搜全站
搜展位

沃特世科技(上海)有限公司(Waters)
 关注
关注
已关注
钻石21年

![]() 已认证
已认证
粉丝量 0


400-875-8210
仪器信息网认证电话,请放心拨打
使用阿片类药物进行超高效合相色谱检测条件的系统方法开发
Jonathan P. Danaceau, Kenneth J. Fountain, 和 Erin E. Chambers
沃特世公司(美国马萨诸塞州米尔福德市)
APPLICATION BENEFITS
■ 系统UPC2
■方法开发策略
■反相色谱的备用选择
■分析速度快
■出色的极性分析物保留性能
■“绿色”流动相
■“绿色”流动相
沃特世解决方案
ACQUITY UPC2™ 系统
ACQUITY UPC2色谱柱
ACQUITY® TQD 质谱仪
MassLynx® 软件
关键词
实验
样品描述
关键词
阿片类药物,疼痛管理,UPC2/MS,方法开发,UPC2,合相色谱
简介
超高效合相色谱(UltraPerformance Convergence Chromatography™, UPC2™)是一种创新型技术,它将UPLC®的优势充分应用至超临界流体色谱(SFC)。UPC2是一种与UPLC正交的分析技术,可将具有高扩散系数的可再生“绿色”溶剂CO2与多种互补型固定相结合使用,用于解决常规LC或GC分析遇到的众多分离难题。由于多数分析化学家们对UPC2缺乏足够的认识,因此有必要为UPC2方法的开发和优化提供直接的开发策略。
本文将UPC2应用于一组天然和合成阿片类药物的分析,演示UPC2方法的开发过程。这些化合物代表一类重要的临床药物。其相关应用包括实验室药品检测、疼痛管理监测和药物治疗方案的合规性。除大约25年前出版的一份报告外,1 我们未曾发现应用SFC分析阿片类药物的任何报道。因此,我们决定通过分析这类重要化合物来评估UPC2
本文将UPC2应用于一组天然和合成阿片类药物的分析,演示UPC2方法的开发过程。这些化合物代表一类重要的临床药物。其相关应用包括实验室药品检测、疼痛管理监测和药物治疗方案的合规性。除大约25年前出版的一份报告外,1 我们未曾发现应用SFC分析阿片类药物的任何报道。因此,我们决定通过分析这类重要化合物来评估UPC2
的实用性。
本应用纪要重点突出了4种不同市售固定相结合4种不同有机辅助溶剂的系统性方法开发策略,该策略可快速确定方法开发的最优初始条件。经过初期筛选,最终通过直接快速的方法优化建立了一种针对19种天然、半合成和合成阿片类药物及其相关药物的分析方法,该方法可用于疼痛管理、成瘾治疗和药物滥用监测。最终方法能够在2 min内完成所有化合物的分析,并获得可接受的峰形,总循环时间为4 min。
实验
样品描述
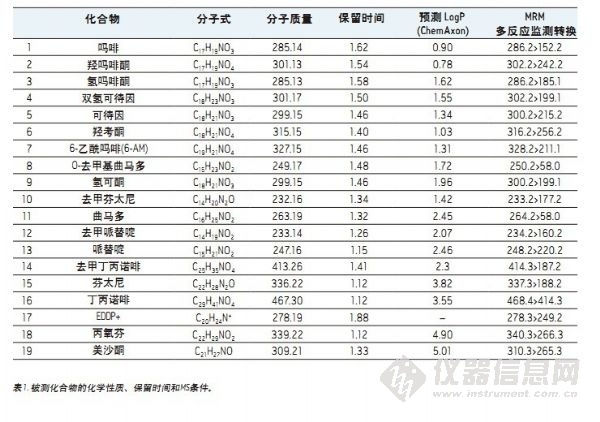
19种经筛选过的化合物列于表1中,构成一组综合性实验药物组,包括用于疼痛管理的天然阿片类药物、半合成阿片类及合成麻醉性镇痛化合物。这些化合物多数为弱碱性,其pKa值大约为8-9。它们的极性范围较广,LogP值涵盖羟吗啡酮的0.78至美沙酮的5.0,如表1所示。MRM多反应监测转换见表1。所有化合物的储备溶液均用甲醇制备,工作溶液则以甲醇或异丙醇或乙腈/甲醇(60:40)为溶剂。工作溶液中所有化合物浓度均为500 ng/mL。
UPC2条件
系统: ACQUITY UPC2
色谱柱: ACQUITY UPC2BEH,2.1×50 mm,1.7 微米(p/n 186006558)
柱温: 55 ℃
进样体积: 2µL
流速: 1.5 mL/min
流动相A: CO2
流动相B: 含0.4%甲酸和40 mM NH4COOH的甲醇
样品瓶: LC/MS认证的12×32 mm螺口最大回收(p/n 600000749CV)
梯度: 初始条件为2%的流动相B。流动相B在2 min内增 加至50%,达到50%后保持0.5min。然后在0.1min内流动相B返回到2%,重新平衡系统1.4 min。整个循环时间为4.0 min。
MS 条件
质谱仪: ACQUITY TQD
补偿泵流速: 含1%甲酸的甲醇(0.25 mL/min)
电离模式: ESI+
电离模式: ESI+
采集模式: MRM(请参阅表1了解离子跃迁)
毛细管电压: 1 kV
毛细管电压: 1 kV
碰撞能量: 针对各化合物优化
锥孔电压: 针对各化合物优化
数据管理: MassLynx软件
结果与讨论
色谱条件优化
结论
结果与讨论
初始条件筛选
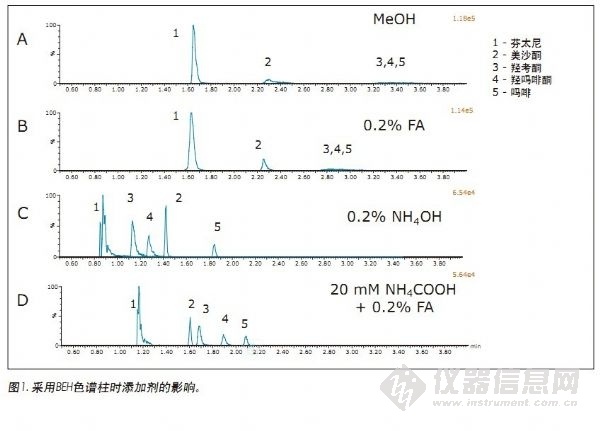
本实验利用色谱柱管理器考察4种不同化学性质填料的色谱柱和4种不同的辅助溶剂,对一系列的运行条件进行了筛选,确定出使用UPC2分析阿片类药物的最佳起始条件。所用色谱柱分别为Waters® UPC2BEH、BEH 2-EP、CSH™氟-苯基和HSS C18 SB。流动相B为甲醇,加有以下添加剂:无(仅甲醇)、0.2%甲酸、0.2% NH4OH或0.2%甲酸+20 mM NH4COOH。初始筛选实验的梯度由5%的流动相B开始,在4 min内从5%增加至75%。然后在1 min内返回到5%,并在初始条件下重新平衡色谱柱1.4 min。每根色谱柱的流速设置为确保系统反压保持在6000 psi限值以下,BEH和2-EP色谱柱的流速为1.5 mL/min,HSS和PFP CSH氟-苯基色谱柱流速为1.0 mL/min。初始条件筛选实验采用包括芬太尼、吗啡、羟吗啡酮、羟考酮和美沙酮在内的几个化合物进行。这几个化合物代表一系列的极性范围,可简化初始筛选过程。
在BEH色谱柱上对添加剂进行的初步评价结果见图1。所有化合物浓度均为500ng/mL。谱图清楚的显示:采用纯甲醇和在甲醇中添加0.2%的甲酸时,羟考酮、羟吗啡酮和吗啡的MS峰形较宽,且峰强度偏低。相反,当以0.2% NH4OH或0.2%甲酸+20mM NH4COOH为添加剂时,多数化合物(本次初始筛选所用化合物)的峰形均可接受。在碱性条件下,分析碱性化合物可获得良好的色谱性能,这在SFC条件下分析阿片类药物表现尤为明显,上述结果与已有的报道相一致。1,2通过对下方的两张色谱图仔细分析可知,采用缓冲盐添加剂相比仅以浓氨水为添加剂可获得更好的保留性能和峰形。
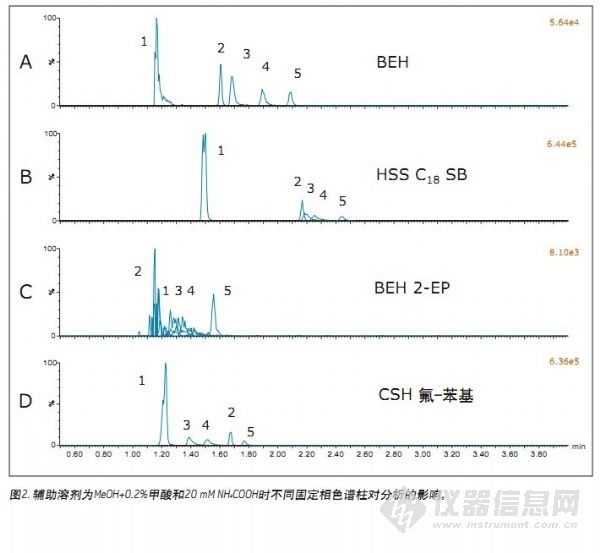
采用图1中所选流动相添加剂对不同色谱柱进行筛选,相关结果见图2。虽然谱图中未显示实验数据(由BEH颗粒填充的色谱柱相关数据),但仍可发现以纯甲醇或含甲酸的甲醇作为辅助溶剂时,所有色谱柱的性能表现均较差。另外,对于所有色谱柱,其分离性能与BEH色谱柱类型,使用0.2%氢氧化铵作为添加剂与仅使用甲醇或含0.2%甲酸的甲醇相比能更好地改善峰形,而在辅助溶剂中使用甲酸和甲酸铵的复合添加剂可获得最佳的峰形、保留时间、分离度和灵敏度。图2展示了在四种色谱柱中采用图1D中所用缓冲流动相添加剂所得的色谱图。使用由2-EP颗粒填充的色谱柱进行分析时,除吗啡外,其它化合物的峰形均不可接受。使用HSS C18 SB色谱柱时,所有被测化合物的峰形均较好,但与BEH或CSH氟-苯基色谱柱相比,峰分离度较差。采用CSH氟-苯基色谱柱时,所有峰均实现基线分离,但选择性与BEH色谱柱略有不同。根据上述结果,我们决定采用BEH和CSH氟-苯基色谱柱进一步优化色谱条件。
色谱条件优化
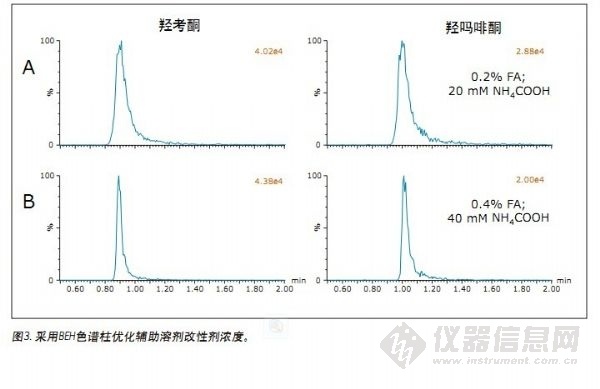
采用已选定的缓冲添加剂(甲酸+NH4COOH)和BEH色谱柱(或CSH氟-苯基色谱柱)对剩余的化合物进行测定分析。在这些条件下,除极少数化合物外,其它所有化合物均获得的良好分离。正如所料,我们还需对混合物中的少数化合物进行额外优化。羟吗啡酮和羟考酮为其中一组化合物。这两种化合物较其它分析物具有严重的峰拖尾和峰变宽现象。两峰的色谱图见图3A。羟考酮和羟吗啡酮色谱峰在5%峰高处的峰宽分别为16.5s和22.5s,而吗啡的峰宽为4s。溶质与固定相之间的相互作用被认为会显著影响超临界条件下的保留机制2,4。据推测,化合物与固定相之间的次级相互作用可能会导致相关化合物的峰形变差。在尝试最大程度降低任何可能的次级相互作用时,我们将辅助溶剂中添加剂的浓度增大了一倍,即0.4%甲酸和40 mM NH4COOH。上述条件的改变确实减小了这两种化合物的峰宽,结果见图3B。羟考酮和羟吗啡酮的峰宽分别减小至7.2s和9.9s,与初始条件下的峰宽相比降低了50%以上。同样重要的是,其它化合物的色谱性能未受到不利影响。
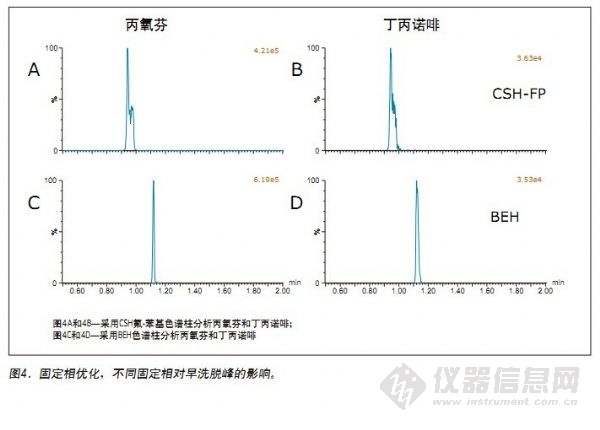
另一组测试化合物展示了许多弱极性化合物洗脱过早的色谱挑战,这些化合物示例包括哌替啶、芬太尼、美沙酮、丙氧芬和丁丙诺啡。根据上述初步筛选的相关数据,采用CSH氟-苯基和BEH色谱柱分析这些化合物非常有希望获得理想的结果;但是,在CSH氟-苯基色谱柱上使用“实验”部分中所述的最终梯度条件进行分析时,出现了分叉峰和较差的色谱结果,如图4A和4B所示。对比之下,采用BEH色谱柱所得所有化合物的峰形均较好,这可能是由于使用此色谱柱可增强分析物的保留性能或减少溶剂的影响。图4显示了在以2% 辅助溶剂为起始梯度比率的最终条件下,采用CSH氟-苯基和BEH色谱柱分析丙氧芬和丁丙诺啡的相关色谱性能。这些谱图清楚地显示采用BEH色谱柱可改善峰形,可能是由于此色谱柱能够增加分析物的保留性能。
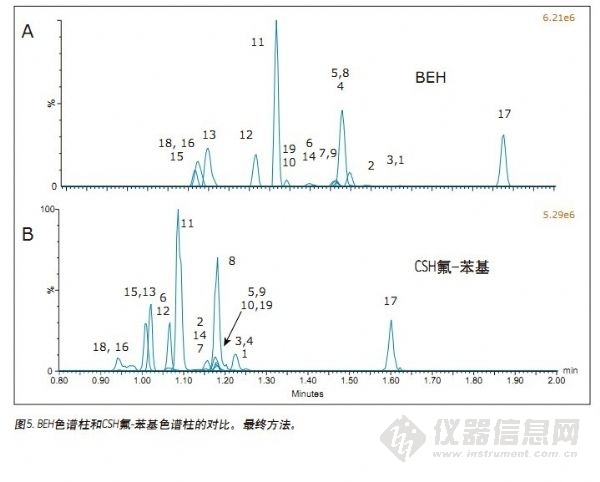
本实验中最后一项评估的参数是样品稀释剂的选择。采用UPC2进行分析的一个优势是其与样品制备所用的溶剂兼容。无论是选择固相萃取、液-液萃取还是蛋白质沉淀,最终提取物常常溶解于可能与反相色谱条件不兼容的有机溶剂中,需对其处理才能进样分析。但是,UPC2可与多种有机溶剂兼容,省略了采用反相色谱系统时通常所需的蒸发和复溶步骤。在这些实验过程中,采用沃特世Oasis® Elution样品板时,通常选择甲醇、IPA和乙腈/甲醇(60:40)复合溶液稀释样品。因此,我们对这三种溶剂进行了考察。对比IPA和ACN/MeOH(60:40)稀释剂时,未观察到溶剂影响示例。仅采用甲醇作为样品稀释剂时可产生一些负面的色谱影响,如早期洗脱化合物(如芬太尼)的谱峰变宽和出现分叉。图5为采用最终方法条件所得组合色谱图。化合物的鉴定和保留时间列于表1中。其中图5A为采用BEH色谱柱所得结果,图5B为采用CSH氟-苯基色谱柱所得结果。色谱图清楚地显示:采用BEH色谱柱时,所有化合物的保留时间更长,且丙氧芬和丁丙诺啡(化合物18和16)的峰形获得改善。该方法仍可在2 min内检出所有化合物,可用于此类化合物的快速筛选。
结论
本应用资料使用一组天然和合成的阿片类药物分析来演示适用于UPC2方法开发的系统筛选策略。同时考察多种
不同化学性质的色谱柱和多种不同的辅助溶剂及添加剂可快速确定首选的初始条件,便于进一步优化。简单快
速的条件优化建立了一种可在2 min内分析19种不同阿片类药物的方法,其中所有化合物的保留时间和峰形结果均较好。另外,此方法进一步突显了UPC2用于分析种类繁多的化合物的潜在适用性。
参考文献
1. Janicot J, Caude M, et al. Separation of opium alkaloids by carbon dioxide
sub- and supercritical fluid chromatography with packed columns;
Application to the quantitative analysis of poppy straw extracts.
Journal of Chromatography. 1988; 437: 351-364.
2. Grand-Guillaume Perrenoud A, Boccard J, et al. Analysis of basic compounds
by supercritical fluid chromatography: Attempts to improve peak shape and
maintain mass spectrometry compatibility. Journal of Chromatography A.
2012;1262(0): 205-213.
3. ChemAxon. Chemicalize.org. Retrieved Dec 15, 2012, from
http://www.chemicalize.org/.
4. Lesellier E. Retention mechanisms in super/subcritical fluid chromatography on
packed columns. Journal of Chromatography A. 2009; 1216(10): 1881-1890.
最新动态
更多![]()

Xevo MRT新应用 | 火眼金睛,让PFAS无处遁形
厂商
2024.08.26

Arc GPC体验:PEO/PEG的SEC分析
厂商
2024.08.26

UPLC二十周年大咖说 | UPLC技术应用于复杂组分的分离分析
厂商
2024.08.26

“悟空”出马,PFAS无处遁形:Xevo MRT智斗污染妖魔
百态
2024.08.22
推荐产品
供应产品
