
 关注
关注
已关注
![]() 已认证
已认证
粉丝量 0

革兰阴性细菌中哺乳动物对微生物的识别机制
LPS可以说是炎症反应最强的刺激剂。20世纪70年代人们普遍认为LPS要发挥作用,必须先插入生物膜脂质双分子层或通过受体作用被吞噬,但这样的猜想一直无法得以证实。Coutinbo发现C3H/HeJ小鼠对LPS无反应性,并将其原因归结为位于常染色体的一个等位基因位点lps的突变。但是C3H/HeJ小鼠对革兰阳性细菌的反应却正常,由LPS介导产生的细胞因子等方面也是正常的。这种表型上的差异可以说是因为对LPS识别的缺陷造成的。lpsd动物实验的研究可以得到一个结论:在哺乳动物存在对微生物的天然识别机制,这种机制识别的范围可以粗略的定义为革兰阴性细菌。这个天然识别机制在对感染的耐受方面起重要作用。对lpsd动物的研究使人们认识到对LPS的识别机制是天然免疫的重要环节,也必然存在一条信号传导途径能将LPS的作用引入胞内,而且lpsd纯合子在LPS作用时信号将完全阻断。可以进一步推论lps编码的产物能识别LPS并引起对革兰阴性菌感染的快速反应。所以lps突变的小鼠对革兰阴性细菌的耐受性增高,而对革兰阳性细菌的反应却正常。为了寻找能编码LPS模式识别受体的基因及相应产物,人们进行了不懈的努力,先后发现了LBP、CD14等能与LPS结合的相关蛋白,但是通过多种方法一直没有找到lps位点及其表达产物。早在1978年,lps位点就定位于小鼠4号染色体的Mup-Ⅰ和Psloci 两个位点之间。20世纪90年代初期,Malo、Schwartz和Beutler分别领导的3个研究小组试图对lps位点进行精确定位。经过大量的工作,1996年前后,3个并不完全相同的结果先后发表。Quresh等认为lps位点局限在2个标志物Ambp和D4MIT178之间;Peiffer-Schneider等将lps界定在一段长度约2.5Mb的片段中;Poltorak等则认为lps位于两个异常标志“B”和“83.3”之间长约2.6Mb的DNA中。3个结果中相互重叠的区域长度约为500kb左右。事实上,lps位点的寻找仍有大量工作要做,目前尚未获得lps位点定位的直接精确数据。在对LPS无反应性的C3H/HeJ和C57BL/10cCr小鼠中发现了TLR4的突变体。在C3H/HeJ小鼠中,TLR4胞内区的一个氨基酸编码发生了点突变;而在C57BL/10ScCr小鼠中,TLR4 mRNA 无法检测到,整个位点被删除。
参数原理
2024.03.12
病原体相关模式分子与模式识别受体
微生物细胞壁的组成成分是天然免疫反应和炎症反应的高效活化分子。这些分子如革兰阴性细菌的脂多糖(lipopolysaccharide,LPS)、革兰阳性细菌的肽聚糖(peptidoglycan)、脂磷壁酸(lipoteichoic acid,TLA)以及真菌的甘露糖(mannans)等称为病原体相关的模式分子(pathogen-associated molecular patterns,PAMPs)。与之相对应的模式识别受体(pattern recognition receptors,PRR)这一概念最早是由Janeway提出的。PAMPs均可被动物作为外来分子进行识别。PAMPs能激发机体细胞因子如 IL -1、TNF-α等以及其他活性分子的合成,对感染的发生发展具有十分重要的作用。细胞因子的过度活化可引起脓毒症休克,是细菌感染患者死亡的首要原因。因此模式识别受体在天然免疫和炎症反应中居于重要地位,通过模式识别受体机体能区别病原体与自身组织,这是免疫反应的起点和根本特征。但过去的研究一直都没有发现能识别PAMPs特别是LPS并引起上述反应的模式识别受体。近年来发现的Toll-like receptors(TLRs)是在研究LPS作用机制方面获得的重要进展,对深入了解模式识别受体作用机制,研究天然免疫反应和炎症反应的传入途径是一个重大突破。内毒素是革兰阴性菌细胞壁中的脂多糖(lipopolysaccharide,LPS)。由亲水性的多糖(О-特异性多糖、核心多糖)及疏水性的类脂A(Lipid A)构成。其中Lipid A是LPS结构中最保守的部分,无种属特异性,是LPS的主要生物活性成分。LPS在细菌生长繁殖、死亡破裂或人工方法裂解后释放。一旦进入人或其他敏感动物体内,将对宿主各系统、器官产生广泛的影响。由于其作用机制广泛而复杂,在过去近20年里尽管针对性抗生素不断问世,革兰阴性细菌感染的病死率仍持续在30%左右居高不下。
参数原理
2024.03.12

内毒素对各种细胞的激活作用
在整体水平认识到内毒素(LPS)对机体损害作用的情况下,随着分子生物学技术的发展,从细胞水平研究LPS的致病机制成为当今相关研究领域的重点与热点之一。现有研究证实,LPS可以激活多种细胞,从而影响细胞生理功能的正常发挥并产生明显的病理损害,内毒素血症是诱发严重创伤、感染患者失控性炎症反应症形成并最终导致死亡的重要原因之一。一、巨噬细胞巨噬细胞是白细胞中功能最为活跃的细胞,在机体的防御中起重要作用,对内毒素具有强烈反应性,是机体产生细胞因子及其他介质的主要细胞。有研究证实,体外培养的巨噬细胞在遭受内毒素攻击时,细胞对病原体的吞噬功能显著下降,而作为炎症介质的主要产生细胞,LPS刺激巨噬细胞可以引起一系列细胞因子、趋化因子的高水平释放,与此同时,巨噬细胞还通过高水平表达的黏附分子介导其与其他细胞间的相互作用,启动炎症反应,巨噬细胞及其他细胞的过度激活可导致致炎因子的过度释放从而引发失控性炎性反应的形成,这些病理生理反应在相关研究中已得到充分证实,在此不作过多阐述。巨噬细胞常因其存在的器官不同而名称各异,如在肝脏称为库普否细胞,在肺脏称为肺巨噬细胞,肺巨噬细胞又可根据所处位置不同分为肺泡间质细胞及肺泡巨噬细胞,新近有研究显示,不同器官或同一器官不同部分的巨噬细胞对内毒素的反应性不尽相同,如肺间质巨噬细胞的反应性明显高于肺泡巨噬细胞,深入探讨这些细胞对内毒素反应不同的机制及其病理生理学意义将有助于对临床感染性疾病发病机制的认识并指导救治方案。二、中性粒细胞中性粒细胞(PMN)是功能较为活跃的白细胞之一,对于中性粒细胞在内毒素血症发生、发展中的作用与地位,人们很早就给予了充分的重视。借由LBP等血清物质的存在,LPS可以直接激活中性粒细胞,产生氧自由基与NO、释放多种蛋白酶与细胞因子、诱导包括黏附分子在内的多种基因的表达,并通过这些因素进而引发炎性介质级联效应,在脓毒症的发生发展中具有十分重要的地位。在内毒素血症早期,末梢血中性粒细胞的明显减少,与LPS对中性粒细胞以及其他细胞如血管内皮细胞的激活密切相关,激活的中性粒细胞与内皮细胞间的黏附能力异常增强,导致中性粒细胞聚积在毛细血管网尤其是肺毛细血管网中。有研究证实,粒细胞在肺部的过度聚集与活化是肺部血液循环障碍及呼吸障碍乃至休克产生的重要原因之一,而粒细胞对骨髓粒细胞的活化所引起的凝血活性增高与弥漫性血管内凝血可能具有一定内在联系。值得一提的是,作为机体的一种主要防御细胞,中性粒细胞在内毒素清除过程中也发挥了重要作用。中性粒细胞对于原发性内毒素血症具有一定的防御作用,PMN可以通过直接吞噬内毒素或者清除遭受内毒素攻击而滞留于血管璧周围的血小板而间接吞噬内毒素,更有研究显示,中性粒细胞还可防御因为继发性内毒素血症而对机体产生的损害,应用放射线照射破坏中性粒细胞的小鼠对由肠道吸收的内毒素的抵抗力明显减弱。三、血小板血小板上存在有LPS受体,对进入血循环的LPS具有清除作用,并运载到网状内皮系统。有研究证实,内毒素血症时,血小板上结合有较多的LPS,给大鼠腹腔注射血小板对内毒素反应具有明显的抑制作用。作为哺乳动物体内对LPS最敏感的有形成分之一,LPS可以通过活化血小板酶、前列腺素环化酶直接激活血小板,引起凝集反应,并释放血小板凝集促进剂ADP与血管活性物质如5-羟色胺等,加速血小板血凝块的形成,促进凝血反应。四、血管内皮细胞近年来对血管内皮细胞的功能有了新的认识,血管内皮细胞不单是具有机械屏障及对营养物质具有通透作用的半透膜功能,在外源性物质的刺激下,内皮细胞可以直接参与炎症反应或对炎症反应具有放大作用。近年来,越来越多的体外实验数据证实,LPS可以直接激活血管内皮细胞,激活细胞内多条信号通路,介导在炎症反应中发挥重要作用的多种细胞因子、黏附分子及其他炎症相关因子的表达,与此同时,细胞形态结构发生明显改变,细胞变形、细胞间连接损害,屏障功能显著受损,此外,LPS还可直接诱导内皮细胞凋亡。五、肠上皮细胞肠上皮细胞( intestinal epithelial cell,IEC)在机体中所处部位特殊,在肠道屏障中具有极其重要的作用。既往一直认为上皮细胞是一类CD14阴性细胞,对LPS不具有反应性或反应低下,但目前部分实验数据支持LPS可以激活IEC的结论。体外实验显示,不同系的肠上皮肿瘤细胞对LPS的反应性存在明显差异,如癌性肠上皮T84、CaCo-2细胞对LPS不具有反应性,而癌性肠上皮细胞SW620、HT29在 LPS的刺激下能分泌IL-8,大鼠IEC-6细胞系在LPS刺激下能诱导细胞因子如IL-6、趋化因子如MCP-1及NO等的产生,也有研究显示LPS刺激也不能诱导人源性原代培养IEC产生IL-6、I1.-1 beta、TNF-alpha等细胞因子。总之,IEC对LPS的反应性问题还有待进一步研究,深入阐明肠上皮细胞对LPS的反应性/耐受性及其调节机制对于认识内毒素激活细胞的分子机制具有十分重要的意义。六、肝细胞肝脏是机体极为重要的器官之一,在内毒素血症时,肝受到损害,尤其是代谢障碍是以肝损伤为中心发生的。先前研究显示,发生肝损害的机制是LPS作用于炎症相关细胞其中包括肝星状细胞后导致多种炎症介质的释放,这些炎症介质反过来作用于肝实质细胞引发肝损伤。与此同时也有相当多的关于LPS直接作用于肝实质细胞而引起肝损伤的证据,LPS可以诱导离体培养肝细胞的凋亡、上调诱导型一氧化氮合酶的表达与NO的产生及一些相关酶学指标的异常改变,但也有一些体外实验数据显示LPS并不能对肝细胞造成直接损害,如LPS单独刺激并不能造成离体肝细胞的脂质过氧化损伤,仅在有D-氨基半乳糖的共同作用下或有库普弗细胞共同培养的情况下才出现明显的肝细胞损害等,关于肝细胞是否是LPS的直接靶细胞的争论至今仍在争论,有待于更进一步的确切证据来证明。七、肌细胞大量实验数据证实,内毒素血症时机体肌肉系统包括骨骼骨、心肌及平滑肌发生明显的病理生理改变,这些病理生理改变包括代谢的紊乱﹑结构的重塑、基因表达的改变等。长久以来,人们一直认为,LPS对肌细胞的激活是通过活化其他免疫细胞导致细胞因子释放以及激活补体等途径而间接发挥作用的。但近年来的研究显示,LPS可以直接活化肌细胞,如微量LPS激活平滑肌细胞c-myc 、c-fos及hsp70等基因的表达、促进其增殖,而较高剂量的LPS则可以直接影响肌细胞的多种代谢过程如氧利用与脂质代谢等,抑制其增殖并降低其生存能力,肌细胞收缩能力也明显受抑。近年在脓毒症情况下骨骼肌过度分解的机制研究上有所突破,即TNF-alpha等炎性介质可以活化肌细胞内蛋白酶小体,从而导致肌细胞的过度降解,而作为IPS活化细胞的主要信号通路NF-kappa B,其激活同样存在蛋白酶小体的过度活化,在此情况下,LPS刺激可否直接引起骨骼肌的降解目前尚未得到证明。八、成纤维细胞作为一种实质细胞,成纤维细胞在维持组织结构及参与损伤修复中具有无可替代的作用,早期体外研究显示,受LPS攻击后的纤维母细胞可消耗葡萄糖,摄取大量的3H-胸腺嘧啶脱氧核糖,增加细胞数量,合成透明质酸的功能也明显增强,这些研究提示创伤条件下LPS与创伤组织的修复以及瘢痕形成有关。近年来的研究显示成纤维细胞的生物学意义远不止如此,它在炎症反应过程中可能也起到非常重要的作用,多种刺激因子均可以激活成纤维细胞,LPS能以血清依赖的方式直接激活成纤维细胞,活化的成纤维细胞与其他炎症细胞一样能够表达多种细胞因子、趋化因子从而启动或扩大炎症反应。九、肥大细胞肥大细胞同样在炎症反应过程中发挥重要作用。既往认为,LPS主要是通过激活其他靶细胞导致炎症因子的产生而间接作用于肥大细胞,如被LPS激活的补体在被消耗的同时裂解出C3a、C5a等,这些成分可以刺激肥大细胞释放组织胺﹑缓激肽、Р物质等,使血管通透性增强,末梢血流减少,是多脏器功能衰竭的启动机制之一。近年来有实验证实,与巨噬细胞等细胞一样,肥大细胞细胞膜上同样存在LPS受体如TLR4等分子,LPS可以通过这些分子的介导直接活化肥大细胞,导致肥大细胞脱颗粒﹑释放组织胺等,并影响多种细胞因子如IL-1beta、TNF-alpha、IL-6等的表达,这些细胞因子的产生一方面可以促使肥大细胞杀灭入侵病原,另一方面也可以作用于其他细胞如中性粒细胞等导致细胞因子释放的级联反应,在失控性炎症反应的发生、发展过程中发挥重要作用。十、造血干细胞早在20世纪50年代就有学者发现,反复注射细菌性致热物质的小鼠照射致死剂量的X线仍可生存下来,此后,Smith及其合作者(1958)证实,Ⅹ线照射前24h给大鼠(Hamster)或小鼠注射LPS,在给予致死剂量Ⅹ线照射后动物仍可存活。以末梢血中血红蛋白量、粒细胞数、淋巴细胞数、血小板数﹑骨髓被动员白细胞人末梢血的能力及骨髓象细胞数等为观察指标,发现预先注射LPS的动物,Ⅹ线损伤轻,尤其是损伤后恢复速度快。这提示LPS能明显激活造血细胞,促进造血,从而促进放射线造成的血液系统障碍的恢复。体外实验已充分证明LPS注射动物后能短暂增加脾细胞、血细胞的细胞集落形成单元,其机制可能与LPS激活其他细胞如T淋巴、B淋巴细胞、单核-巨噬细胞等产生造血生长因子最终活化造血干细胞有关,目前尚未有明确证据显示LPS可以直接激活造血干细胞,对内毒素血症时造血细胞活化的病理生理意义也有待进一步认识。
参数原理
2024.03.11

内毒素对蛋白质代谢的作用
创伤、大手术、脓毒血症和其他严重感染疾病可引起机体蛋白质代谢的明显变化。其最主要指征是外周肌肉蛋白质分解加强,出现尿氮排出增多,引起负氮平衡。另方面则是肝脏蛋白质合成增加表现急性相反应。结果引起多种血浆蛋白水平异常,即某些正急性相反应蛋白增多,而某些负急性相反应蛋白则减少。(一)对肌肉蛋白分解的影响脓毒血症或注射内毒素,出现体重减轻,骨骼肌蛋白质消耗加速。肌肉蛋白质分解代谢增强主要是由于肌原纤维蛋白大量降解,大量3-甲基组氨酸﹑脯氨酸和谷氨酰胺的释放。由外周肌肉组织释放的氨基酸大部分向中央器官(或组织),如肝脏与小肠转移和(或)再分配,用于提供葡萄糖再生成的碳源和急性相反应蛋白合成的氮源。骨骼肌蛋白分解是尿素氮的主要来源。另外从肌肉蛋白分解释放的谷氨酰胺可由免疫细胞和小肠黏膜细胞摄取,提供重要的能源和保护肠黏膜上皮细胞完整性及免疫细胞功能。因此谷氨酰胺有“病理性必需氨基酸”之称。创伤,脓毒血症骨骼肌蛋白质分解增强同样是宿主对感染、细菌毒素反应的一种防御机制。至少在早期对中央组织提供足够量氨基酸有积极意义。当然,此时肌肉蛋白质分解如果继续失控,大量肌肉蛋白质耗损最终可转变为自身消蚀(autocannibalism)而将加重和延误脓毒血症过程。由于肌肉蛋白占全身蛋白质的大部分,持续的肌肉蛋白质耗损将引起全身蛋白质丢失和能量消耗。尤其是呼吸系统相关肌肉蛋白质的分解,导致呼吸功能障碍可进一步增加肺部感染和血凝失控等一系列并发症的危险。内毒素促进肌肉蛋白质分解增强,是由多种介质相互作用的结果。其中有两方面重要介质参与脓毒血症肌肉蛋白质降解的调节。它们是糖皮质激素和致炎细胞因子。脓毒血症与创伤患者血中皮质醇及皮质酮水平迅速升高。肌原纤维蛋白对糖皮质激素极为敏感。用糖皮质激素受体拮抗剂RU38486处理脓毒血症大鼠,可以明显抑制外周肌肉总蛋白质分解,酪氨酸和3-甲基组氨酸释放排出明显减少。糖皮质激素通过特异性受体介导,活化的糖皮质激素受体复合物作为配基应答转录因子刺激泛素基因的转录活性,增加肌肉细胞的泛素连接蛋白表达,活化ATP-泛素依赖的26s蛋白酶辉解系统,节脓毒血症肌肉蛋白质分解。致炎细胞因子TNF、IL-1和 IL-6以自分泌、旁分泌或内分泌方式参加脓毒血症肌肉蛋白质降解的调节。给人注射重组TNF(hrTNF),出现全身外周组织蛋白质分解增强,同时血中皮质醇浓度显著增高。前臂氨基酸流量增加,以丙氨酸和谷氨酰胺为主。表明TNF促进外周肌肉蛋白质分解以便为时相蛋白质合成。内毒素和细胞因子促进肌肉蛋白质分解,同时还抑制蛋白质合成,抑制肌肉细胞对氨基酸的摄取。注射内毒素或TNF可抑制肌原纤维的肌凝蛋白和肌动蛋白的mRNA表达,降低核糖体蛋白13S和20S亚基蛋白含量。慢性注射重组IL-1α,亦可引起肌原蛋白含量和mRNA水平降低。(二)对肝脏蛋白质合成的影响严重创伤和感染以及内毒素血症﹐除了引起肝组织细胞氨基酸摄取障碍和氨基酸代谢紊乱外,肝脏蛋白质合成代谢表现为明显的急性相反应(acute phase responses,AP-RS),包括血浆多种急性相反应蛋白浓度改变和许多行为、生理、生化和营养的变化。引起急性相蛋白升高(称正急性相蛋白>和降低(称负急性相蛋白)。其中C-反应蛋白和血清淀粉样蛋白A水平可升高上千倍。下表列出炎症反应时人血清急性相蛋白质变化情况(表5-1)。此外,内毒素血症时由肝脏合成的一系列代谢相关酶蛋白,如金属疏基三甲组氨酸酶(metallothionein),诱生型一氧化氨合酶(iNOS),Mn-超氧物歧化酶(Mn-SOD)和组织金属蛋白酶抑制物明显增多,而PEPCK蛋白(活性)则明显降低。同时还出现低锌血症,低转铁蛋白血症和铜蓝蛋白血症以及血浆视黄醇及谷胱甘肽浓度升高。内毒素同样通过活化单核/巨噬(枯否)细胞,激活NF-κB和JAK-STAT两条细胞内信号偶联通路,诱导炎症细胞因子,主要是IL-6、IL-β、TNFα、IFNγ和 TGFβ作为肝细胞最主要的天然或炎症依赖刺激的细胞因子在诱导肝细胞合成急性相蛋白起关键作用。LPS诱导多种细胞因子通过它们彼此之间的信号级联放大和信号网络,相互联系、相互协同及相互抑制,从转录和翻译水平以及翻译后修饰的不同阶段调节各种急性相蛋白的合成与分泌。另外某些激素(如皮质激素,生长激素和胰岛素及胰岛素样生长因子-1)亦参与细胞因子对肝脏急性相蛋白质合成的调节。其中糖皮质激素可增强细胞因子促进急性相蛋白合成;胰岛素则减少某些急性相蛋白的合成。由于各种细胞因子有不同来源,不同靶细胞和不同功能,从而起着不同的细胞间信号通讯(cellular communication),因此急性相反应可依据不同个体或不同病理状况而有不同的表现。在肝细胞急性相蛋白质合成中,枯否/巨噬细胞是肝细胞功能主要调节物。当无炎症刺激时,枯否细胞可刺激肝细胞普遍的蛋白质合成。在用LPS(10μg/ml)或重组IL-6(300μg/ml)刺激枯否细胞时,则抑制枯否-肝共培养的肝细胞白蛋白合成,另外分子量2.3万,3.8万和6万蛋白质合成不变。LPS对单纯培养的肝细胞蛋白质合成不影响。认为在内毒素攻击(insult)下产生LI-6特异性增强肝巨噬细胞-肝细胞之间的信号传递,由肝巨噬细胞提供信号引起肝细胞某些分泌型蛋自质合成受抑制和线粒体呼吸功能紊乱,而某些急性相蛋白质生成被激活。有人认为这些分泌型蛋白质合成减少,其意义可能是为急性相蛋白合成提供原料。肝脏蛋白质合成变化也是急性相反应的防御措施之一,具有重要的生物学意义。例如C-反应蛋白作为固有(innate)免疫系统成分,可识别某些外来病原,促进靶物质排出;还可活化补体系统和吞噬细胞,引起炎症灶的趋化性,血浆蛋白渗出和细胞损伤,表现致炎作用。但C-反应蛋白更主要作用是阻止中性粒细胞对内皮细胞的黏附,减少L-选择素表达和抑制中性粒细胞产生过氧化物,刺激IL-1受体拮抗物(IL-1Ra)表达而发挥抗炎症作用。而淀粉样蛋白A作为载脂蛋白家族成员,其合成后迅.速结合HDL,影响胆固醇代谢外,还可引起吞噬细胞和淋巴细胞黏附和趋化作用,增强ox-LDL生成,在急性和慢性炎症和动脉粥样硬化过程起重要作用。其他一些急性相蛋白,如触珠蛋白可对抗自由基生成,刺激血管新生,对创伤组织修复有利。α1-蛋白酶抑制剂(包括α1-抗糜蛋白酶,α1-抗胰蛋白酶,α2-巨球蛋白和α1-酸性糖蛋白),除拮抗蛋白水解酶活性外,亦能抑制超氧阴离子产生,拮抗TNFα诱导的致死作用。纤维连接蛋白可促进内皮细胞黏附、播散和增生,促进损伤组织的修复。
参数原理
2024.03.08

感染和内毒素血症对脂类代谢的影响
感染和内毒素血症可引起脂类代谢改变。这亦是机体急性相反应的组成部分。(一)对三酰甘油代谢的影响动物注射内毒素2h内,血清三酰甘油(TG)水平迅速升高并至少保持24h。TG升高是伴随于vLDL的升高,尽管apoE可能减少和apoSAA(血清淀粉样蛋白A)升高,但LDL颗粒成分相对正常。低剂量注射LPS(0.1~3μg/100g体重)的大鼠血清TG增高,主要是由于刺激肝脏合成TG和分泌vLDL;同时亦增加脂肪组织的脂解作用,以提供肝脏TG合成所需的原料,虽是无效的脂酸底物循环,却是一种脂肪的再分配。较高剂量(50μg/100g或)LPS可由另一种机制诱导高三酰甘油血症。此时LPS不是促进vLDL的合成与分泌,而是通过抑制脂肪和肌肉等肝外组织的脂蛋白脂肪酶(LPL)活性,减少富含TG的脂蛋白的清除。此外,vLDL颗粒中apoE含量相对降低或是巨噬细胞LDL受体相关蛋白(LRP),又称apoE受体表达减少,易影响相应脂蛋白如vLDL,CM或IDL的清除,而导致高三酰甘油血症。炎症细胞因子,如TNF、IL-1和IL-6既是急性相反应蛋白合成的主要介质,又能模仿内毒素作用,介导内毒素引起脂类代谢变化,其中最主要是由 TNF刺激肝脏脂酸合成和激活脂肪组织激素敏感脂肪酶,促进脂解作用。增加肝脏TG的合成。与此同时,内毒素和细胞因子可下调整体的肝和心肌特异性的脂肪酸结合蛋白(L-FABP与H-FABP)合成,减少宿主组织细胞对脂肪酸的摄取﹑转移和氧化利用,升高血中TG水平。内毒素还可通过刺激交感-肾上腺素系统活性,释放儿茶酚胺通过β-肾上腺素能受体介导低剂量的内毒素增加肝脏TG分泌和高剂量的内毒素对LPS活性的抑制,引起高三酰甘油血症。(二)对胆固醇代谢的影响感染和内毒素血症时,降低人和其他灵长类动物血清胆固醇含量。而其他非灵长类动物(大鼠﹑兔、狗)对内毒素刺激,可增加其血清胆固醇水平,但胆固醇浓度增加迟于三酰甘油的增加。内毒素增加肝脏胆固醇合成,主要是特异性刺激HMG-CoA还原酶mRNA转录和蛋白质合成。另外内毒素抑制7-α羟化酶基因表达和酶活性,从而减少胆固醇转变成胆汁酸。内毒素增加LDL-ch水平,但它不影响LDL受体mRNA或蛋白水平。所以内毒素增加LDL-ch水平对增加LDL合成的作用大于对LDL清除的作用。TNF和IL-1仍然是内毒素对胆固醇代谢的主要介质。它们可模仿内毒素增加血中胆固醇含量和增加肝脏合成胆固醇的能力,亦是通过增加HMG-CoA还原酶mRNA水平。抗TNF抗体可明显避免内毒素对胆固醇代谢的影响,而IL-1拮抗剂只表现很弱的作用。(三)对 HDL代谢的影响内毒素可迅速(2~4 h)降低灵长类和非灵长类血中HDL-ch水平,并持续24h。内毒素减少HDI中胆固醇酯(ChE)和apoA-Ⅰ含量;增加游离胆固醇(FCh)和apoSAA以及apoJ的含量。增加HDL颗粒中apoE的比例。上述HDL颗粒成分的变化直接影响HDL代谢功能的改变:由于apoA-Ⅰ是HDL与细胞膜之间相互作用的主要连接物,当apoA-Ⅰ减少,将减少细胞胆固醇的流出,从而影响胆固醇逆向转运。另外,由于富含apoSAA的HDL生成,HDL很快从血中被清除,这是内毒素降低血中胆固醇的重要原因。同时富含apoSAA的HDL降低对肝脏的亲和力,增加对巨噬细胞的亲和力,当SAA 与ch结合后很快被肝外细胞所摄取。因此在感染﹑脓毒血症急性相反应时,HDL-Ch可直接进入巨噬细胞。内毒素及相关的细胞因子还通过减少肝细胞LCAT和胆固醇酯转运蛋白(CETP)的mRNA表达,降低循环中LCAT和CETP活性,影响HDL的胆固醇酯化及HDL与vLDL之间的胆固醇和三酰甘油的交换,使HDL颗粒中ChE降低而FCh升高,从而减少外周组织细胞的胆固醇流出。感染和注射内毒素可降低肝脂肪酶(HL)活性,而HLmRNA不受影响,提示内毒素作用于HL蛋白翻译和翻译后的修饰阶段。HL促进HDL3中TG和Ch在肝脏的清除,使HDL3转变成HDL2和前β-HDL。HDL2和前β-HDL再返回肝外组织介导细胞膜FCh的流出。内毒素降低HL活性,实质上是减少胆固醇的逆向转移,结果使更多的Ch在外周细胞堆积。由上可见,内毒素通过诱发HDL组成成分的改变和代谢关键酶活性降低,对体内脂质代谢产生明显的影响,其结果一方面降低HDL胆固醇逆向转运作用,减少外周组织细胞Ch流出;另方面降低血浆HDL-ch水平同时减少HDL与vLDL之间Ch及TG的交换,不仅使FCh在HDL中贮留,同时亦抑制HDL和vLDL在肝中的清除,结果血中大量Ch和TG被巨噬细胞吞噬,除了提供能源物质外,可以引发系列慢性炎症的并发症发生。内毒素对脂质代谢的影响,仍然是机体急性相反应的重要组成部分,其积极的防御作用主要有:①脂肪从脂肪组织动员到被巨噬细胞摄取,达到细胞营养成分的重新分配,对宿主防御功能有利;②提高血浆脂蛋白(HDL、vLDL以及CM)含量可结合内毒素和其他生物试剂以减少其毒性作用;③某些载脂蛋白(apoE, apoSAA)具有中和病毒和裂解寄生菌的作用,对保护宿主生存有积极意义。
参数原理
2024.03.07

胰岛素抵抗内毒素血症的详细机制
内毒素血症,糖代谢异常最主要原因与机体对胰岛素作用的耐受(tolerance)相关。给予健康自愿者静脉注射LPS(20 U/kg体重)的高胰岛素血症的血糖钳夹试验,观察LPS对组织胰岛素敏感性和葡萄糖利用的影响,发现开始120 min除了模仿对细菌感染的发热、心悸、中度动脉血压降低的全身炎症反应综合征(SIRS)外,同时葡萄糖摄取利用增加64.1±12.0%,但以后逐渐降低,并持续420min后。葡萄糖利用减少与非氧化葡萄糖消耗和血乳酸产生增加相关。同时对胰岛素依赖的剂量增加,出现胰岛素耐受(抵抗)。内毒素血症引起胰岛素抵抗的详细机制尚未完全清楚,可能涉及三个不同层次:(1)内毒素可干扰体内糖激素之间的平衡和刺激炎症细胞因子产生引起细胞对胰岛素的敏感性降低,进而影响糖代谢。狗及大鼠给予内毒素后血浆胰岛素、胰高血糖素和儿茶酚胺水平明显升高,但胰岛素与胰高血糖素比值则降低。胰高血糖素和儿茶酚胺可以明显拮抗组织对胰岛素的敏感性。(2)外周组织(尤其是骨骼肌)对胰岛素的反应性明显降低。急性内毒素血症大鼠骨骼肌葡萄糖利用的胰岛素剂量反应曲线明显右移。胰岛素刺激肌肉对葡萄糖摄取减少,葡萄糖清除能力降低,肝糖原合成减少。而糖原分解正常。临床资料表明,Ⅱ型糖尿病并发感染时,胰岛素注射剂量往往增大,表明胰岛素抵抗加重。(3)胰岛素受体信号传递缺陷。脓毒血症大鼠肌肉胰岛素剂量效应曲线下移而胰岛素受体数目往往不变,表明存在胰岛素受体后缺陷。内毒素休克大鼠骨骼肌胰岛素受体(IR)、胰岛素受体底物-1(IRS-1)和丝裂原活化蛋白激酶(MAPK)含量不变。基础状态其磷酸化能力正常,但胰岛素刺激的磷酸化程度明显下降。内毒素血症出现胰岛素受体信号传递缺陷与多种细胞因子产生有关,特别是TNFα和NO在胰岛素抵抗中起重要作用。TNFα可激活PKC等多种丝/苏蛋白激酶阻止IR和IRS-1磷酸化或是活化特异性磷蛋白磷酸酶使IR或IRS-1去磷酸化,从而干扰胰岛素受体信号传递。NO对内毒素血症糖代谢异常和胰岛素抵抗有重要意义。NO可抑制胰岛素刺激的肝脏糖原合成作用,同时还能抑制胰岛素刺激的肌肉葡萄糖摄取。TNFα和(或)NO均能诱导GluT1表达,刺激基础状态葡萄糖摄取,但抑制胰岛素敏感的GluT4表达,从而抑制肌肉和脂肪组织胰岛素刺激的葡萄糖摄取。NO还可选择性对胰岛β-细胞损伤,通过对DNA亚硝基化和脱氨基作用引起DNA断裂或使脱氧核糖核酸还原酶R2亚基的Fe氧化,使该酶活性受抑制而影响 DNA修复,引起β-细胞凋亡。内毒素一方面可诱生胰岛β-细胞促因子(β-cell tropic factor)促进胰岛素分泌引起高胰岛素血症,另方面内毒素又可诱导居留于胰岛的单核/巨噬细胞表达iNOS。产生过量NO,损伤β-细胞胰岛素合成与分泌的功能,最终导致胰岛素抵抗和糖代谢障碍。
参数原理
2024.03.07

注射内毒素可引起高血糖和糖原耗竭?
注射内毒素可引起高血糖和糖原耗竭。许多研究发现,大鼠注射亚致死量的内毒素,肝糖原含量迅速降低;而肾上腺切除的大鼠,(每天注射0. 1mg醋酸可的松或0.5mg醋酸脱氧皮质酮),给予同样剂量的内毒素﹐肝糖原含量无显著变化,明显避免了内毒素引起的肝糖原分解作用。认为亚致死量的内毒素并不直接作用于肝脏,而是通过刺激交感神经兴奋,促进肾上腺素的释放,进而引起肝糖原的分解。实际上亚致死量内毒素能引起狗肾上腺静脉血儿茶酚胺浓度显著升高。用中等剂量的内毒素(LD30)可使血浆去甲肾上腺素升高8倍,肾上腺素升高20倍。动物经内毒素处理后﹐肾上腺髓质和交感神经系统的活性亦明显增强。因此,内毒血症早期出现高血糖为特征的糖代谢变化中,儿茶酚胺升高是重要因素。内毒素血症早期出现高血糖,随后逐渐转为低血糖,尤为内毒素休克晚期常伴有严重低血糖症。其主要原因是内毒素一方面通过促进肾上腺素分泌,加速肝糖原分解而引起肝糖原耗竭;另一方面内毒素可明显抑制肝糖原合成酶和糖异生关键酶的活性,减少肝糖原的合成与储存。内毒素休克晚期低血糖发生与糖异生作用受抑制有直接关系。动物注射LD50剂量的内毒素,发现14C-丙氨酸向葡萄糖的转变明显受阻(-40%~-50%)。由乳酸、丙氨酸、丙酮酸为底物的糖异生作用也明显减弱。用低剂量的内毒素同样抑制以乳酸、丙酮酸、丙氨酸、门冬氨酸、脯氨酸和谷氨酰胺为底物的糖异生作用。内毒素休克肝脏生成葡萄糖能力受损是造成低血糖症的直接原因。革兰阴性细菌感染或注射致死量内毒素大鼠的肝组织切片显示其糖原生成能力严重受损。肝脏葡萄糖-6磷酸酶(G-6-Pase)活性降低,而磷酸果糖激酶-1(PFK-1)活性增强3~4倍。结果使糖异生两种底物(葡萄糖-6磷酸和果糖6-磷酸)浓度降低。LPS还明显抑制丙酮酸羧化酶和磷酸烯醇式丙酮酸羧激酶(PEPCK)两种糖异生关键酶的基因表达和蛋白质含量。LPS还可抑制胰高血糖素和糖皮质激素对PEPCK的诱导表达。注射LPS大鼠,发现iNOS活性升高与PEPCK 活性受抑制明显相关。用外源NO供体SNAP(S-nitroso-N-acetyl penicillamine)与大鼠肝切片孵育,发现肝脏葡萄糖生成受抑制与NO生成有极好的相关性。NO抑制糖异生所需浓度L。
参数原理
2024.03.06

内毒素对葡萄糖代谢动力学的影响
应用[6-3H]葡萄糖和14C-乳酸示踪技术观察内毒素对葡萄糖代谢动力学的影响发现,大鼠注射LPS后15 min,血中葡萄糖出现率(Ra)和消失率(Rd)显著加快,葡萄糖代谢清除率(MCR)也显著加快。30 min后血中乳酸的Ra和Rd明显增快,血中乳酸浓度显著升高,来源于乳酸的葡萄糖更新显著增强。进一步发现骨骼肌对葡萄糖摄取增加57%,而乳酸和丙酮酸的释放分别增加217%和82%,骨骼肌的氧耗量并未改变。表明内毒素促进组织细胞对葡萄糖的摄取和非氧化性葡萄糖利用(糖酵解和碳-碳循环)增强,加快葡萄糖的清除。然而不同器官对葡萄糖的消耗明显不同。如大鼠注射内毒素(0. 1 mg/100g 体重)3h,不同组织细胞内磷酸-2-脱氧葡萄糖的水平升高分别为:肝增加4.8倍、脾和皮肤增加2.9倍、肺增加2.4倍,肠和肾增加2.1倍。表明富含单核/巨噬细胞的肝和脾对葡萄糖摄取增加最明显。而且富含单核/巨噬细胞和肝、脾组织对葡萄糖的利用显著增加与延长。内毒素引起机体葡萄糖消耗加速主要是引起组织细胞(尤其是炎症的单核/巨噬细胞)对葡萄糖的基础摄取增加,而肌肉等大量消耗葡萄糖组织对胰岛素刺激的葡萄糖摄取明显降低,葡萄糖的有氧氧化亦明显受抑制。注射LPS大鼠肝实质细胞主要表达的葡萄糖转运载体-1(GluT1)表达增加7倍。内毒素血症时,巨噬/枯否细胞摄取大量葡萄糖主要通过磷酸戊糖途径明显增强,用于生成更多的NADPH以维持细胞内还原型谷胱甘肽(G-SH)浓度,参与处理呼吸爆发产生的过氧化自由基(H2O2和O₂-)。
参数原理
2024.03.06

内毒素对血管内皮细胞膜的促凝作用
几乎所有细胞的质膜均由脂质双分子层组成,细菌也是如此。脂质双分子层是不对称的。细胞膜的外层主要由磷脂酰胆碱和含胆碱的鞘磷脂组成,而邻近胞浆侧的内层主要由氨基磷脂(包括磷脂酰丝氨酸、磷脂酰肌醇﹑磷脂酰乙醇胺等)组成。氨基磷脂由外层向内侧的主动转运是通过ATP依赖性氨基磷脂转位酶(又称反转酶,flippase)实现的。这种不对称分布的维持也是一个ATP依赖过程,这与胞内骨架活动也有一定联系。在细胞受损破坏时,氨基磷脂就会向周围暴露。带负电荷的氨基磷脂(特别是磷脂酰丝氨酸)实际上是凝血中间过程的催化表面,因为维生素依赖因子(凝血酶原、FⅦ、FⅨ、FⅩ)和FⅪ、FⅧ及FⅤ都需要装配在它的表面,才能充分发生反应。因此,输入多聚血红蛋白溶液、ABO血型不合输血﹑夜间阵发性血红蛋白尿,大剂量抗体或抗生素造成细菌破损等情况均可造成氨基磷脂出现在血液循环中,则有可能引起血管内凝血。作者等曾将塑料导管由股静脉插入直到下腔静脉,并给狒狒使用TNFα和蛋白C抗体,结果仅仅引起下腔静脉和骼静脉等处局部血栓形成;但若同时注入磷脂酰胆碱/磷脂酰丝氨酸混悬液,则出现典型的DIC表现。鉴于氨基磷脂入血与DIC发生密切相关,故从一定意义上可以认为氨基磷脂是败血症休克的一种自身毒素(autotoxin)。内毒素可以引起氨基磷脂由膜内层向膜外层翻转,其原因可能包括:细胞受损高浓度内毒素本身可以引起细胞严重损伤。细胞受损时,由于细胞内钙离子浓度升高以及ATP依赖性氨基磷脂转位酶活性降低,氨基磷脂可以发生向外层翻转现象。细胞凋亡内毒素刺激单核/巨噬细胞和枯否细胞合成并释放TNFα和IL-1β等促炎性细胞因子。这些细胞因子作用于血管内皮细胞,激活其中的酸性鞘磷脂酶(acidsphingonyelinase,ASMase);ASMase引起鞘磷脂水解而产生神经酰胺(ceramide),神经酰胺介导血管内皮细胞凋亡。ASMase基因敲除小鼠可以防止内毒素引起的细胞凋亡。以碱性成纤维细胞生长因子(bFGF)阻制神经酰胺产生,同样也可防止细胞凋亡。实验证明在细胞凋亡过程中,氨基磷脂翻转现象远较DNA裂解出现在前,从而使这些走向凋亡细胞的细胞膜变成促凝的;与此同时,这些血管内皮细胞膜上的凝血酶调制素、硫酸乙酰肝素以及TFPI都明显减少,表明抗凝特性丧失。总之,内毒素对凝血系统具有激活作用,主要是由于内毒素能诱导单核细胞和血管内皮细胞表达TF,并使血管内皮细胞膜变为促凝性质所致。
参数原理
2024.03.05
内毒素对血管内皮细胞膜的促凝作用
几乎所有细胞的质膜均由脂质双分子层组成,细菌也是如此。脂质双分子层是不对称的。细胞膜的外层主要由磷脂酰胆碱和含胆碱的鞘磷脂组成,而邻近胞浆侧的内层主要由氨基磷脂(包括磷脂酰丝氨酸、磷脂酰肌醇﹑磷脂酰乙醇胺等)组成。氨基磷脂由外层向内侧的主动转运是通过ATP依赖性氨基磷脂转位酶(又称反转酶,flippase)实现的。这种不对称分布的维持也是一个ATP依赖过程,这与胞内骨架活动也有一定联系。在细胞受损破坏时,氨基磷脂就会向周围暴露。带负电荷的氨基磷脂(特别是磷脂酰丝氨酸)实际上是凝血中间过程的催化表面,因为维生素依赖因子(凝血酶原、FⅦ、FⅨ、FⅩ)和FⅪ、FⅧ及FⅤ都需要装配在它的表面,才能充分发生反应。因此,输入多聚血红蛋白溶液、ABO血型不合输血﹑夜间阵发性血红蛋白尿,大剂量抗体或抗生素造成细菌破损等情况均可造成氨基磷脂出现在血液循环中,则有可能引起血管内凝血。作者等曾将塑料导管由股静脉插入直到下腔静脉,并给狒狒使用TNFα和蛋白C抗体,结果仅仅引起下腔静脉和骼静脉等处局部血栓形成;但若同时注入磷脂酰胆碱/磷脂酰丝氨酸混悬液,则出现典型的DIC表现。鉴于氨基磷脂入血与DIC发生密切相关,故从一定意义上可以认为氨基磷脂是败血症休克的一种自身毒素(autotoxin)。内毒素可以引起氨基磷脂由膜内层向膜外层翻转,其原因可能包括:细胞受损高浓度内毒素本身可以引起细胞严重损伤。细胞受损时,由于细胞内钙离子浓度升高以及ATP依赖性氨基磷脂转位酶活性降低,氨基磷脂可以发生向外层翻转现象。细胞凋亡内毒素刺激单核/巨噬细胞和枯否细胞合成并释放TNFα和IL-1β等促炎性细胞因子。这些细胞因子作用于血管内皮细胞,激活其中的酸性鞘磷脂酶(acidsphingonyelinase,ASMase);ASMase引起鞘磷脂水解而产生神经酰胺(ceramide),神经酰胺介导血管内皮细胞凋亡。ASMase基因敲除小鼠可以防止内毒素引起的细胞凋亡。以碱性成纤维细胞生长因子(bFGF)阻制神经酰胺产生,同样也可防止细胞凋亡。实验证明在细胞凋亡过程中,氨基磷脂翻转现象远较DNA裂解出现在前,从而使这些走向凋亡细胞的细胞膜变成促凝的;与此同时,这些血管内皮细胞膜上的凝血酶调制素、硫酸乙酰肝素以及TFPI都明显减少,表明抗凝特性丧失。总之,内毒素对凝血系统具有激活作用,主要是由于内毒素能诱导单核细胞和血管内皮细胞表达TF,并使血管内皮细胞膜变为促凝性质所致。
参数原理
2024.03.05
内毒素对能量代谢的作用
严重创伤感染、脓毒血症(sepsis)可引起机体包括体温升高、体重降低和能量耗损的高(超)代谢反应。其主要致病因素是由细菌内毒素,即脂多糖(Lipopolysaccharide,LPS)刺激体内多种髓系细胞(单核/巨噬细胞)和(或)非髓系细胞(内皮细胞/平滑肌细胞)活化,产生一系列致炎介质(proinflammatory mediator),如TNF、IL-1、IL-6、NO和PGE,等参与机体急性相反应(acute phase responses,APR)的结果。内毒素对细胞代谢的作用是多方面的,对机体具有双重意义,既有利又有弊。严重感染或脓毒血症时,机体出现进行性能量代谢障碍,组织摄取氧能力显著降低,结果组织细胞缺氧而静脉血氧浓度升高;细胞内氧利用障碍,导致循环血中乳酸含量增多。给大鼠静脉注射LPS(3mg/kg体重)4h,肝线粒体超微结构发生明显损伤,第3态呼吸功能明显受抑制。LPS及炎症细胞因子诱导NO生成增多,可抑制平滑肌细胞、肝实质细胞、巨噬细胞和星形胶质细胞以及心肌细胞线粒体电子传递呼吸链功能,使氧化磷酸化中断。NO可直接损伤呼吸链中的铁-硫中心,又可对呼吸链的复合物Ⅰ、复合物Ⅱ产生不可逆的抑制作用。NO还可与细胞色素氧化酶竞争结合O2,使ATP生成受抑制,同时引起呼吸链产生氧自由基(H2O2、O₂-)增多。促使NO进一步与O₂-形成过氧化硝基化合物(ONOO-)。这种相对稳定的强氧化剂除了对细胞造成强烈的损害作用,并可反馈损伤电子传递链和线粒体功能。ONOO-还可抑制超氧物歧化酶(SOD)活性,造成更多的O₂-堆积,从而形成一种自身催化的能量代谢障碍的恶性循环。ONOO-除了抑制巨噬细胞、血管内皮细胞和心肌细胞的呼吸功能,同时还激活多聚(ADP-核糖)转移酶(PARS),催化ADP-核糖化反应,消耗细胞内NAD+。NO抑制三羧酸循环中的顺-乌头酸酶和苹果酸脱氢酶活性,引起三羧酸循环障碍。最近证明LPS促进多种组织线粒体解偶联蛋白(uncoupling protein,UCP)基因表达,取消线粒体内膜的跨膜质子梯度。造成细胞能量代谢障碍,ATP生成减少,最终导致细胞死亡。
参数原理
2024.03.05

内毒素与组织因子途径的关系
由于接触激活不参与生理性止血的凝血过程和绝大多数病理条件下血栓形成,组织因子途径成为体内凝血过程的启动环节。因此内毒素激活凝血系统的作用,也集中在探讨它与组织因子途径的关系。动物实验证明,在给予内毒素或大肠杆菌之前,事先给动物使用抗TF单克隆抗体或重组突变TF(sTFAA,具有与FⅦa结合能力但无辅因子活性)或抗FⅦ/FⅦa单克隆抗体,或重组失活FⅦa(rFⅦai:具有与TF结合能力但已丧失催化活性),结果虽对动物休克或成人呼吸窘迫综合征的发生没有明显影响,但却有效地阻断了DIC的发生。这些事实表明内毒素引起DIC的确是由组织因子途径启动的。动物实验发现事先以免疫方法耗竭TFPI,再注入对正常动物不致引起DIC的内毒素剂量或凝血活酶剂量便可造成典型急性DIC;反之,若事先给予大剂量TFPI,则可明显地抑制致死剂量内毒素或大肠杆菌引起的DIC。由此可见,内毒素是否引起 DIC,主要取决于TF/TFPI平衡的倾向。我室与Taylor等实验室都观察到注入内毒素后,血浆中TF非常显著升高;但对TFPI的报道并不尽同,然而TF/TFPI比值明显增大是一致的。对正常人注入小剂量内毒素同样地观察到血浆中TF升高,TF/TFPI比值显著增大。所有这些资料都充分支持内毒素引起DIC是通过组织因子途径启动的观点。应当指出,内毒素虽不能通过血小板表达TF,但由于血小板活化因子和凝血酶等诱导血小板活化,活化的血小板膜表面的P-选择素可以上调单核细胞表达TF。
参数原理
2024.03.01
内毒素究竟诱导哪些细胞表达TF?
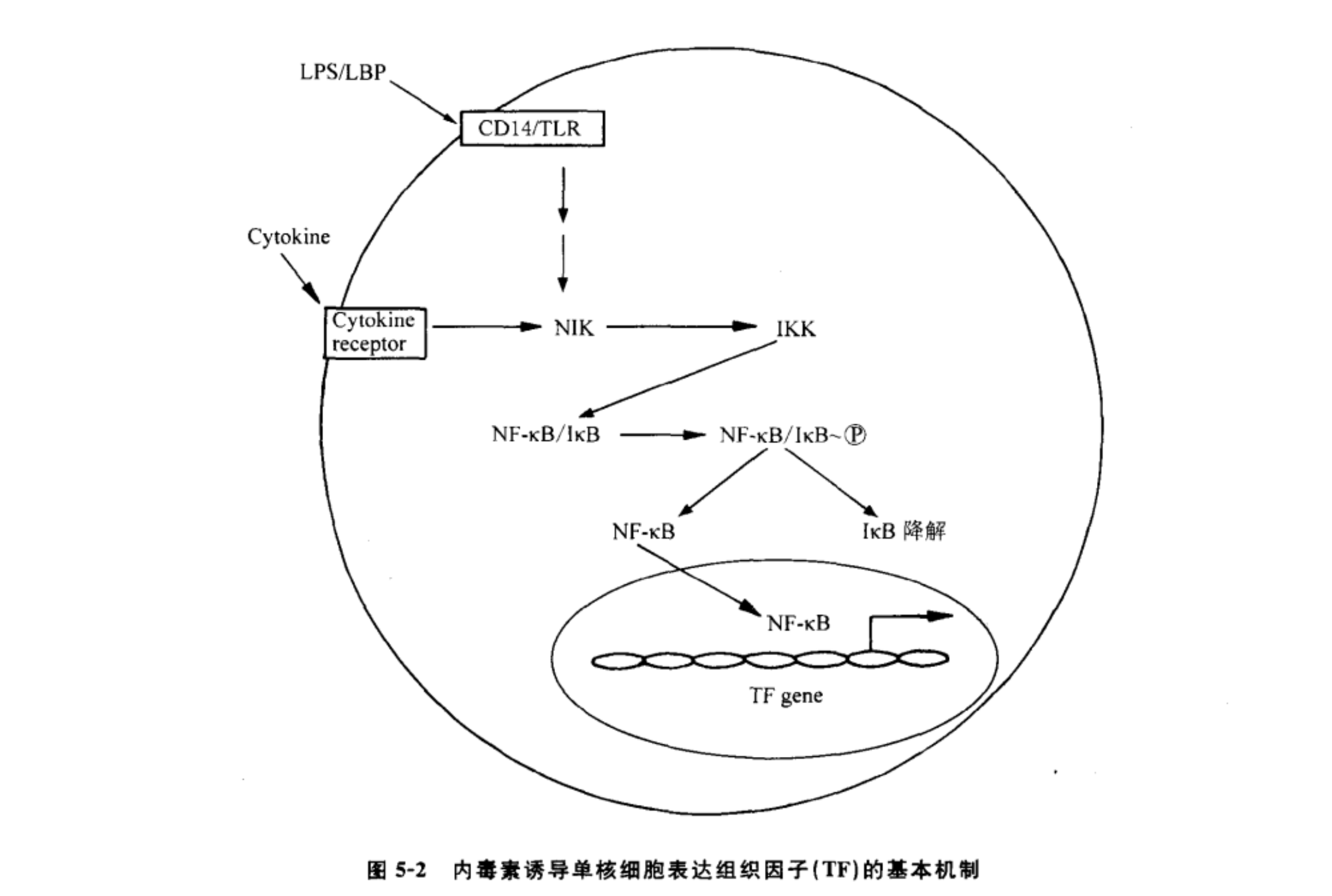
既然内毒素可以引起组织因子途径启动,那么内毒素究竟诱导哪些细胞表达TF引起血管内凝血呢?根据TF合成情况,体内细胞可分为三类:①固有性表达TF细胞,如脑部星状胶质细胞,胎盘滋养层细胞等TF固有性表达极为丰富;其他大部分组织细胞如血管平滑肌细胞﹑成纤维细胞等也有TF的固有性表达,尤以血管外膜、器官包膜、皮肤表皮及黏膜等处相当丰富,它们犹如袖套状围绕血管与被套状包绕器官,在体内构成一个分布广泛的止血屏障。②不表达TF的细胞,如淋巴细胞、血小板、红细胞等。③诱生性表达TF的细胞,如血管内皮细胞、单核细胞、巨噬细胞等。一般认为固有性TF表达是以嵌膜蛋白形成存在,它主要与生理性止血有关;诱生性表达可以嵌膜蛋白或小囊泡形式存在,它主要与病理条件下血栓形成有关。现在普遍认为内毒素引起血管内凝血,这是由于它能诱导单核细胞与血管内皮细胞大量表达TF所致。以往所谓组织凝血活酶,实质上包括TF与磷脂两部分,曾误认为TF为脂蛋白。现已明确TF是一个跨膜糖蛋白,又称CD142。成熟的TF为一单链跨膜蛋白,由263个氨基酸组成。按氨基酸组成计算分子量为2.96万,按SDS-PAGE推算为4.2万~4.7万,可能与膜外部分糖基化有关。膜外区由氨基酸残基(AA)1~219组成,构成二个β折叠片呈V形排列。跨膜区由AA220~242组成。胞浆区由AA243~263组成。胞浆区通过硫酯键与盐键,使TF锚定在胞膜上,胞浆尾在信号传递中起着重要作用。TF在功能上既是FⅦ与FⅦa的受体,又是FⅦa激活FⅩ和FⅨ的必需辅因子。FⅦ只有与TF结合才有可能被激活成为FⅦa ;FⅦa也只要与TF结合才能有效地激活FⅩ和FⅨ。这是由于在磷脂和Ca2+存在的条件下,TF与FⅦ喱或FⅦa以1∶1方式结合,一旦结合立即引起FⅦ和FⅦa发生构象变化,这种构象改变有利于FⅦ的活化及FⅦa充分发挥催化活性的缘故。新近实验表明TF是一个真正的受体,FⅦa 作为配体,FⅦa 与TF结合立即引起胞内信号转导。从已有资料看来,它主要是通过丝裂原活化蛋白激酶途径,上调多个基因表达(如Poly(A)聚合酶、尿激酶受体、TF等)。TF基因剔除可以导致小鼠在胚胎期死亡。这主要与卵黄囊血管生成和凝血障碍有关。人的TF基因位于染色体1P21-22,全长12.4kb,含6个外显子,5个内含子。转录起始点距TATA盒26 bp。在TATA盒上游含有多个转录因子结合位点,包括SP-1、EGR-1、AP-1,AP-2和NF-B。其中在单核细胞和血管内皮中涉及内毒素及细胞因子反应的转录因子最重要的是NF-κB。那么内毒素究竟是如何启动TF表达的?目前多数学者认为内毒素首先与血浆中脂多糖结合蛋白结合,然后以复合物形式与CD14结合。CD14一般称为内毒素受体。实际上,单核/巨噬细胞表面的确含有较多的CD14,内毒素-脂多糖结合蛋白复合物(LPS-LBP)可以直接与CD14结合。但血管内皮细胞表面并无CD14,而是内毒素-脂多糖结合蛋白复合物与血浆中可溶性CD14结合,其后这个大复合物再与血管内皮细胞结合。由于CD14并不是一个跨膜蛋白,它仅以糖磷脂酰肌醇形式锚定在细胞膜表面,本身不可能将内毒素信号转导进人细胞内,肯定还需通过其他跨膜蛋白。新近发现单核/巨噬细胞和血管内皮细胞存在钟样受体(toll-like recepor,TLR),它们是跨膜蛋白。其中TLR2主要识别并结合革兰阳性细菌壁上的肽聚糖和脂蛋白,TLR4主要识别并结合内毒素-CD14复合物。从信号转导角度考虑,可以认为TLR4才是内毒素的真正受体。内毒素-CD14复合物与TLR4结合后,相继通过髓样细胞分化因子88(My88 )→白介素-1受体相关激酶(IRAK)→肿瘤坏死因子受体相关因子(TRAF)6-NF-κB诱导激酶(NIK)→抑制性NF-κB激酶(IKK)→NF-κB/Ⅰ-κB中的Ⅰ-κB磷酸化→NF-κB与Ⅰ-κB分离。于是NF-κB穿过核孔进人核内,NF-κB(实际上是cRel和P65源二聚体)与TF基因DNA的结合部位结合,从而启动TF基因的转录(图5-2)。其后TF mRNA相继通过翻译与随后蛋白修饰,最后TF蛋白以跨膜形式存在或以小囊泡形式释放。应当强调指出:NF-κB是指由Rel蛋白家族成员以同源或异源二聚体组成的一组核转录因子。内毒素也引起其他基因转录,从而促进许多蛋白表达,诸如急性期蛋白(如C-反应蛋白、血管紧张素原、α1-酸性糖蛋白、补体C3等)、细胞因子(如TNFα、IL-1β、IL1、IL-8等)黏附分子(如VCAM-1、ELAM1、ICAM等)以及单核细胞趋化蛋白(MCP)-1和纤溶醇原活化素抑制物(PAI)-1等等。新近发现内毒素还可通过NF-κB途径促进血管内皮细胞TLR4和TLR2的表达。此外,内毒素诱导表达的细胞因子(TNFα、IL-6、IL-1等)又可通过NF-κB途径促进血管内皮细胞TF表达。因此,在内毒素引起TF表达过程中,可能通过多重正反馈方式而得到放大效应。还应当看到:注入内毒素或大肠杆菌使微血管体系中单核细胞增加,在其TF表达明显增加的同时,血管内皮细胞表面TFPI和凝血酶调制素减少。这种促凝和抗凝活性之间的失衡,则可能促进DIC的发生。
参数原理
2024.03.01

内毒素引起接触激活的生物学意义
动物实验证明内毒素注入可引起接触激活,表现为血浆中FⅫ、PK与HMWK消耗性降低,激肽释放酶-C1抑制物复合物以及激肽释放酶-α2-巨球蛋白复合物升高,并且HMWK降低程度与存活时间呈反变。临床观察革兰阴性败血症患者血浆中接触激活的3个因子亦明显减少,同样HMWK水平变化与预后密切相关。曾经认为这与内毒素的组分脂质A激活FⅫ,启动内在凝血途径,从而导致弥散性血管内凝血(dissminated intravascular coagulation,DIC)有关。随后,在狒狒大肠杆菌输注诱发败血症的模型中,事先以抗FⅫ的单克隆抗体阻断接触激活,结果有效地阻遏低血压的发生,但仍然发生DIC。若动物事先被给予激肽释放酶抑制物,而后再注入内毒素,同样依然发生DIC,但可防止成人呼吸窘迫综合征的发生。因此内毒素血症时引起的接触激活主要涉及休克和全身性炎症反应综合征发生,并不参与DIC发生。在人的实验性内毒素血症中,可以观察FⅫ激活,但未发现接触激活的变化。因此接触激活并不参与内毒素引起的DIC发生,这已基本成为共识。至于内毒素通过接触激活引起低血压,一般认为主要与缓激肽大量形成有关。实验表明带负电荷的硫酸右旋糖酐激活接触系统,可以使动物产生低血压,β2受体阻滞剂(HOE140)能够显著阻断低血压反应,但β受体阻滞剂并无此作用。由此可见,内毒素引起的低血压,可能是缓激肽通过β受体实现的。由于缓激肽刺激血管内皮细胞释放PGI2和NO,二者均可引起阻力血管舒张,并使血管通透性增高,一般认为这就是内毒素通过接触激活引起低血压的基本原因。此外,内毒素通过接触激活还可激活补体系统,其中C5a是一个较强的致炎物质,它可使血管通透性明显增高,这也与内毒素休克的发展有关。
参数原理
2024.02.28

机体对内毒素的致热耐受现象研究进展
机体对内毒素的致热耐受现象已有较多研究。发生耐受机体的单核细胞CD14和CD18表达并无明显异常,LPS与CD14的亲和力也无改变,但产生的内源性致热原包括IL-1,IFN,TNFα,IL-6明显减少。进一步研究发现,耐受机体的单核细胞并非因为在LPS反复刺激下发生了功能耗竭而失去反应能力,因为此时IL-10,TNF Ⅱ型受体及NF-κB的P50等的表达均增强。一般认为,在细胞水平上,发热耐受的机制可能包括:①反复注射LPS可刺激机体产生中和抗体,脂蛋白等,加速LPS的清除。②虽然单核细胞的CD14和CD18表达正常,但LPS的生物信号传入发生了变化。表现在正常情况下LPS激活产致热原细胞,诱导胞浆内蛋白磷酸化,使NF-κB抑制因子IκB从复合体上脱离,P50-P65-κB异二聚体进入核内,与编码基因上游启动子的相应序列结合,启动细胞因子基因表达内源性致热原。而发生耐受时,NF-κB的P50表达大大增加,入核的不是P50-P65-κB异二聚体而是P50-P50-κB同源二聚体。P50-P50-κB无激活细胞因子基因表达的作用,反而对多种内源性致热原基因的表达产生抑制作用。③IL-10是TNFα的内源性拮抗物,耐受细胞IL-10表达增强能抑制促炎性细胞因子的生成并拮抗其生物活性。④耐受细胞的糖皮质激素受体上调,对促炎细胞因子的自身激活级联反应有阻断作用。⑤由于可溶性TNFⅡ型受体增加,结合与灭活了循环的TNFα,降低了其致热效应。由内毒素诱导产生的内源性致热原特别是IL-1不存在致热耐受性,给动物反复注射均能保持相似的发热反应,早年常用此作为鉴别是否有内毒素污染的简易方法。目前已有多种内毒素定性和定量方法在实验研究和临床检验中广泛使用,但作为热原检定的兔法,仍然是最经典和最广为接受的方法,在各国药典中保留其权威地位。
参数原理
2024.02.28

内毒素发热的体温调节机制
一、发热时体温正负调节学说内毒素发热的体温调节机制涉及到中枢神经系统多个部位的共同作用。李楚杰根据国内外研究积累的丰富资料提出了发热体温正负调节学说,该学说认为,发热时的中枢体温调节机制至少应当由两个部分组成,一是负责正调节的中枢机制,其神经解剖定位有POAH、OVLT等区域;另一方面是发热的体温负调节中枢机制,其神经解剖定位包括 VSA,MAN等。致热信号通过某些途径传入中枢后启动中枢体温正负调节机制,一方面通过升温介质和神经整合作用使体温上升(正调节),另一方面又通过内源性解热物质的作用限制体温升高(负调节),这两方面的整合过程及其相互作用的结果决定了调定点上移的水平,发热的幅度和时程,传统上把体温调节中枢局限于POAH的观点应予以修正。二、其他发热信息的传导机制在内毒素双相热动物实验中使用内源性致热原拮抗剂,如 TNF结合蛋白(TNF-binding protein)或IL-1受体拮抗物(IL-1receptor antagonist)只能减弱双相热第2热相的发热反应,第1热相无明显变化,提示内毒素尚有其他早期的发热信息传导机制。有报道迷走神经的中枢传导作用与腹腔免疫炎症反应有密切的关系。门静脉灌注IL-1可见迷走神经肝支传入冲动发放加强。切除迷走神经肝支可部分阻断腹腔注射LPS引起的发热。由于肝脏是内毒素进入机体后的主要靶器官,肝迷走神经节旁神经上有IL-1受体存在,肝脏Kupffer细胞又是机体产生这类细胞因子的主要细胞,是否存在内毒素作用下肝脏产生的化学信号,例如IL-1等激活迷走神经传入神经从而将发热信号向中枢传递的机制,有待深入研究。值得注意的是,迷走神经切除术涉及到机体的许多功能改变,另外手术方式,术后恢复及功能代偿等均对实验结果产生影响,目前有限的资料可见某些相互矛盾之处,对此尚难作出明确判断。
参数原理
2024.02.27
内毒素中致热原细胞是如何进行信息传递的?
一、CD14非依赖性途径较大剂量的LPS可通过CD14非依赖性途径启动内源性致热原基因的表达,其确切的细胞信息传导通路目前尚不清楚。根据Ulevitch提出的多构件受体(multicomponent receptor)模型,脂质A是LPS分子负责向靶体传递生物信息的重要结构。当LPS-LBP复合物与CD14结合后,脂质A可直接与信号传递蛋白交连。由于CD14的存在及其相互作用,此时的结合亲和力与信号转导能力很高。当无CD14参与的情况下,脂质A仍可与信号传递蛋白结合只是亲和力与信号转导能力都大大降低。在可溶性CD14的参与下,某些不表达CD14的细胞如内皮细胞可在LPS作用下表达TNFα等细胞因子。CD11/CD18也能作为LPS受体参与单核巨噬细胞的活化与细胞信息传递。有证据显示,作用于单核细胞上的LPS并非仅停留在细胞膜上,用胶体金免疫电镜可观察到LPS在CD14存在的条件下,可循至少两种不同途径进入细胞。至于进入细胞内的LPS是被代谢清除或是传递生物信息尚待深人研究。二、下丘脑视前区的作用通常公认的体温调节中枢位于下丘脑(hypothalamus),主要是前下丘脑视前区(preoptic area of the anterior hypothala-mus,POAH)。该区含有温度敏感神经元,不仅对该区或附近的血温有感受性,而且对来自外周和深部的温度信息起整合作用,损伤该区可导致正常体温调节障碍。早年已发现在POAH注射微量的内源性致热原可引起明显的发热反应,所需内源性致热原的剂量不到静脉注射引起发热所需剂量的1%,微量内源性致热原注射到其他脑区,或不引起发热,或发热潜伏期较长、效应较弱,提示POAH是内源性致热原作用的敏感部位。静脉注射内毒素引起发热时下丘脑区的发热介质前列腺素E(prostaglandin E,PGE)、环磷酸腺苷(cyclic adenosine monophosphate,cAMP)等的含量明显增多,向POAH注入PGE2、二丁酰环磷酸腺苷(db-cAMP)可引起明显的发热反应,微量的环氧酶抑制剂(抑制PGE的合成)注入POAH显著抑制静脉注射LPS引起的发热反应。因此,传统上一直公认POAH是参与发热体温调节的主要中枢部位。位于第3脑室壁视上隐窝处的血管终板器(organum vasculosumlaminae terminalis,OVLT)是血脑屏障的特殊结构部位,紧靠前下丘脑视前区。该处存在有孔毛细血管,对大分子物质有较高的通透性,外周循环的一些肽类物质如血管紧张素等可从此进入中枢。近来发现OVLT区域也有对温度敏感的神经元分布,有资料说明OVLT区也是发热时体温调节的重要中枢结构。三、腹中隔和中杏仁核的热限作用内毒素发热时体温并非无限制地上升,在体温调节功能正常的情况下,内毒素达到一定剂量后,发热反应不再随内毒素剂量的加大继续升高,而是稳定在一个高水平,形成“热限”。研究表明,致热信号传入中枢后,除了启动中枢升温机制使体温上升外,同时也通过内源性解热物质,如精氨酸血管加压素(arginine vasopressin,AVP)、α-黑素细胞刺激素(α-melanocytc stimulating hormone,a-MSH)等的作用限制体温升高。然而,这些内源性解热物质发挥限热或抑热作用的主要中枢部位并不是下丘脑,而是腹中隔( ventral septal area,VSA)和中杏仁核(medial amygdaloid nucleus,MAN),其限制体温升高的通路可能是终纹床核(bednucleus of stria terminalis,BST)-腹中隔神经通路。
参数原理
2024.02.27

内源性致热原与细菌内毒素的区别简述
内源性致热原是引起调节性体温升高的基本信息分子。目前已知的内源性致热原均属于促炎性细胞因子(proinflammatory cytokines),例如白细胞介素-1 (interleukin 1,IL-1),干扰素(interferon,IFN),IL-1),肿瘤坏死因子(tumor necrosis factor,TNF),白细胞介素-6(interleukin 6,IL-6)等,对机体除了等,对机体的产生致热效应外还介导与发热又密切关系的急性期反应,包括中性粒细胞数量的变化,低铁血症、低锌血症,高铜血症,多种急性期反应蛋白合成增多等。此外,这些细胞因子又对免疫系统发挥重要的调节作用。早在1948年,Beeson等从家兔腹腔渗出白细胞培养上清液中提取出一种不耐热、无致热耐受性、能被蛋白水解酶灭活的致热物质,与常称为致热原(pyrogen)的发热激活物细菌内毒素有本质的不同,取名为白细胞致热原(leukocytic pyrogen)。现已明确,这种内源性致热信息分子主要由激活的单核巨噬细胞产生,为蛋白质成分,加热 70℃、20min可丧失致热活性,一般剂量静脉注射引起单相热,其发热潜伏期明显短于细菌内毒素,且无致热耐受现象,注射于已对细菌内毒素产生耐受的家兔仍能引起发热反应,在部分种属的动物中有交叉致热性。
参数原理
2024.02.20

发热激活物与细菌内毒素的关系阐述
内毒素在人类的生活空间广泛存在,污染内毒素的情况极易发生,多种感染性疾病的发热反应都与细菌内毒素有关。虽然将微量内毒素注入下丘脑可引起发热,至今尚无证据表明内毒素能通过血脑屏障直接作用于体温调节中枢。相反,内毒素等能引发机体发热反应的物质,包括多种细菌及其毒素和代谢产物、病毒、真菌、螺旋体等,它们的主要致热机制是激活机体的产致热原细胞,主要是单核巨噬细胞产生内源性致热原(endogenous pyrogen),通过后者引起机体调节性体温升高,笼统称之为致热原容易造成概念上的混淆,文献上常常对内毒素等物质使用外源性致热原(exogenous pyrogen)一词加以区别。严格来说,这些物质应称之为发热激活物(activators),即能激活机体的产致热原细胞产生内源性致热原的物质。存在于革兰阴性细菌细胞壁上,由O-特异性多糖侧链,核心多糖和脂质A(lipid A)组成的脂多糖(lipopolysaccharide,LPS)是细菌内毒素的主要成分,是最有代表性的发热激活物,脂质A是决定LPS致热性的主要化学结构。根据目前公认的看法,细菌内毒素进入机体后,作用于产致热原细胞(主要是单核巨噬细胞)产生内源性致热原,内源性致热原是各种发热激活物引起发热反应的基本信息分子,它们以不同的途径将发热信号传入体温调节中枢,使控制中心温度的体温调定点上移,通过传出途径调节效应器对全身产热和散热作出反应,直至深部体温对应于体温调定点在较高水平上出现新的平衡,是为发热(fever)。机体在此过程中并无体温调节能力的不足或障碍,这与中暑等体温调节失控或调节障碍,又称过热(hyperthemia)的病理过程有着本质的区别。多年来,许多学者认为多种发热激活物包括内毒素都是致热原,发热是它们直接作用于机体体温调节机构的结果。在这方面,前苏联学者提出的“反射始动机制假说”可作为代表。他们认为外致热原(指致病微生物及其毒素等)和内致热原(指病灶产物)首先作用于病灶组织的内感受器,反射性地引起体温调节中枢兴奋,加强调节而引起发热。致热原作用于不同部位引起不同的发热反应是由于各部位的感受器不同所决定的。我国学者李楚杰用大量的实验事实证明该假说并不成立。首先它把外来致病因子都当作致热原是不恰当的,许多致病因子本身并无致热性,它们能引起发热的根本原因在于激活产致热原细胞产生内源性致热原。用细菌内毒素做发热动物实验表明,所谓反射始动机制并不存在。例如把细菌内毒素注人去神经的兔耳皮下,所引起的发热与注射同量细菌内毒素于神经支配完整的兔耳皮下时所引起的发热相比并无差别。至今尚无证据显示不同病灶存在对温度调节反应不同的内感受器。相反局部炎症灶产生的内源性致热原只有吸收入血才能发挥作用。在家兔背部造成无菌性炎症肉芽囊,囊内渗出液存在大量内源性致热原。在开始阶段囊内渗出液可被吸收入血,可见炎症引起明显的发热反应。但到6~9d,囊壁纤维化完成后,囊内渗出液中的内源性致热原无法进入血流,发热也随之消退。此时从囊中取出渗出液制成无细胞滤液(内含大量内源性致热原)给同一动物静脉注射,可引起明显发热。这一实验结果表明,在炎症肉芽囊的发展过程中,发热的消退是炎囊屏障阻碍了内源性致热原吸收入血的结果。
参数原理
2024.02.20

内毒素的致热性特点有哪些?
细菌内毒素最常见的生物活性是它的致热性,以寒战发热为主要临床表现的输液反应,多数是输液制剂污染了细菌内毒素所致。内毒素的致热性有如下特点:①存在种系差别,哺乳类中,人、牛和家兔等对内毒素较为敏感,而大鼠小鼠等则不太敏感。人可能是所有动物对内毒素最敏感的,按2ng/kg剂量静脉注射可使体温上升达2℃。新西兰白兔对内毒素的致热性非常敏感,是各国药典规范使用的热原检定标准化实验动物。②在一定范围内,发热高度与时程呈剂量依赖性。家兔对内毒素的发热反应,小剂量引起单相热,较大剂量引起双相热,第一热峰出现在注后90min左右,第二热峰于3h左右出现,形成极有特征的热型。若给予超大剂量,动物出现感染性休克的典型症状,此时体温不升反降。③内毒素发热有较明显的潜伏期(指从注射内毒素起到体温升至高于基础体温0.3℃时所需要的时间)。家兔的潜伏期依静脉注射剂量的不同常在15~30min,而人静脉注射2ng/kg剂量时发热潜伏期可长达90min。④容易产生耐受性。家兔若连续接受内毒素注射,其发热反应逐日减弱,首先是双相热第2峰消失,第1峰也逐渐消退,相同剂量的内毒素失去了原有的发热反应。若动物停止注射内毒素,3周后可恢复正常发热反应。⑤致热活性不易清除,一般的灭菌方法(如常规高压灭菌)不能将内毒素彻底破坏,需160℃干热2h以上才能将其灭活。
参数原理
2024.02.19

机体防御系统对内毒素代谢的影响
机体对内毒素作出应答时,一方面触发炎症反应,另一方面产生或激活能够清除、灭活内毒素的物质,其中包括抗О特异性多糖抗体和抗核心多糖抗体。两种抗体与内毒素结合后,再与细胞膜上的Fc受体结合,介导内毒素内源化,从而使内毒素在胞内灭活。抗内毒素抗体也能干扰内毒素与LBP结合,从面阻止LBP将内毒素转运合CD14。令入遗憾的是,抗某一菌株内毒素的抗体,对同一种属其他菌株的内毒素无中和作用;内毒素血症,革兰阴性杆菌菌血症动物模型显示,抗内毒素抗体具有保护性作用,但两种抗体在入体内却未显示出明显的保护怍用。总之,机体防御系统能有效防止外源性内毒素入侵及内源性内毒素移位,即使在因革兰阴性杆菌轻微感染而释放少量内毒素或少量肠源性内毒素发生移位时,机体防御系统也能有效清除、灭活内毒素。但是,当出现大量外源性内毒素释放或大量肠源性内毒素移位时,机体防御系统不但不能有效地清除、灭活内毒素,反而在内毒素的作用下,其自身的防御功能受到了明显抑制,与此同时,天然免疫细胞由防御性细胞转化为效应性细胞,合成、释放大量的炎性介质,从而导致脓毒症发生,严重者可以并发脓毒性休克甚至导致病人死亡。由此可见,机体防御系统对内毒素代谢的影响是有限的,若想有效地抑制内毒素的生物学活性,必须寻找有效的内毒素拮抗措施,但令人遗憾的是,目前我们对内毒素在肝脏内代谢的具体过程等重要内容还知之甚少,严重阻碍了内毒素拮抗措施的研究。虽然迄今为止尚无特效的内毒素拮抗剂应用于临床,但我们深信,随着对机体防御系统与内毒素代谢等相关内容研究的不断深入,人类一定会找到有效的内毒素拮抗措施。
参数原理
2024.02.19

AOAH如何水解内毒素?
AOAH是重要的内毒素解毒物质,是白细胞产生的一种分子量为5.2万~6.0万的糖蛋白,由5.0万的大亚基和1.4万~2.0万的小亚基组成,大、小亚基之间由二硫键共价连接。AOAH的大、小亚基由单一的mRNA编码,在翻译时,首先形成的是7.0万的单链AOAH前体多肽,并且在链内形成二硫键。AOAH前体存在两种代谢方式:其一,AOAH前体进入溶酶体并被水解成大,小亚基,而二硫健仍被保留并连接于大、小亚基之间,从而形成成熟的AOAH:其二,未经处理的AOAH前体直接分泌到细胞外。AOAH作为一种水解内毒素的脂肪酶,能够选择性地水解脂质A酰氧酰基基团上的次级酰基链。在水解内毒素时,AOAH的大、小亚基均起着重要作用,二者缺一不可。大亚基具有内毒素水解位点,同许多脂肪酶一样,其活性位点具有特征性的Gly-X-Ser-x-Gly 共同氨基酸序列,其中的丝氨酸在维持酶的活性方面起着关键性作用,当该丝氢酸被去除后AOAH水解内毒素的活性下降了99%以上。虽然大亚基在AOAH水解内毒素时起着主要作用,但仅有大亚基面没有小亚基,AOAH水解内毒素的活性也会大大下降。小变基的作用主要表现在以下几个方面:①AOAH前体中的小亚基区,能够促进AOAH前体进入溶酶体并被水解:②有助于维持AOAH的结构稳定性:③能够增强AOAH对内毒素的识别;④增加脂质A的溶解性,从而有利于大亚基与脂质A之间发生相互作用。在组织器官发生炎症时,AOAH可以在局部浸润的单个核细胞(包括单核细胞、巨噬细胞、淋巴细胞等)及多形核白细胞中表达,但单个核细胞的内毒素去酰基化活性明显高于多形核白细胞。单个核细胞内的AOAH去酰基化作用迅速而广泛,该作用是CD14依赖性的LBP与内毒素结合后,将内毒素转运给单个核细胞的CD14:而BPI、脂蛋白等则能抑制LBP向单个核细胞转运内毒素。AOAH也可以释放至胞外炎性渗液中,并且能够降解没有被单个核细胞等摄取的内毒素,该过程是CD14非依赖性的。由于渗出液中的AOAH水解内毒素的活性仅为胞内AOAH的1/10~1/20,因此,该过程速度较为缓慢。除了能够直接灭活内毒素外,AOAH水解内毒素后形成的去酰基化内毒素,也参与了AOAH对内毒素的解毒机制,该物质能够在细胞外积聚并抑制内毒素诱导的炎症反应。但由于局部浸润的白细胞产生,分泌的AOAH 数量有限,因而AOAH对内毒素的解毒作用也是有限的。
参数原理
2024.02.18

Cathelicidin、乳铁蛋白、防御素在内毒素中有哪些作用?
阳离子抗菌肽是天然免疫系统进化过程中的一个古老的成分﹐包括 BPI、Cathelicidin、乳铁蛋白、防御素等多种物质,既具有抗革兰阴性杆菌活性,又具有结合内毒素的能力。阳离子抗菌肽主要出现在哺乳动物的皮肤、消化道、呼吸道等经常与病原体接触的部位,在血液、分泌液及中性粒细胞颗粒中固有表达或受病原体及其产物诱导而表达。下面介绍Cathelicidin、乳铁蛋白、防御素的作用。CathelicidinCathelicidin是一个既具抗菌活性又具有结合内毒素能力的阳离子多肽家族,hCAP-18是该家族在入体内的唯一成员,存在于中性粒细胞特异性颗粒中以及炎性皮肤疾病的鳞状上皮细胞、角化细胞内,在血浆中也有较高的浓度,hCAP-18的短基端高度阳离子化并具疏水性,因而具有抗菌及结合内毒素的能力,这同时也决定了hCAP-18能同入体细胞膜相结合,从而具有细胞毒性。血浆中的脂蛋白能够与hCAP-18 的羧基端结合。有效抑制了hCAP-18的细胞毒作用。其他的阳离子抗菌肽与hCAP-18相比·虽然氨基酸序列明显不同,但都具有疏水性且富含阳离子,因而也能与靶细胞的细胞膜结合并插入膜内,产生细胞毒性。通过对hCAP-18的研究,结合其他阳离子抗菌肽也能与脂蛋白结合的事实,可以推测,脂蛋白与抗菌肽或其前体蛋白结合,既有利于阻止抗菌肽的细胞毒作用,又有利于这些抗菌肽在血浆中保持较高水平。乳铁蛋白乳铁蛋白是机体防御系统中的一个组成部分,是一种高度阳离子化的单体糖蛋白,等电点为8.4~9.0.分子量为7.6万~8.0万,其氨基端因富含精氦酸而带正电荷,因而具有杀灭革兰阴性杆菌的活性及结合内毒素的能力。乳铁蛋白在许多黏膜表面及乳汁中具有较高浓度(1~10mg/ml)。中性粒细胞内的乳铁蛋白是细胞特异性颗粒中的一种主要成分,可以与被中性粒细胞吞噬的革兰阴性杆菌及内毒素相互作用,也可释放至胞外面发挥其生物学效应。肠黏膜表面的乳铁蛋白有利于防止肠道细菌及内毒素移位。防御素人体内的防御素可分为α-防御素和β-防御素两种不同的亚型,其中α-防御素、β-防御素分别具有6种、2种不同的分子。α-防御素主要分布于中性粒细胞及肠腺潘氏细胞(Paneth eell),而β-防御素则主要分布于皮肤及黏膜的上皮细胞。生理条件下,上皮细胞仅有低水平的,防御素表达,病理状态下,内毒素、细菌及致炎细胞因子等可强烈诱导β-防御素表达。
参数原理
2024.02.18

肝脏能有效清除内毒素吗?
生理情况下,虽有少量内毒素不断向肠外移位,经门静脉进人肝内,但并不引起内毒素血症;在轻度革兰阴性杆菌感染时,虽然细菌不断向组织或血液中释放内毒素,但并不引起强烈的炎症反应,以上情况均有赖于机体内存在有效的内毒素清除与解毒机制。肝脏是清除内毒素的主要器官﹐脾﹑肠也是清除内毒素的重要器官;机体内的脂蛋白、阳离子抗菌肽(cationic antimicrobial peptides,CAP)、酰氧酰基水解酶(acyloxyacyl hydro-lase,AOAH)以及抗内毒素抗体是重要的内毒素解毒物质。肝脏对血液中的内毒素的清除功能,主要是通过库普弗细胞、肝细胞对内毒素的内吞作用而实现的,但具体代谢过程并不完全清楚。介导库普弗细胞吞噬内毒素的可能是清道夫受体,也可能是分子量11.9万和8.3万的蛋白质;介导肝细胞吞噬内毒素的结构可能是其凝集素样受体。内毒素与肝细胞血窦面细胞膜上的受体1:1结合后,被肝细胞以内吞的方式摄入细胞内,以微管依赖的囊泡运输方式,穿过肝细胞,运至肝细胞胆小管面,以胞吐方式排入胆小管中,再经胆道系统排至肠腔内。
参数原理
2024.01.30

几种重要细胞对内毒素的识别
一、巨噬细胞巨噬细胞除了能够在细胞表面表达CD14、TLRs以及清道夫受体等内毒素相关受体外,胞浆内也能表达蛋白质Nod1。CD14、TLRs是介导内毒素激活巨噬细胞的重要受体;清道夫受体与巨噬细胞清除、灭活内毒素有关;Nod1是胞浆内识别内毒素的分子。二、库普弗细胞库普弗细胞是肝脏吞噬、清除内毒素的主要细胞。在生理条件下,虽然仍有少量细菌及内毒素经门静脉进入肝内,但库普弗细胞都将之清除。库普弗细胞是肝内定居的巨噬细胞,是数量最多的定居组织内的巨噬细胞。理论上推测,如果库普弗细胞和其他巨噬细胞一样对内毒素十分敏感,细胞将会处于持续活化状态,但事实上库普弗细胞吞噬、清除内毒素时,其本身并未被内毒素所活化。说明在处理内毒素时,库普弗细胞与其他巨噬细胞有着不同的机制:库普弗细胞处理内毒素主要依靠其吞噬作用。在无血清存在时,库普弗细胞吞噬内毒素的作用也能正常发挥;而且随着内毒素浓度的适当增加,库普弗细胞的吞噬活性增强,该效应与分子量分别为11.8万和8.3万的两种蛋白质酪氨酸残基磷酸化事件有关。CD14是介导内毒素激活库普弗细胞的主要受体,清道夫受体是库普弗细胞重要的防御性受体,介导库普弗细胞清除和灭活内毒素。库普弗细胞激活共分四个阶段,其中CD14是细胞活化及功能变化的特征性标志。①静止期:表现为库普弗细胞数量少,形态小,多位于肝窦内,CD14染色多为阴性;②反应期:表现为局部库普弗细胞刺激性增生及全身单核-巨噬细胞肝内聚集﹔③预激期:即库普弗细胞表型发生转化期,表现为CD14等细胞膜受体出现,库普弗细胞功能发生改变;④激活期;表现为核转录因子NF-κB活化,细胞分泌各种细胞因子。通常库普弗细胞表达CD14水平较低、CD14阳性细胞仅占3.3%,而且CD14散在表达于这些细胞表面;与此相反,库普弗细胞普遍表达清道夫受体,且细胞表面清道夫受体呈弥漫性分布。有趣的是,随着内毒素浓度的不断提高,表达CD14的库普弗细胞明显增多,甚至高达96.1%。且细胞表面CD14表达呈弥漫性分布;与之相反,清道夫受体表达则明显下调,且与CD14表达上调呈显著负相关。与此同时,血浆ALT、总胆红素和肝组织内丙二醛水平均明显升高,以上三指标的变化与CD14表达水平呈显著正相关,而与清道夫受体表达水平呈显著负相关,说明随着内毒素水平的提高,库普弗细胞防御功能减弱,而引起肝功能损害的致炎作用增强,提示库普弗细胞由防御性细胞逐步转化为效应性细胞,而CD14表达上调、清道夫受体表达下调则是库普弗细胞功能转变的重要机制。三、中性粒细胞中性粒细胞表面能够表达膜CD14(membrane-bound CD14,mCD14)和TLR4,内毒素通过与该类受体结合而激活中性粒细胞。中性粒细胞表面除了表达高亲和力内毒素受体CD14外,还表达低亲和力内毒素受体L-选择素,并经过该受体介导细胞活化。此外,整合素CD11b/CD18也被认为是中性粒细胞表面的一种低亲和力内毒素受体。在低浓度内毒素刺激时,由mCD14介导中性粒细胞活化;而高浓度内毒素刺激时,则由低亲和力内毒素受体介导中性粒细胞活化。游离内毒素激活中性粒细胞及与中性粒细胞结合均由CD14介导;而与红细胞等颗粒结合的内毒素,激活中性粒细胞由CD14介导,但后续的与中性粒细胞结合则由cD11b/CD18介导;完整的革兰阴性杆菌表面的内毒素与中性粒细胞结合,既非由CD14介导,也非由CD11b/CD18介导。四、内皮细胞一般认为,内皮细胞表面无mCD14表达,血清中的可溶性CD14(soluble CD14,sCD14)是介导内皮细胞识别内毒素的分子,同时TLR4参与内毒素诱导的内皮细胞激活。内毒素结合蛋白(Lipopolysaccharide—binding protein,LBP)将内毒素转运给sCD14,LPS/ sCD14复合物与内皮细胞膜上的TLR4结合并激活内皮细胞。sCD14除了能够介导内皮细胞活化外,还能介导内皮细胞清除内毒素,LPS/ sCD14/LBP形成三聚体并结合至内皮细胞上,继之LPS/sCD14发生内源化,从而将内毒素清除。最近,Jersmann等研究发现,内皮细胞表面也有mCD14表达,但其含量仅约为单核细胞表面的1/20。在无血清(无sCD14)存在的情况下,mCD14能够介导内毒素所致的内皮细胞活化,而且该过程是mCD14依赖性的;在此基础上加入血清,能使内皮细胞对内毒素的应答增强。先前已有研究发现,sCD14能够快速,高效地将内毒素转运给巨噬细胞、中性粒细胞表面的mCD14,从而使巨噬细胞、中性粒细胞对内毒素的应答增强。根据以上结果,Jersmann等认为,在内毒素激活内皮细胞的过程中,mCD14是必不可少的,sCD14在该过程只是起着将内毒素加速转运给mCD14的作用。五、肠黏膜上皮细胞肠黏膜上皮细胞始终与细菌及其产物发生持续性接触,这些细菌及其产物能够刺激其他类型的细胞并诱发炎症反应,但并不诱导肠上皮细胞产生防御反应,这一特点对于结肠上皮细胞来说显得尤为重要,因为如果结肠上皮细胞能够对肠道正常菌群产生反应,则会对机体造成不良影响。但是这并不意味着肠上皮细胞是免疫缺陷细胞,当遭受致病菌及其产物侵袭时,肠上皮细胞能够产生正常的应答反应,说明:①肠上皮细胞具有天然区分正常菌群和致病菌的能力,这与其识别系统的亚细胞定位有关。一些病原体识别分子定位于胞浆内(如Nod1)或基底侧细胞膜(如TLR5),只有当病原体及其内毒素、鞭毛蛋白等成分进人细胞内或到达细胞基底侧时,才能被识别并导致细胞活化;正常菌群不但不引起肠上皮细胞活化,反面能够通过与上皮细胞相互作用,抑制IκB降解,从面阻止NF-κB活化,表现出抗炎效应。②肠上皮细胞存在着与髓系细胞不同的内毒素识别机制。在正常人肠组织活检标本中,TLR2、TLR4的表达水平几乎测不出来;Caco-2、T84及HT29等肠上皮细胞系虽有TLR4表达,但无CD14及共受体MD-2表达,这也很好地解释了肠上皮细胞为什么对正常细菌及其产物无反应性。但也有研究发现,即使无CD14表达,TLR4表达阳性的肠上皮细胞也能识别内毒素并对之做出反应,前提是必须有别的细菌毒力因子存在。尿路致病性大肠杆菌(uropathogenic Escherichia Coli)的Ⅰ型菌毛是该菌的毒力因子之一,单独作用可引起肠上皮细胞产生细胞因子,此外Ⅰ型菌毛还能将内毒素呈递给肠上皮细胞,诱导肠上皮细胞释放细胞因子,该效应的实现有赖于肠细胞膜上TLR4的表达。
参数原理
2024.01.30

内毒素机体防御系统的抑制成分之肠黏膜免疫学屏障
肠道黏膜免疫学屏障是防御肠道病原体和内毒素入侵的重要防线,slgA在肠道黏膜免疫中发挥重要作用。sIgA是保护肠黏膜的一个重要成分,既能阻止肠腔内细菌在黏膜表面定植,也能中和内毒素。有研究发现,在肠黏膜遭受O157大肠杆菌感染时,抗内毒素核心多糖的特异性slgA分泌增加,在恢复期病人表现得尤为明显,提示slgA对防止内毒素向机体内转移具有保护性作用。另外,有研究提示NO在肠黏膜局部形成的氧化屏障具有防止内毒素移位的作用。生理条件下,iNOS仅在呼吸上皮,妊娠子宫及回肠黏膜等少数部位表达。但在内毒素的诱导下,正常的结肠上皮细胞也表达 iNoS并催化合成NO,在局部形成氧化屏障,从而防止结肠细菌移位,因此有效地防止细菌的移位,也间接防止了内毒素的移位。
参数原理
2024.01.23

内毒素机体防御系统的抑制成分之肠黏膜机械屏障
在机体防御系统中,对肠道内毒素移位起抑制作用的成分包括肠黏膜机械屏障、肠黏膜免疫屏障、肠道正常菌群以及肝脏的肝细胞和库普弗细胞,其中肠黏膜机械屏障,肠黏膜免疫屏障、肝细胞和库普弗细胞起着直接抑制作用,而正常菌群则起着间接抑制作用。肠道是巨大的“内毒素库”,特殊的解剖部位决定了肠黏膜必须是一道有效的防御屏障。肠黏膜屏障由黏膜上皮细胞形成的机械屏障和分泌型IgA(sIgA)等形成的免疫学屏障组成。生理条件下,肠上皮细胞一般不吸收肠腔内的内毒素。有实验证明,经静脉注入的内毒素可以出现在肠上皮细胞中,但向肠道内灌注的内毒素只在肠腔内靠近肠黏膜的部位聚集,而不进入上皮细胞内。肠上皮细胞及细胞间紧密连接形成的黏膜机械屏障,是防御肠腔内毒素移位的一道重要屏障,保持完整性是其发挥防御作用的重要保证。严重创伤﹑烧伤时,体液大量丢失,血容量不足,导致机体缺血缺氧。为维持血压、保证心脑等重要器官的血液供应,内脏血管代偿性收缩,其中胃肠缺血缺氧时间较其他器官更长,即使休克病人经液体复苏血流动力学恢复正常后,胃肠道仍处于隐匿性休克状态。因此,当肠璧微血管恢复灌流时,肠发生缺血/再灌注损伤,上皮细胞产生大量的活性氧等介质,导致肠上皮细胞凋亡、细胞间紧密连接破坏,肠黏膜通透性因而迅速增加,机械屏障功能削弱,从而促使肠腔内的内毒素经肠璧吸收并向肠外组织移位。活性氧是导致肠黏膜损伤的最重要因素。进入血中的内毒素可以引起血管内凝血、微血管通透性增加及血压下降等病理生理变化,从而导致全身多个器官发生缺血缺氧性损害,而胃肠道又是最先受损的靶器官之一,在此情况下,肠上皮细胞及肠固有层巨噬细胞内的诱生型一氧化氮合酶(induciblenitric oxide synthase,iNOS),黄嘌呤氧化酶(xanthine oxidase,XO)活化,一氧化氮(NO),氧自由基大量产生、NO与超氧阴离子结合生成强氧化剂过氧化亚硝酸盐。过氧化亚硝酸盐及超氧阴离子,是介导内毒素血症诱发肠损伤的主要介质,它们通过作用于脂质﹑蛋白质及DNA,导致脂质过氧化、酶失活﹑蛋白质功能抑制以及DNA损伤,从而导致肠上皮细胞、固有层微血管内皮细胞损伤甚至凋亡;大量产生的NO也可能直接诱导肠上皮细胞凋亡;另外局部浸润的白细胞能够释放氧自由基等活性物质,也参与肠组织的损害。肠黏膜营养不良也是促进肠黏膜损伤的重要因素之一。首先,肠上皮细胞对营养物质的需求量较大。肠上皮细胞具有增殖周期短、生长旺盛等特点,因而具有较强的自我修复能力,对营养物质的需求量也较大。其次,肠上皮细胞对营养物质的需求具有自身特殊性,即需要营养物质和肠黏膜直接接触。直接接触不仅可以提供肠上皮增殖所需的营养物质,而且还能促进局部生长因子的分泌,尤其是肠三叶因子的分泌。肠三叶因子由肠绒毛杯状细胞所分泌,对肠道具有特殊保护作用。在严重创伤、烧伤情况下,虽然已建立静脉营养途径,可为全身提供足够的营养成分,但肠黏膜营养物质仍然缺乏,因此肠黏膜的损伤仍然严重,反映黏膜完整性受损的指标——血浆二胺氧化酶活性明显升高、肠黏膜跨膜电位差显著降低,与此同时,反映黏膜细胞增殖能力的增殖细胞核抗原值明显降低,说明肠上皮细胞增殖﹑移动速度减慢,这就使得受损的肠黏膜不能及时修复。
参数原理
2024.01.22

内毒素的释放和内毒素的移位简述
一、内毒素的释放革兰阴性杆菌菌体自溶或被裂解时释放内毒素,但这并非是细菌释放内毒素的惟一途径。在革兰阴性杆菌生长繁殖过程中,内毒素也不断从细胞壁上脱落下来并释放到周围的介质中去。即使在营养物质贫乏的生理盐水中,细菌也能生长,加之内毒素性质比较稳定,如100℃时1h尚不能被破坏,必须加热至160℃经2~4h,或用强酸、强碱或强氧化剂加温煮沸30min才能灭活﹐因此,内毒素几乎无处不在。二、内毒素的移位肠道中寄存着数目庞大,种类繁多的细菌,其中的大肠杆菌等革兰阴性杆菌不断向肠腔中释放内毒素。因此,肠腔不仅是“储菌所”,也是“内毒素库”,肠腔中的内毒素含量足以导致宿主死亡,但由于肠粘膜具有良好的屏障功能,故生理情况下仅有少量内毒素向肠外移位,而且这些内毒素很快被肝脏所破坏。但是,当化疗、休克引起的缺血缺氧、严重创伤、烧伤时的应激等因素导致肠黏膜屏障功能受损时,内毒素向周围组织器官中或血液中移位,其移位途径包括:①经门静脉、肝脏进入体循环;②经肠道淋巴管进入淋巴系统;③穿过肠壁进入腹膜腔进而吸收入血。肠道细菌、呼吸道细菌等产生的内毒素引起的内毒素血症,称为内源性内毒素血症;创面感染灶或体内感染灶的内毒素也可以向血液中移位,来自于感染灶的内毒素﹑输注的液体中污染的内毒素等引起的内毒素血症,称为外源性内毒素血症。
参数原理
2024.01.22

内毒素在机体内的代谢过程及机体防御系统
在人类赖以生存的自然环境中,生活着数以万计的微生物,它们不断地与人体发生接触,或黏附于皮肤表面,或被摄入消化道、吸入呼吸道,有的甚至穿越皮肤、黏膜屏障而侵入机体,与此同时,病原体的某些特定的保守结构向机体发出感染危险信号,从而激活机体防御系统产生防御应答,限制病原体扩散,清除、杀灭病原体;清除、中和病原体释放的产物。革兰阴性杆菌感染时,作为感染危险信号的内毒素,同时也是最主要的机体防御应答诱导剂。机体防御系统主要由天然免疫系统和获得性免疫系统组成。天然免疫系统包括皮肤、黏膜的机械屏障及其表面的正常菌群、杀菌和(或)中和毒素的物质、补体等体液成分、单核一吞噬细胞系统等。天然免疫系统对细菌及其产物的识别和应答是非特异性的,因而能够对入侵的病原体立即做出应答,从而构成机体防御系统中的第一道防线。获得性免疫系统由抗体介导的体液免疫及T细胞介导的细胞免疫构成。获得性免疫系统对病原体及其产物的应答是特异性的,即只对特异性抗原作出特异性免疫应答,而且只有在受到特异性抗原刺激或诱导后,获得性免疫系统才能够建立。尽管健康人体已经具有了完善的获得性免疫系统,但对病原体及其产物的识别仍有赖于古老的天然免疫系统。巨噬细胞、树突状细胞等天然免疫细胞识别病原体特定成分,并将病原体摄入细胞内加以杀灭,将病原体降解后形成的特异性抗原提呈给抗原特异性T细胞,从而形成激活抗原特异性T细胞的第一刺激信号;天然免疫细胞受内毒素等病原体特定成分刺激后,能够分泌细胞因子并在细胞表面表达共刺激分子,从而形成激活抗原特异性T细胞的第二刺激信号,促使T细胞活化,产生特异性细胞免疫。特异性免疫反过来又可以加强天然免疫系统,从而有效地抵抗病原体感染。内毒素在机体内的代谢过程主要包括:内毒素的释放和移位、内毒素的清除和解毒。机体防御系统对内毒素产生应答时,起主要作用的是天然免疫系统,它几乎参与了内毒素在机体内代谢的整个过程:肠道黏膜屏障能够抑制肠道内毒素移位;清道夫受体(scavenger receptors,SR)等介导吞噬细胞识别、吞噬、清除内毒素;肝脏库普弗细胞(Kupffer cell,KC)清除内毒素;杀菌性/通透性增加蛋白(bactericidal/ permeability in-creasing protein,BPI)等中和内毒素。
参数原理
2024.01.18

内毒素分子的跨膜转运和O-特异多糖链合成的遗传学研究
一、内毒素分子的跨膜转运LPS分子合成后从周质间隙转运至外膜的机制不清。与磷脂的跨膜转运不同,LPS的跨细胞外膜转运是不可逆的。ABC转运装置中的MsbA可能参与LPS分子的跨膜转运,该过程需要ATP参与。Muhlradt等提出LPS分子可能通过内、外膜黏附点处的“Bayer桥”(Bayers bridges)实现跨膜转运,因为新合成的LPS分子都位于此处的细胞外膜的外叶,通过外叶中LPS分子的横向运动,这些胞膜皱缩呈散在分布。每个细胞大约有50处这种内外膜间的黏附点。二、O-特异多糖链合成的遗传学研究与O-抗原合成有关的基因分三类(表3-2):①NDP-单糖合成有关的基因,此类基因的命名按其参与合成的糖命名,如与CDP-阿比糖合成有关的基因命名为abe;②NDP单糖转移有关的基因,其命名为wb-,它的产物为糖基转移酶,负责О-抗原重复单位合成中的糖基转移;③多糖链加工有关的基因,命名为wz-,如参与O-抗原链长度调节的基因命名为wZz。综上所述,LPS的生物合成是在细胞膜的胞浆面独立合成core-Lipid A和О-抗原,二者在周质间隙面连接形成完整的LPS分子,经跨膜转运分布于细胞表面。由于LPS分子的多样性,迄今仍有许多尚未阐明。如短波单胞菌Lipid A的分子具有一个2,3-二氨基-2,3-双脱氧-D-葡萄糖(DAG)二糖而不是葡萄糖胺二糖结构;而空肠弯曲菌则含有一个由DAG和葡萄糖胺组成的杂二糖结构等,因此完整揭示内毒素的分子结构,尚需进一步的研究工作积累。
参数原理
2024.01.18
