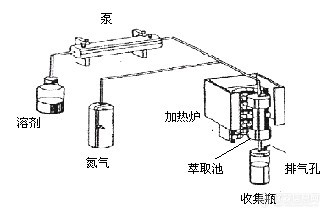
加速溶剂提取的工作流程如图所示,先将固体或半固体的样品放入不锈钢萃取池中,若未加满则填入适量的硅藻土,然后萃取池被转入加热炉内,溶剂被输液泵(或依靠氮气的压力)输送到萃取池内。此时,加热炉开始加热升温,在达到设定的温度和压力后进行静态萃取。萃取结束后,萃取池中的溶剂经由滤膜进入到收集瓶中,并用溶剂清洗管路,再用氮气将其一并吹入收集瓶中。加速溶剂提取主要利用了有机溶剂在高温、高压下,其粘度降低,能更好的浸润样品基体从而能提高溶剂提取的效果。加速溶剂提取很容易实现自动化,因此其应用非常广泛。[img]http://ng1.17img.cn/bbsfiles/images/2006/04/200604081423_16448_1613333_3.jpg[/img]微波辅助提取是利用溶剂的极性分子(如甲醇、丙酮等)在微波电磁场中快速旋转和离子在微波场中的快速迁移,相互摩擦而发热,从而加热与固态样品接触的极性溶剂,使所需要的化合物从样品中分配到溶剂里。微波辅助提取一般是在密闭或敞开的微波-透明容器中进行,萃取时将提取溶剂和样品混合在提取器中。目前国内外使用最多的是密闭的萃取罐,在较高的温度和较高的压力下进行提取的,而敞开式的微波萃取系统应用的却很少。用微波萃取时,将样品和溶剂都放入特制的密闭的聚四氟乙烯萃取罐中,在设定的条件下进行微波萃取。
样品处理时用国药集团的95乙醇(050608)处理的,对照用色谱级乙醇配制的。因含量低复测,乙醇不够,换了另一个厂家的(020930),结果比原来的高2个百分点,排除对照误差,排除系统误差(重现性很好),称量也在范围内,只有溶剂有差别,紫外扫了一下发现确实有差别,050608的乙醇247nm吸收值为0.1多点,020930的乙醇吸收值在0.05,跟色谱级的乙醇差不多。而且200-300nm的出峰情况也不一样。020930的乙醇跟色谱级的乙醇差不多。我的疑问是1,溶剂有吸收对样品的影响应该是叠加还是消减?2,是溶剂在工作波长下有吸收,还是是溶剂本身带入的杂质对样品影响的?望各位大侠帮我解解惑吧!!!
微波快速溶剂萃取技术主要有以下优点:1)溶剂用量少。10g样品仅需10~30mL溶剂,试剂量只为常规萃取的1/15左右。2)微波萃取法回收率和重复性普遍优于其他萃取方法。3)微波萃取仪萃取容器每罐可达50-100mL容积,加入样品量可达10~50g,每批样品处理只需要5~20分钟即可达到非常好的萃取效果。4)微波萃取法不受含水量的影响。EPA用MARSX对不同含水量的土壤中农药的回收率进行了分析(见图1),认为微波萃取前可以无需样品干燥,这大大提高了分析的工作效率。微波萃取免除了萃取前的样品干燥处理,因此样品含水量比其他的萃取技术对基体影响更小。
[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]或ICP测试助焊剂或有机溶剂中的重金属含量时能不能直接进样?如果不能,怎么处理?能用微波消解吗?一起讨论好不好?
[b][font=宋体]问题描述:用紫外分光光度计测叶黄素含量,国标中用无水乙醇最佳波长是[/font]445nm[font=宋体],文献中用正己烷最佳波长是[/font]474nm[font=宋体],这是因为溶剂不同造成的吗?[/font][font=宋体]解答:[/font][/b][font=宋体]([/font]1[font=宋体])紫外光谱是用紫外光待测物紫外吸收波长与所采用的溶剂密切相关,不同溶剂对紫外吸收峰波长和强度影响不同,特别是对波长影响更大。溶剂极性对溶质最大吸收峰[/font]λ[sub]max[/sub][font=宋体]的影响与溶剂的介电常数和溶质分子的电子跃迁性质有关。[/font] [font=宋体]([/font]2[font=宋体])当用光照射时,基团中的电子吸收光能发生能级改变,形成特征的强吸收带,这些基团称作发色团,它们都含有不饱和键或未共用电子对,能产生[/font]π-π*[font=宋体]及[/font]n-π*[font=宋体]的跃迁。溶剂极性越强,由[/font]π-π*[font=宋体]跃迁产生的谱带向长波方向移动越显著。这是因为发生[/font]π-π*[font=宋体]跃迁的分子激发态的极性总是大于基态,在极性溶剂作用下,激发态能量降低的程度大于基态,从而实现基态到激发态跃迁所需能量变小,致使吸收带发生红移;所用溶剂极性越强,由[/font]n-π*[font=宋体]跃迁产生的谱带向短波方向移动越明显。这是因为发生[/font]n-π*[font=宋体]跃迁的分子都含有未成键[/font]n[font=宋体]电子,这些电子会与极性溶剂形成氢键,其作用强度是极性较强的基态大于极性较弱的激发态。因而基态能级比激发态能级的能量下降幅度大,实现[/font]n-π*[font=宋体]跃迁所需能量也响应增大,致使吸收谱带发生蓝移。[/font][font=宋体]([/font]3[font=宋体])在选择测定吸收光谱曲线的溶剂时应注意如下几点:[/font]a[font=宋体].[/font][font=宋体]能很好地溶解被测物,并形成良好化学和光化学稳定性的溶剂;[/font][font=宋体]b.[/font][font=宋体]在溶解度允许范围内,尽量选择极性较小的溶剂;[/font][font=宋体]c.[/font][font=宋体]溶剂在样品的吸收光谱区无明显吸收。[/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font][color=red] [/color]

利用微波无溶剂萃取快速提取精油精油 精油是通过各种方法从植物的根、茎、叶、花、果皮等中提取的一种具有挥发性的油状烃类物质,主要有萜烯烃类、芳香烃类、醇类、醛类、酮类、醚类、酯类和酚类等组成。精油又称香精油、挥发油和芳香油。精油的挥发性较强,一般需要密封保存。精油可分为三大类:单方精油、复方精油以及基础油,通常复方精油是由单方精油和基础油按一定的配比和工艺精确调和而成。 植物精油是一类天然植物次生代谢物,由于其具有增香、杀菌、抗病毒和抗氧化等生物活性,在香料、化妆品、食品工业、制药、医疗及农业害虫等方面得到了广泛应用。目前最流行的水疗法(SPA),其精髓就是天然植物香精油,也是受欢迎之处。精油提取方法1 传统方法 1.1 溶剂浸提:是用挥发性有机溶剂将植物原料中某些成分浸提出来,主要特点是有机溶剂残留、有毒、萃取时间长效率低并且不能自动操作。 1.2 水 萃 取:常温常压下利用睡进行提取植物精油,主要特点就是过于慢,不能自动操作并且不能定量。 1.3 压 榨 法:将原料粉碎压榨,从植物组织中将挥发油挤压出来,然后静置分层或离心出油分,即得粗品。此方法所得产物不纯,可能含有水分、叶绿素、粘液质以及细 胞组分等杂质呈浑浊状态,同时很难将挥发油完全压榨出来。 1.4 水蒸气蒸馏:目前为止应用最广泛的一种方法,适用于挥发性的、水中溶解度不大的成分提取。设备简单,容易操作,成本低,一套加热装置,一套蒸馏装置即可。此 方法提纯率高,但传统加热时间长,效率低。 1.5 同时蒸馏萃取:同时蒸馏萃取(SDE)的工作原理是样品蒸气和萃取溶剂的蒸气在密闭的装置中充分混合,反复萃取。操作简单,只需要几十克样品,适用于色谱分 析 ,但不适合提取工艺的放大,另外此方法萃取率低,并不能成比例的萃取,不适合定量分析。2 新技术 2.1 微波辅助萃取:微博辅助萃取(MAE)是近几年发展的从天然物中提取香料的一种方法,利用微波辅助加热有机溶剂提取,该法最大的特点是萃取时间短,产物收率高,节省时间和所用溶剂,广泛用于有机化学和分析化学制备样品。但利用了有机溶剂,毒性较大。 2.2 超临界二氧化碳萃取(SC-CO2):超临界流体萃取(SFE)是利用流体在临界点以上某区域(超临界区)所具有的高渗透性、高扩散性和高溶解能力选择萃取目标组分。由于SFE所提取的植物精油往往还需要进一步分离提纯,所以不少研究者尝试将SFE技术与其他分离技术相耦合,来提高分离效率,另外,SFE需要高压,设备投资和维护费用较大。 2.3 微波无溶剂萃取:利用微波加热原理和水蒸气蒸馏原理相结合的一种最新技术,无需有毒溶剂微波直接作用于待提取物,数十分钟内可提取结束,操作简单,整个过程自动完成,精油得率高,时间快,无需其他成本,费用低。微波无溶剂萃取应用实例 微波无溶剂萃取装置: http://ng1.17img.cn/bbsfiles/images/2017/10/2015072208584909_01_3024284_3.png http://ng1.17img.cn/bbsfiles/images/2015/07/201507220858_556571_3024284_3.png 微波无溶剂萃取系统 http://ng1.17img.cn/bbsfiles/images/2015/07/201507220858_556572_3024284_3.png http://ng1.17img.cn/bbsfiles/images/2015/07/201507220859_556573_3024284_3.png 精油提取中精油提取中 http://ng1.17img.cn/bbsfiles/images/2015/07/201507220859_556574_3024284_3.png 收集器中精油和水明显分层 1 薰衣草精油的提取 取器: 200ml萃取器装入样品之最大刻度线(大约150g) 蒸馏水: 300g,与样品搅拌均匀 收集器: 将收集器中装好蒸馏水至刻度线下方 水循环冷却器: -15℃采用以下功率程序微波萃取样品: 消解程序: Step TIME E (max) Step1 20min 400W Step2 20min 250W 提取精油量:1.5ml2 薄荷精油的提取 取器: 200ml萃取器装入样品之最大刻度线(大约200g) 蒸馏水: 无需添加 收集器: 将收集器中装好蒸馏水至刻度线下方 水循环冷却器: -15℃采用以下功率程序微波萃取样品: 消解程序: Step TIME E (max) Step1 20min 400W Step2 20min 250W 提取精油量:2.5ml 3 橙皮精油的提取 取器: 200ml萃取器装入样品之最大刻度线(大约600g) 蒸馏水: 无需添加 收集器: 将收集器中装好蒸馏水至刻度线下方 水循环冷却器: -15℃ 采用以下功率程序微波萃取样品: 消解程序: Step TIME E (max) Step1 20min 400W Step2 40min 250W 提取精油量:2.0ml 4 烟草花(半开、全开、花蕾)精油的提取 样品处理:将样品用1mol/l的氢氧化钠溶液浸泡30min 萃取器: 200ml萃取器装入样品之最大刻度线 蒸馏水:浸泡后无需蒸馏水 收集器: 将收集器中装好蒸馏水至刻度线下方 水循环冷却器: -15℃采用以下功率程序微波萃取样品: 消解程序: Step TIME E (max) Step1 20min 400W Step2 40min 250W 提取精油量:0.01ml以上方法均比传统方法精油得率高10%以上,但时间仅是传统方法的几分之一甚至几十分之一。微波无溶剂萃取特点: w无需有机溶剂,精油直接提取 w利用微波加热,提取效率高 w采用水蒸气冷凝原理,提取纯度高 w一键式操作,解放人力微波无溶剂萃取应用领域: http://ng1.17img.cn/bbsfiles/images/2015/07/201507220859_556576_3024284_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507220859_556577_3024284_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507220859_556578_3024284_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507220859_556579_3024284_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/07/201507220859_556580_3024284_3.png[img=,106,85
[color=#444444]最近在做液相,用对香豆酸辅酶a标准品进样,流动相是1%乙酸+乙腈,选择标准品的峰查看光谱,发现最大吸收波长在296nm,但是文献上说对香豆酸辅酶a的最适吸收波长在333nm,会不会对实验产生影响,标准品使用的溶剂是乙醇[/color]
测定化合物的紫外吸收光谱时选择溶剂的原则是:(1) 样品在溶剂中溶解良好,能达到必要的浓度以得到吸光度适中的吸收曲线;(2) 溶剂不影响样品的吸收光谱,因此在测定的波长范围内溶剂应当是紫外透明的,即溶解本身没有吸收。透明范围的最短波长称为透明界限,测试时应根据溶剂的透明界限选择合适的溶剂;(3) 为了降低溶剂与溶质分子间的作用力,减少溶剂对吸收光谱的影响,应尽量采用低极性溶剂;(4) 溶剂挥发性小、不易燃、无毒性、价格便宜;(5) 所选用的溶剂应与待测组分不发生化学反应。
普析[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]火焰,前处理微波法,检测乳粉钙结果偏低理论值,前处理:称样0.3克,加10毫升硝酸,微波消解最高温度200,然后160度赶酸剩余1毫升,用水定容25,再稀释25倍上机,稀释时加20g/L氧化镧0.125毫升,上机条件:曲线用5%硝酸配置,同样加入氧化镧,曲线最高点5ug/mL吸收值0.16-0.2左右,检测结果偏低,希望大家给给建议
用Cary 50测定一个染料,其吸收范围在250-500之间,最大在400nm左右,能否用丙酮作溶剂测定?理论上,用丙酮作参比,扣除背景后,应该可以得到一个完整曲线,但实际却做不到为什么?是否不能用丙酮作溶剂,只能用吸收波长小于250nm的其它溶剂。
同一个化合物用不同的溶剂溶解,进行紫外扫描,最大波长一般会有多大的差别呢?同一化合物用同一种溶剂,只是浓度大小不同,扫出的最大吸收有变化没?
我们做污水水质分析,用原子吸收及原子荧光测金属元素,想买一台微波消解仪做水样前处理,不知道什么牌子哪个型号的好用,大家给提点建议啊,谢谢
用CEM的MARS-5微波消解仪做萃取试验的时候,能用正己烷+丙酮(1+1,体积比)做萃取溶剂吗?有讨论说微波系统中禁止使用酮、烃类物质,其中就包括正己烷和丙酮。可是标准SN/T1877.2-2007《塑料原料及其制品中多环芳烃的测定方法》中,前处理方法就是用的正己烷+丙酮(1+1,体积比)做溶剂的。会不会有危险?
单位新研究了一批色份,用了丙酮、甲苯等做溶剂溶解度均很小。请教各位这些有机溶剂的沸点都小于100度,可以用微波消解吗?急!十分感谢。
[b]微波消解溶样技术在冶金化学分析中的应用[/b][i]李洁,张穗忠,宋卫良[/i][u]钢铁研究 2006 年第34卷,第2期[/u]摘 要:微波溶样技术是世界上最先进的样品预处理技术之一。介绍了微波消解溶样技术的基本原理、优点及其在冶金化学分析中的应用。关键词:微波消解溶样技术 样品预处理 冶金化学分析 应用近年,微波消解技术取得了长足的进步。实验室微波样品处理系统具有能几十至几百倍地加快化学反应速度,成为现在最重要的实验室设备。微波消解溶样技术作为一种新型的试样预处理技术,已经成为无机元素分析中试样预处理的理想方法之一。在化工、塑料、催化剂、能源、合金、矿渣、难溶陶瓷、地矿、药品、生物等领域的分析检测得到了广泛的应用。1 微波消解溶样技术的基本原理1. 1 微波的特性微波是一种电磁波,是频率在300 MHz~300 GHz 的电磁波。为了防止民用微波功率对无线电通讯、广播、电视和雷达等造成干扰,国际上规定工业、科学研究、医学及家用等民用微波的频率为(2 450 土50) MHz 。因此,微波消解仪器所使用的频率基本上都是2 450 MHz。(1) 金属材料不吸收微波,只能反射微波。如铜、铁、铝等。用金属(不锈钢板) 作微波炉的炉膛,来回反射作用在加热物质上。(2) 绝缘体可以透过微波,它几乎不吸收微波的能量。如玻璃、陶瓷、塑料(聚乙烯、聚苯乙烯) 、聚四氟乙烯、石英、纸张等,它们对微波是透明的,微波可以穿透它们向前传播。这些物质都不会吸收微波的能量,或吸收微波极少。微波密闭消解溶样罐用的材料是聚四氟乙烯、工程塑料等。(3) 极性分子的物质会吸收微波,如: 水、酸等。它们的分子具有永久偶极矩(即分子的正负电荷的中心不重合) 。极性分子在微波场中随着微波的频率而快速变换取向,来回转动,使分子间相互碰撞摩擦,吸收了微波的能量而使温度升高。
[b]影响微波消解和微波萃取的因素[/b] 微波消解和微波萃取的效率受多种因素的影响。采用微波消解和微波萃取的方法处理样品时.要同时考虑列样品的种类、萃取溶剂、萃取温度、微波消解和萃取的功率和时间等多种因素的影响。 一、酸和萃取溶剂的影响 在元素总量分析中,一般是利用强酸〔硝酸。硫酸、双氧水,王水和盐酸〕或强氧化剂如酸性溴化钾一溴酸钾作消解介质对待测元素进行消解。而在有机金属化合物的形态分析中,为了避免酸对化合物的破坏作用,一般是采用较稀的酸和有机溶剂(异辛烷、苯、丙酮和甲醉)进行萃取。 在线消解全血样品时,选择稀盐酸和稀硝酸作为萃取溶剂口酸的浓度稍高.在消解管内会产生人量的泡沫。影响液体在管内的流动方式,降低重复性。在消解生物组织如肾、肝脏、鱼以及污水淤泥和沉积物时,只用硝酸就足以将目标分析物萃取出来。但在萃取鱼组织中的硒时,只用硝酸不能将其定耸萃取,只有加入双氧水和硫酸后才能将硒定量萃取出来,可能是由于双氧水和硫酸的加人使酸混合物具有较高的沸点和较强的氧化能力,而只硒化合物易挥发、在氧化条件下,能将硒化合物最大程度的保存在酸混合物中。一般情况下,萃取样品基体中的硒不采用盐酸作为萃取溶剂。因为硒在盐酸介质中易挥发。 在分析特定的元素时,如钙和硫,应避免使用硫酸,以防生成不溶的硫酸钙和硫元素测定的不准确性一。在某些情况下,萃取溶剂的体积影响萃取效率,如沉积物中甲基汞的萃取效率与萃取溶剂中盐酸的体积有关。利用微波辅助萃取技术处理样品时所选择的萃取溶剂一般情况下和传统的萃取方法选择的萃取溶剂相同。“相似相溶”,的原理在微波辅助萃取中仍然适用。但微波萃取中所用的萃取溶剂应具有适当的介电常数(ε)来吸收微彼能并将其转化为热能。Ganzler等的研究成果表明萃取溶剂的电导率和介电常数大时,在微波萃取中可显著提高萃取效率。然而,在有些情况下,萃取溶荆的选择还应考虑到所萃取物质的稳定性,以防止快速加热引起化合物降解。Xiong等比较了不同萃取溶剂在相同的加热条件下压力升高的速 度,其结果是:甲醉>丙酮>水≥二氯甲烷,但正己烷的压力几乎没有变化,而压力升高的速度又和溶剂吸收微波的能力有关。所以溶剂吸收微波的能力大小与上述顺序相同。二、消解和萃取温度的影晌消解和萃取温度是保证萃取效率的重要因索,在通常情况下高的消解和萃取温度会提高萃取效率。例如,密闭系统中多环芳烃类化合物在室温下的萃取效率只有52%。在115℃的萃取效率可达到75% ;酚类化合物在130℃萃取能得到较好的回收率;三嗪类化合物在密闭系统中的温度达到80-120℃时也可得到较好的回收率。但高的萃取温度可能会便多种化合物同时萃取出来,降低萃取的选择性,对待测化合物造成干扰,所以萃取湿度的选择应同时兼顾高的萃取效率和高的萃取选择性。提高萃取温度还可能会导致所萃取的化合物的降解。例如,有机氯杀虫剂二氯萘醌在115℃降解。一般来说,萃取温度的设置应在萃取溶剂的沸点附近以使萃取洛剂允分搅动起来增大萃取效率。在敞口微波装置中。消解和萃取的温度是根据所选择的酸和有机溶剂的种类决定的。难消解的样品一般加入高沸点强氧化性的酸,如硫酸使样品彻底消解。
不吸收微波的材料有哪些?比如金属
想请问一下各位微波科学的大侠们,氢氧化铁对于微波的吸收能力大吗?如果得到氢氧化铁的凝胶,它能吸收微波脱水吗?本人对于这一领域知之甚少,希望得到各为的解答,谢了!
国内知名分析仪器品牌:奥谱勒公司最近又出新成果,熟悉微波消解仪的用户都清楚,微波消解方法消解速度快,使用溶剂少等多种优越性,但使用过程中才发现,她也有很大的缺点,常规湿法消解或者干法消解的样品取样量都在1克以上,但目前国内外常规的微波消解消解罐容积在50-100ml,一般的取样量都在0.1-0.5克。由于取样量较小,代表型不强,最重要的是这么低的量定容后,对[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]是个考验,一般火焰法无法测定,石墨炉重现型不好等众多问题。奥谱勒研发的大容量消解罐容积有200ml,300ml等,粮食类样品的取样量可以到1克,根据用户需要可以定做500ml的消解罐,这在微波消解技术是个突破。
[em09511]微波加热固体材料(该固体材料对微波吸收很少),加热时间长会不会损坏微波加热设备?
各位大侠,小生最近在学习紫外光谱的基础知识,有一个问题看了很多教材还是不明白:在做紫外吸收的时候,溶剂极性对吸收的最大波长有影响,这个我是知道的,但是溶剂极性对吸光度的大小有影响吗?如果有影响,那么是什么机理呢?希望各位高手不吝赐教,谢谢了![em0808]
废气吸收的溶剂选择有什么讲究吗?一般是按什么原则进行呢?我们要测化工尾气的成分,应该怎么选择吸收溶剂呢?
1、 往主控罐插入光纤:光纤应竖直地插入主控罐;不要用力过猛,以免折了光纤。如果插入时阻力较大,重新检查装配主控罐。光纤一定要插入套管底部,否则会造成测温错误。如果不能确定,是否插到套管底了,可以记住插入的长度,取出光纤,在罐子外面比较插入是否到位。 2、 往转盘上固定主控罐:应首先安装主控罐,然后安装标准罐,只要主控罐在微波腔体内,必须一只手拿主控罐,另一只手在主控罐顶部保护光纤,以免主控罐顶到仪器顶板而夹到光纤。(具体操作请查看安装拆卸光纤操作录像) 3、 往仪器安装光纤: 要竖直的往上推,听到嘎噔一声即可。 4、 从仪器取下光纤:取时,把手的虎口顶住仪器顶板,用大拇指和食指捏住光纤探头黑色部分,用手指垂直向下使力,同时保持虎口不离开仪器顶板。 5、 从主控罐取出光纤:应首先取出标准罐,然后取主控罐。从转盘上取出主控罐时,必须用一只手拿主控罐,另一只手在主控罐顶部护住光纤,再从转盘取下主控罐,直到把主控罐取出仪器外。以免把主控罐顶到仪器顶板而夹到光纤。(具体操作请查看安装拆卸光纤操作录像)从主控罐取出光纤,要保证光纤与主控罐螺钉上表面垂直,然后顺出光纤,否则会拉断光纤。从仪器中取出主控罐时,一定要保证光纤从主控罐顺出或光纤已经从仪器顶接头取下。 6、 光纤的干燥清洁:要注意保持光纤干燥清洁。特别要避免光纤接触酸、溶剂。每次运行完后,都要清洁光纤。使用前也要检查光纤是否干燥清洁。 7、 反应的试剂反应体系,特别是萃取、合成反应时,一定要保证反应体系对微波有较强的吸收。萃取溶剂:正己烷、甲苯、二氯甲烷、石油醚等溶剂对微波的吸收非常弱,如果使用上述溶剂萃取,必须加入一定比例的丙酮等对微波吸收较好的溶剂(强吸收微波与弱吸收微波溶剂比例一般为1:1),或者在反应体系中使用加热子来吸收微波。 8、 主控罐的紧密每次装配反应罐时,一定要注意罐子特别是主控罐的紧密,每次都应拧紧盖子上所有的螺帽。注意定期观查主控罐的紧密情况:温控套管内壁有污垢,主控罐弹片中心孔有腐蚀迹象都说明有漏气。如果有漏气迹象,检查装配是否正确;如装配没有问题,更换光纤套管顶部螺帽,同时检查白色垫片是否正常。萃取时,仪器频繁溶剂报警最可能的也是主控罐漏气,这时首先确认主控罐的装配;必要时更换顶部螺帽,检查白色垫片是否正常。
采用微波消解、氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定食物中的汞,研究了微波消解样品的最佳条件,并和国家标准消解方法进行了比较,结果令人满意。本方法简便、快速,重现性好,准确度高,灵敏度为0.43μg/L,检测限为0.35μg/L,相对标准偏差为2.8%,回收率为93.5%~103.0%。 主题词 微波消解, 氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]3 食物,汞 前 言 汞是环境中重要的有毒元素[1],在自然界中,汞由于其性质活泼易于蒸发而造成对环境、生物及食品等的污染,因此,微量汞的测定直接关系到人们的健康。要准确测定样品中的汞,关键之一是样品的消解。采用干法消化法或湿法消化法消解样品,因其为间接、敞开式加热,不仅费时费电,还容易损失易挥发的汞元素,带进干扰。采用微波消解,由于微被辐射引起的内加热和吸收极化作用所达到的较高温度和压力,使消解速度大大加快,消解效率大大提高,并减少了氧化剂的用量[2]:又由于是在密闭的溶样罐中消解,避免了汞的挥发损失。 本文介绍了微波消解、氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱[/color][/url]法测定食物中汞含量的方法,研究了微波消解样品的最佳条件,并和国家标准消解方法进行了比较。本方法试剂用量少、溶样速度快、样品分解完全、待测元素无挥发损失、无污染、空白值低、灵敏、准确、精密度好、检测限低,特别适合于汞的测定。1 实验部分1.1基本原理汞蒸气对波长253.7nm的共振线有强烈的吸收作用。样品经酸消解使汞转化为离子状态,在酸性介质中与硼氢化钾发生强还原反应,生成气态汞原子、由载气(高纯氧气〉将汞原子导入石英管,在常温下,对录空心阴极灯发射的特征谱线产生吸收,在一定浓度范围内其吸收值与汞含量成正比,与标准系列比较定量。1. 2仪器 AA4701型[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收光谱仪[/color][/url](日本岛津),带HVG-1氢化物发生器,COMPAQ486微机工作站,user Jet 5L打印机,汞空心阴极灯 MK-1型压力自控微波溶样系统。1.3试剂实验用水为去离子水,试剂为优级纯。1.硝酸-重铭酸钾溶液(5+0. 05+94.5) 称取0.05g重铬酸钾,溶于水中,加入5ml硝酸,用水稀释至l00mL。2.汞标准储备液: 准确称取0.1354g经干燥过的二氧化汞,溶于硝酸-重铬酸钾溶液中,并移入l00mL容量瓶中,以硝酸-重铬酸钾溶液稀释至刻度,摇匀。此溶液每毫升含汞1.0mg。3.汞标准中间液:将2中液用硝酸-重铬酸钾溶液稀释,使含汞为10.0μg/mL。4.硝酸。5.过氧化氢。6.0.4%硼氢化纳溶液:将2.5g氢氧化钠和2g硼氢化纳依次溶于水中,再加水定容到500mL。7. 5moI/L盐酸:取208.3mL盐酸,加水定容到500mL。1.4仪器工作条件波长253. 7nm,灯电流4mA,光谱通带宽0. 5nm,燃烧器高度16mm,原子化温度为室温,高纯氩气流量1.0L/min,转速30转/min,积分时间8s,峰高吸光度定量,氘灯扣除背景。1.5实验方法1.样品处理: 准确称取0.5~1.0g样品于溶样杯内,依次加入5mL浓硝酸,2mL过氧化氢,将溶样杯放入可控密闭溶样罐,再置入微波炉内。开启微波炉,1档5min,2档3min,3档3min,4档3mine消解完毕,微波炉自动关熄。稍等片刻,打开炉门,取出罐体,待冷却至室温,开盖,将杯内样液转移到1OmL容量瓶中,并用水定容至刻度,摇匀。同时做试剂空白。 2.标准曲线的绘制=取l00mL容量瓶5只,分别加入汞标准中间液0.00、0.05、0.10、0.20、0.30此,用水定容至刻度,摇匀。此标准系列汞浓度为0.0、5.0、10.0、20.0、30.0μg/L。按仪器工作条件,进行氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定,以吸光度对汞的浓度绘制标准曲线。 3.样品测定: 将试剂空白及消化后的样品溶液按仪器工作条件进行氢化物发生[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测定。 4.计算:按下式计算样品中汞浓度: C =m/G C一样品中汞浓度,μg/g: m一由标准曲线上查得的汞的含量,μg: G一测定所取样品重量,g。2结果与讨论2.1微波消解条件的选择2.1.1消解液的选择[3] 本实验比较了硝酸及硝酸-过氧化氢两种消解液,结果表明仅用硝酸,不仅所需试剂量多,且样品不易消解完全 而用适宜比例的硝酸一过氧化氢,不仅消解时间短,消解效果好,而且减少试剂用量及带入的干扰。因此,本方法选用了硝酸-过氧化氢作消解液。2.1.2硝酸及过氧化氢用量对消解效果的影响 样品不同,消解条件也不同。本方法以0.5g虫草哈士膜胶囊1. 为例,分别一次性加入不同配比及总量的硝酸、过氧化氢,其消解效果有一定差异,结果见表1。从表1可见,加入5mL硝酸和2mL过氧化氢时消解效果最好。 Tab.1Digestion effect of smpIe序号 加入试剂量 消解时间/min 消 解效 果 HNO3 H2O2 1档 2档 3档 4档 1 5 0 5 3 3 3 稍浊液 2 5 1 5 3 3 3 稍浊液 3 5 2 5 3 3 3 无色透明清液 4 5 3 5 3 3 3 无色透明清液 5 3 0 5 3 3 3 稍浊液 6 3 1 5 3 3 3 稍浊液 7 3 2 5 3 3 3 无色透明清液 8 3 3 5 3 3 3 无色透明清液 2.1.3消解压力和消解时间对消解效果的影响 消解压力过低,反应速度慢,消解时间长:但消解压力过高,反应过剧而致溶样罐压力过大,引起溶样罐泄漏,影响消解效果。本方法所用自控微波溶样系统,是通过控制压力来控制反应温度的。所以本方法采用梯度加压方式来消解样品。但具体采用怎样的梯度加压的压力档和时间对应关系,得根据具体样品来定。本方法以0.5g虫草哈干膜胶囊为例,采用一档5min,二档3min三档3min,四档3min的梯度加压方式,使样品消解完全,获得满意结果。2.2硼氢化钾浓度对汞测定的影响 硼氢化纳浓度对汞的测定有较大影响。若硼氢化纳浓度过高,由于发生大量氢气对汞原子产生稀释作用而使灵敏度降低:若硼氢化钠浓度过低,由于氢化反应不完全而使灵敏度降低。按法试验了0.4%~1.0%硼氢化纳与20.0μg/L的汞标准溶液的反应,结果表明: 硼氢化纳浓度为0.4%时灵敏度最大,故本方法选用硼氢化钠浓度为0.4%。2.3盐酸浓度对汞测定的影响 氢化反应宜在酸性介质中进行,以盐酸介质为好。按法试验了20.0μg/L的汞标准溶液在1.0~5.0mo1/L的盐酸介质中的反应,结果表明:盐酸浓度为5.0mol/L时,灵敏度最大。故本方法选用盐酸浓度为5.0moIml/L。2.4标准曲线的线性关系 标准曲线的回归方程为Y=0.008 4X+0.017 7,相关系数r=0.9999,可见标准曲线在汞浓度0.0~30.0μg/L范围内,峰高和浓度之间呈良好线性关系。2.5方法的灵敏度和最低检测限 对10.0μg/L的汞标准溶液按法10次测定,得出灵敏度(1%吸收)和最低检测限(DL=c ×(3S)/(A))分别为0.43μg/L和0.35 μg/L。2.6精密度和准确度试验 对含汞为10.0μg/L的加标样品,按法8次测定,吸光度均数为0.102,相对标准偏差为2.8%。为了确证分析结果的可靠性,应用本法对样品进行加标回收试验,结果见表2。由表2可知,本方法的回收率在93.5%~103%之间,平均回收率为98.2%。2.7本方法与国家标准消解方法[4]的比较 用本方法与GB5009;17-85标准消解方法对十二种不同食物中的汞进行测定,对测定结果进行统计学处理,经 t 检验,P〉0.05,两种方法无显著性差异。同时,由实验可知,采用国家标准方法消解样品,不仅需要冷凝固流装置,而且需要化十多个小时才能将样品消解完全。采用本方法,仅需十四分钟,样品即消解完全。可见,本方法比国家标准消解方法简便、快速得多。 样品 本底值μg/L 加入汞量μg/L 测得值μg/L 回收率% 赐尔皇降 0 10 10.2 102.0 脂胶囊 0 20 18.7 93.5 蓝亿健螺 0 10 9.42 94.2 旋藻胶囊 0 20 20.6 103.0
为什么说极性分子物质会吸收微波?欢迎讨论
我有看过用微波消解有机溶剂来提取鱼肉中的菊酯类农药,他说鱼肉微波消解主要是为了除去鱼肉中的蛋白质,而茶叶是植物性的,没有蛋白质,用微波消解来处理是不可行的,这种说法正确吗,为什么
1、什么是微波?微波是指是频率在300MHz—300GHz的电磁波(即波长在100cm至1mm范围内),也就是说波长在远红外线与无线电波之间。微波波段中,波长在1-25cm 的波段专门用于擂达,其余部分用于电讯传输。为了防止民用微波功率对无线电通讯、广播、电视和雷达等造成干扰,国际上规定工业、科学研究、医学及家用等民用微波的频率为2450 土5OMHz。因此,微波仪器所使用的频率基本上都是245OMHz,家用微波炉也如此。2.微波的特性(1)金属材料不吸收微波,只能反射微波。(2)绝缘体可以透过微波,它几乎不吸收微波的能量。(3)极性分子的物质会吸收微波(属损耗因子大的物质),如:水、酸等。3、微波加热的原理和特征目前,对微波加热机理的探讨很多,大多数都是从传统的电磁波物理学理论出发对其加以解释的,可简单地描述如下:分子在微波的辐射下(电场的作用下) ,转向偶极矩发生变化,由於摩擦产生热量。在微波加热的情况下,热量来自分子本身,这和传统的加热方式--热量来自热源并经过物质的热传导有明显的区别。微波加热具以下显著的特点:1) 和传统的加热方式相比,所用有机溶剂更少,甚至可以不采用有机溶剂2) 热传导、对流性质不好的物质可以在短时间内的以加热,均匀性更好;3) 可以对目标物“选择性” 地进行加热,加热效率高、更节省能量;4) 可以对热损失系数较大的物质选择性地进行加热;5) 热传导较差和几何形状不规则的物质可以在短时间内得以加热;6) 可以通过感应器来对温度进行控制,反应自动化程度得以提高;7) 密闭加热,可以进行有压力反应和排除空气干扰。4、微波与化学合成微波技术用于化学合成最早可追溯到1986年,当时加拿大的R.Gedye等实验中发现:和传统的加热方式如电加热、油浴加热相比,微波辅助化学合成的反应速度大大的得以提高。此外,由于微波反应还具有重现性高、环保、选择性高等诸多特点,迅速引起了人们的广泛关注。自90年代后半期以来,有关微波合成的报导逐年呈上升趋势,至今已有1000多篇相关报导。事实上,现在有机合成类代表性杂志如Tetrahedron Letters,Synlett等基本上每期上都刊登有微波合成的文章。
微波萃取的机理可从两方面考虑:一方面是微波射线自由透过透明的萃取介质,深入到生物材料的内部维管束和腺胞系统。由于吸收微波能,物料内部温度突然升高,在天然物料中的维管束和腺胞系统升温更快,保持此温度直至其内部压力超过细胞壁膨胀的能力,细胞破裂。位于细胞内的有效成分从细胞壁自由流出,传递到萃取溶剂里。另一方面,由于不同物质的tanδ值不同,对微波能的吸收程度也不同,微波可以对体系中不同组分进行选择性加热,从而使被萃取物质从基体或体系中分离出来, 进入到萃取溶剂中。是否有版友利用微波做萃取呢?
1 引言微波辐射技术用于促进化学反应始于1986年Gedye R等在微波炉内进行的酯化、水解和氧化反应,而微波辐射技术在环境工程中的应用潜力直到最近几年才逐渐被人们注意到。截止到目前,微波辐射技术已被成功地用于环境监测、废气治理、污水处理和固体废弃物处理等各个环境工程研究领域,在环境监测中的应用研究则主要集中在微波萃取和微波消解等样品预处理方面。2 微波萃取2.1 微波萃取原理微波萃取的基本原理是利用介质吸收微波辐射能程度的差异,通过选择不同溶剂和调节微波加热参数,对物料中目标成份进行选择性萃取,从而使试样中的目标物(如有机污染物)和基体物质有效地分离。微波萃取已经广泛地应用于土壤、沉积物和各种有机体中目标物的萃取分离。2.2 微波萃取特点1、快速高效 样品及溶剂中的偶极分子在高频微波的作用下,以109/s圈的速度变换其正、负极,产生偶极涡流、离子传导和高频率摩擦,从而在短时间内产生大量的热量。偶极分子旋转导致的弱氢键破裂、离子牵移等加速了溶剂分子对样品基体的渗透,待分析成分很快溶剂化,使微波萃取时间显著缩短。2、加热均匀 微波加热是透入物料内部的能量被物料吸收转换成热能对物料加热,从而形成独特的物料受热方式,整个物料被加热,无温度梯度,即微波加热具有均匀性的优点。3、微波加热具有选择性 微波对介电性质不同的物料呈现出选择性的加热特点,介电常数及介质损耗小的物料,对微波的入射可以说是“透明”的。溶质和溶剂的极性越大,对微波能的吸收越大,升温越快,促进了萃取速度。而对于不吸收微波的非极性溶剂,微波几乎不起作用。所以,在选择萃取剂时一定要考虑到溶剂的极性,以达到最佳效果。4、生物效应(非热效应) 由于大多数生物体内含有极性分子,在微波场的作用下引起强烈的极性震荡,从而导致细胞分子间氢键松弛,细胞膜结构电击穿破裂,加速了溶剂分子对基体的渗透和待提取成分的溶剂化。因此,利用微波萃取从生物基体萃取待分析的成分时,可以提高萃取效率。2.3 微波萃取技术与其它技术的比较任何一种萃取技术都是为了从基体中快速、高效地分离出待分析成分,但是由于基体的复杂性及萃取技术的不同特点,常常在选取方法的时候必须考虑到分析的目的和分析方法的费用、操作的繁简、时间的多寡等因素。其它方法如超声波萃取、超临界流体萃取和加速溶剂萃取等特性比较见表1。索氏抽提是一种历史悠久的经典萃取方法,该法在对那些活性物质的提取中比较常用,但该法有费时、工作强度大且耗费溶剂量大,在浓缩时易造成环境污染等不足。表1 不同萃取方法的比较索氏提取 超声波萃取 微波萃取 超监界流体萃取 加速溶剂萃取时间 24~48h 30~60min 4~20min 30~60min 15min预分离 不过滤 过滤和溶剂蒸发 洗脱 不过滤 不过滤溶剂用量 大 大 小 小 大费用 低 低 高 高 高工作强度 大 大 低 低 低污染程度 大 大 小 小 小3微波消解3.1微波消解的原理微波消解的基本原理是利用样品的微观粒子在微波场中可产生电子极化(原子核周围电子的重新排布)、原子极化(分子内原子的重新排布)、取向极化(分子永久偶极的重新取向)和表面极化(自由电荷的重新排布)。在这4种极化中,与微波电磁场的振动周期(10-9~10-12s)相比,前2种极化要快得多(驰豫时间分别为10-15~10-16s和10-12~10-13s),所以不会产生介电加热,而后2种极化则与之相当,可以产生介电加热,即通过微观粒子的这种极化过程,将微波能转化为热能。3.2微波消解的特点1、样品分解完全由于分解反应是在高温、高压的密闭容器里进行,在应用合理的酸和溶剂,控制最佳压力和微波加热时间的条件下,使样品在无污染和无损失的情况下达到完全分解。2、溶样速度快由于样品和溶剂的反应是在瞬间吸收微波辐射能量后即产生的,不需传热过程,瞬时可达较高的温度,消除了热量传导过程中能量的损失,因而样品分解所需时间比常规法大大缩短,一般溶样所需时间不超过20分钟。3、经济密闭消化消除了溶剂的挥发,最大限度地发挥了溶剂的作用,因此消耗的试剂量少,加热时间短,操作简便,降低了分析成本和减轻了分析者的劳动强度。4、简便只需把样品及溶剂放入消解罐内,调整好所需要的压力,设定好加热时间即可进行微波消解。5、污染少 由于样品消解是在密闭容器里进行的,没有酸雾的泄漏,消除了对环境和人员的污染。4 小结微波辐射技术在环境监测中的应用起步较晚,但发展较快。在美国,微波消解正在逐渐成为环境样品分析的标准方法。从1983年起,我国环境监测领域开始涉足微波辐射技术,目前已经取得了较为可喜的进展。随着科学技术的进步,有关微波技术的基础研究必将取得较大突破、微小辐射技术得到不断完善,微波辐射技术也必将在环境监测领域取得更广泛的应用。

微波消解-石墨炉原子吸收光谱法测定明胶空心胶囊中的铬含量 作者:孙 梅http://ng1.17img.cn/bbsfiles/images/2015/12/201512071106_576579_2904170_3.jpg摘要:本文采用敞开湿法消解法和微波消解法分别对明胶空心胶囊进行了前处理,并比较了两种前处理方法的优劣;最后利用石墨炉原子吸收光谱法对其中的总铬含量进行了测定。发现微波消解-石墨炉原子吸收光谱联用法具有分析快速、线性关系好和样品的加标回收率高等特性,该方法适合明胶空心胶囊样品中微量铬的含量测试。关键词:石墨炉原子吸收分光光度法;微波消解;明胶空心胶囊;铬 明胶空心胶囊是由药用明胶加辅料精制而成,由主料药用明胶和食用色素等添加剂勾兑成流质状的胶体液体,然后放到成型的模具中压塑成型,再进行修剪包装便可完成。 我国的药品管理法中规定,生产药品所需的原料、辅料,必须符合药用要求,即用于胶囊生产的明胶必须符合药用要求,不得使用工业明胶。工业明胶与药用明胶的区别主要是工业明胶中的铬含量要比药用明胶高很多。我国《中国药典》(二部)对明胶空心胶囊中的含量进行了限量要求,不得超过百万分之二。 本文采用微波消解法对明胶空心胶囊进行了前处理,并利用石墨炉原子吸收光谱法对其中的总铬含量进行了测定。实验结果发现:所建立的方法快速、准确,适合于药品生产企业和明胶空心胶囊生产企业等控制胶囊中的铬含量。1 仪器与试剂1.1 仪器与条件 美国Perkin Elmer原子吸收光谱仪(型号:AAnalyst 800),AS-800型石墨炉自动进样器;铬空心阴极灯;石墨炉原子吸收光谱仪测试条件见表1,石墨炉程序升温程序见表2。CEM MARS 6微波消解仪(美国);MS105DU型Mettler Toledo电子天平;BHW-09 C恒温加热器(上海博通化学科技有限公司,上海);Advantage A10 Milli-Q纯水器。http://ng1.17img.cn/bbsfiles/images/2015/12/201512071108_576581_2904170_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/12/201512071108_576582_2904170_3.jpg1.2 试剂 硝酸(MOS级,天津市风船化学试剂科技有限公司);铬标准储备溶液(1000 mg×mL-1,Inorganic Ventures公司,美国);Milli Q超纯水(18.2 MW×cm);Mg(NO3)2(分析纯,上海试剂四厂,上海)。2 方法与结果2.1 标准工作溶液的配制及测定 从铬标准储备溶液(1000 gmL-1)移取一定的量,利用2 %硝酸溶液逐级稀释配制成浓度为20 ngmL-1的标准工作溶液。 实验中利用仪器自动进样器分别吸取20 ngmL-1的标准工作溶液0,5,10,20 L,配制的浓度分别为0,5,10,20 ngmL-1,注入石墨管中,按照石墨炉升温程序进行加热,测定其吸光度值。仪器自动绘制工作曲线,获得的标准曲线的线性方程为: Y=0.00618+0.01563×X,r=0.9994 上式中:Y:代表吸光度; X:代表待测溶液的浓度; r:代表工作曲线的线性系数。2.2 不同消解方法的比较 实验中对直接敞开湿法消解法、微波消解法两种前处理消解方法进行了比较。2.2.1 直接敞开湿法消解法 称取0.5 g左右的样品到玻璃烧杯中,加入10 mL的MOS级HNO3,加盖置于电热板上加热消解。样品缓慢溶解,加热约4小时后,样品中仍有不溶物(疑是Si),取下、冷却,定容到10 mL。2.2.2 微波消解法 称取0.5 g左右的样品到微波消解罐中,加入10 mL的1:1(V/V)的MOS级HNO3,先浸泡15 min后盖好盖子,放置到微波消解仪中,按下列程序(见表3)进行消解,消解步骤完成后,用恒温加热器赶酸,最后定容到10 mL。http://ng1.17img.cn/bbsfiles/images/2015/12/201512071110_576585_2904170_3.jpg2.3 测试结果样品前处理完成之后,仪器先预热稳定,再按照仪器的操作规程进行样品的分析。样品分析结果见表4http://ng1.17img.cn/bbsfiles/images/2015/12/201512071111_576586_2904170_3.jpg3 结论 通过上面的实验结果可以看出采用直接敞开湿法消解法和微波消解法这两种前处理方法,所获得的样品检测结果并没有太明显的区别。但是直接敞开湿法消解法所需要的时间更长,消耗的酸更多,所以推荐使用微波消解法进行样品的前处理。







 我要推广仪器
我要推广仪器
 下载APP
下载APP