair A, Liu A, Li L, Ye X. Macromolecules, 2019, 52: 1173-1187. doi:10.1021/acs.macromol.8b01784
55
Hao N, Duan X, Yang H, Umair A, Zhu M, Zaheer M, Yang J, Li L. Macromolecules, 2019, 52: 1065-1082. doi:10.1021/acs.macromol.8b02364
56
Löf D, Schillén K, Jönsson B, Evilevitch A. Phys Rev E, 2007, 76: 011914. doi:10.1103/physreve.76.011914
57
Balog S, Rodriguez-Lorenzo L, Monnier C A, Obiols-Rabasa M, Rothen-Rutishauser B, Schurtenberger P, Petri-Fink A. Nanoscale, 2015, 7: 5991-5997. doi:10.1039/c4nr06538g
原文链接:
http://www.gfzxb.org/thesisDetails#10.11777/j.issn1000-3304.2021.21184&lang=zh
DOI:10.11777/j.issn1000-3304.2021.21184
《高分子学报》高分子表征技术专题链接:
http://www.gfzxb.org/article/doi/10.11777/j.issn1000-3304
[来源:高分子学报]
2021.12.28

2024.07.26

首届江西南昌高校联合检测机构大型仪器操作技能大赛在南昌成功举办
2024.07.25

第3届测量仪器国际会议暨第13届精密工程测量与仪器国际会议通知
2024.07.24

2024.07.24

AI时代,质谱软件也被卷到了——访科迈恩(北京)科技有限公司总经理/创始人田润涛
2024.07.24
品牌合作伙伴






























版权与免责声明:
① 凡本网注明"来源:仪器信息网"的所有作品,版权均属于仪器信息网,未经本网授权不得转载、摘编或利用其它方式使用。已获本网授权的作品,应在授权范围内使用,并注明"来源:仪器信息网"。违者本网将追究相关法律责任。
② 本网凡注明"来源:xxx(非本网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责,且不承担此类作品侵权行为的直接责任及连带责任。如其他媒体、网站或个人从本网下载使用,必须保留本网注明的"稿件来源",并自负版权等法律责任。
③ 如涉及作品内容、版权等问题,请在作品发表之日起两周内与本网联系,否则视为默认仪器信息网有权转载。
![]() 谢谢您的赞赏,您的鼓励是我前进的动力~
谢谢您的赞赏,您的鼓励是我前进的动力~
打赏失败了~
评论成功+4积分
评论成功,积分获取达到限制
![]() 投票成功~
投票成功~
投票失败了~