我今天按照国标GB/T5750.10-2006中的9测定二氯乙酸,进行的是加标回收。因为是用甲基叔丁基醚做的介质,质谱分析时有很多峰,我无法确定哪一个是二氯乙酸甲酯,自带的质谱库内也没有。请问哪位能帮我找一下二氯乙酸甲酯的质谱分析参数,谢谢。
求乙酰氯,二溴丁烷,对甲氧基苯胺,硫代乙酸钾,溴苯含量的分析方法,那个大哥大姐知道的帮帮忙
溴乙酸乙酯样品,用DMSO溶解,进样Agilent 7890 + DB624柱 + FID样品不出峰,何解?bp 溴乙酸乙酯159°,DMSO189°难道溶剂与样品发生了什么反应?
活性炭管同时采了甲苯与乙酸甲酯,二硫化碳解吸。甲苯我一直用非极性柱做,乙酸甲酯极性柱做。这要分两次进样麻烦。岛津2014c自动进样那台装的是OV101非极性柱。我想用OV101柱(25m*0.20mm*0.25um)同时做甲苯和乙酸甲酯。最大的麻烦是二硫化碳与乙酸甲酯分离的问题,有谁成功的用非极性柱分离过二硫化碳溶剂峰和乙酸甲酯吗?
[color=#444444]请教一下谁用过GC-FID检测溴乙酸乙酯呀?是用的什么色谱柱呀?色谱条件是什么呀?有没有相关的文献介绍呀?谢谢各位大神!!![/color]
二硫化碳做解析夜分级乙酸甲酯,柱温已经降到40了,出峰还是这样~请问如何能减少二硫化碳的影响~第二张为单独纯二硫化碳峰,文件不能大于5Mhttp://ng1.17img.cn/bbsfiles/images/2015/11/201511061013_572465_3054588_3.jpg
请大家看看,一般乙酸二氢香芹酯是几个峰,我这里认为是后面两个大峰,但不知道前面的一个9.592的峰是什么,大家看看。
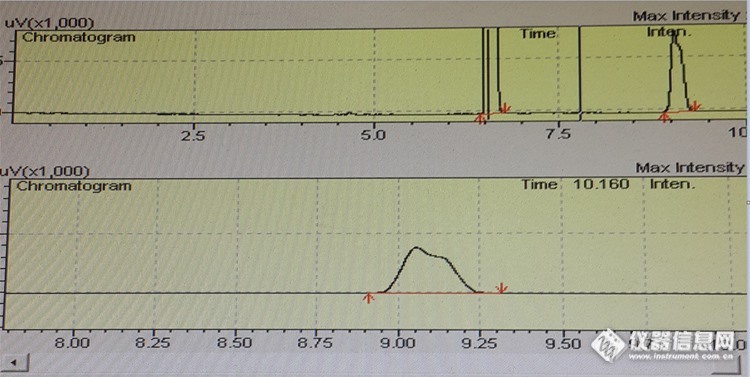
职业卫生 乙酸甲酯与甲酸乙酯同事用HP-INNOWAX 试过了说分不开,这次我用DB-FFAP来试试也分不开。因为聚乙二醇类柱子在职业卫生比较好用,哪种聚乙二醇柱子可以分开乙酸甲酯与甲酸乙酯?我的色谱条件 DB-FFAP(30m*0.53mm*1.0um) 柱温40℃ 线速度12cm/s 分流比50 检测器 进样器均为200℃http://ng1.17img.cn/bbsfiles/images/2016/05/201605051534_592369_2103464_3.png
谁做过二硫化碳中乙酸甲酯,柱温多少,分享下呗好友回复:60大家说说看~
做偶氮二异丁腈,发现有资料用乙酸甲酯:水=1:1做流动相,反相色谱,所以想问问乙酸甲酯是否可以用于反相色谱?乙酸乙酯是用于正相色谱,所以求助
各位老师,NYT1723标准测食品中富马酸二甲酯。缓冲溶液用的乙酸钠,溴化四丁基铵,乙酸调PH=6.0。实验室现没有溴化四丁基铵,用别的缓冲液可以吗?有代替方法吗?感谢!!
如题,皮革中的富马酸二甲酯检测,谁用过5A分子筛使乙酸乙酯脱水,怎么用?5A分子筛选什么样的?

前几天同事扩项工作场所空气中饱和脂肪酸酯类物质包括乙酸甲酯,乙酸乙酯,乙酸丙酯,乙酸丁酯,标准是GBZ/T160.63-2007。柱子是SH-Rtx5(30m*0.32mm*0.25um),同事欲恒温同时分离这四种酯类,我提示乙酸乙酯,乙酸丙酯,乙酸丁酯混一起恒温做没事,如果乙酸甲酯也混一起做那么会与溶剂二硫化碳峰难分离,于是他计划先做乙酸乙酯,乙酸丙酯,乙酸丁酯再另外单独做乙酸甲酯。 乙酸乙酯,乙酸丙酯,乙酸丁酯色谱条件:岛津气相色谱GC2010PLUS 柱温60℃ 检测器进样器均为200℃ 分流比50 恒线速度22cm/shttp://ng1.17img.cn/bbsfiles/images/2017/01/201701191702_673938_2103464_3.jpg三种乙酸酯峰型还不错。接下来他说想试试乙酸甲酯在这个条件下出峰会怎么样?因为我之前用OV101做过二硫化碳中乙酸甲酯,它是紧挨着在二硫化碳前出峰,同时也用DB-FFAP做过它是在二硫化碳之后。SH-Rtx5极性比OV101强些 比DB-FFAP弱很多,那么在二硫化碳之前还是二硫化碳之后出峰呢? 看到甲乙丙丁突然有了一想法:不是有碳数规律吗?利用碳数规律推测乙酸甲酯的保留时间:保留时间:乙酸乙酯 2.854min 乙酸丙酯 3.562min 乙酸丁酯5.162min 二硫化碳2.612min碳数规律:lnt‘=An+C t’为调整保留时间 n为同系物中碳个数 A, C均为常数首先精确计算死时间:间隔均匀同系物 精确死时间计算公式:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616020890_01_2103464_3.pngtm=(2.854*5.162-3.562*3.562)/2.854+5.162-3.562-3.562=2.292min调整保留时间代入碳数规律公式:乙酸乙酯ln0.562=4A+C 乙酸丙酯 ln1.27=5A+C 求得乙酸甲酯调整保留时间lnt’=3A+C t’=0.249min乙酸甲酯的预测保留时间0.249+2.292=2.541min.这个保留时间在二硫化碳(2.612min)之前,两者仅仅相差0.07min。于是预测同条件下乙酸甲酯在二硫化碳之前出峰并且分离度不好!同条件实验做二硫化碳中乙酸甲酯3000ug/ml来验证:http://ng1.17img.cn/bbsfiles/images/2017/10/2016090616132954_01_2103464_3.jpg实验结果乙酸甲酯保留时间是2.571min与预测的2.541min比较符合,外推是有误差的,而且本例碳数不多,碳数多些会更准确。降低柱温至40℃,线速度15cm/s 乙酸甲酯与二硫化碳分离达到定量要求!http://ng1.17img.cn/bbsfiles/images/2016/09/201609061616_608604_2103464_3.jpg 结论:碳数规律还是比较准的,可以预测出峰情况。
各位专家,大家好,我现在用GC112A分析乙酸丁酯和2,3-丁二醇,柱子用的是SE-30和SE-54,2,3-丁二醇沸点为181左右,乙酸丁酯为126.1,可是乙酸丁酯的峰在2,3-丁二醇后面,而且分离度不够,不能大于1.5,我采用了各种升温程序都不行,有谁能告诉我怎么办么??
(1)原理试样经处理后,在pH6左右的溶液中,镉离子与二硫腙形成配合物,并经乙酸丁酯萃取分离,导入原子吸收仪中,原子化以后,吸收228.8nm共振线,其吸收值与镉含量成正比,与标准系列比较定量。(2)试剂 氨水、混合酸、1g/L二硫腙-乙酸丁酯溶液(称取0.1g二硫腙,加10mL三氯甲烷溶解后,再加乙酸丁酯稀释至100rnL,临用时配制)、2mo1/L柠檬酸钠缓冲液(称取226.3g柠檬酸钠及48.46g柠檬酸,加水溶解,必要时加温助溶,冷却后加水稀释至500mL,临用前用1g/L二硫腙-乙酸丁酯溶液处理以降低空白值)、镉标准储备溶液和标准使用液的配制与碘化钾-4-甲基戊酮-2法中的相同。(3)仪器原子吸收分光光度计。(4)分析步骤①试样处理对于谷类要去除其中的杂物及尘土,必要时除去外壳。对于豆类,取可食部分洗净晾干,切碎充分混匀。②样品消化称取5.00g上述试样,置于250mL高型烧杯中,加15mL混合酸,盖上表面皿,放置过夜,再于电热板或电砂浴上加热。消化过程中,注意勿使干涸,必要时可加少量硝酸,直至溶液澄明无色或微带黄色。冷后加25mL水煮沸,除去残余的硝酸至产生大量白烟为止,如此处理两次,放冷。以25mL水分数次将烧杯内容物洗入125mL分液漏斗中。取与处理样品相同量的混合酸、硝酸按同一操作方法做试剂空白试验。③萃取分离 吸取0、0.25mL、0.50mL、1.50mL、2.50mL、3.50mL、5.0mL镉标准使用液(相当于0、0.05μg、0.1μg、0.3μg、0.5μg、0.7μg、1.0μg镉)。分别置于125mL分液漏斗中,各加盐酸(1+11)至25mL。向试样品处理溶液、试剂空白液及镉标准溶液各分液漏斗中各加5mL柠檬酸钠缓冲液(2mol/L),以氨水调节pH至5~6.4,然后各加水至50mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),以氨水调节pH至5~6.4,然后各加水至501mL,混匀。再各加5.0mL二硫腙-乙酸丁酯溶液(1g/L),振摇2min,静置分层,弃去下层水相,将有机层放入具塞试管中,备用。④测定测定方法与碘化钾-4-甲基戊酮-2法中的相同。⑤结果计算 样品中镉的含量按下式进行计算。X=/(m×1000)式中,X为试样中镉的含量,mg/kg;A1为测定用试样液中镉的质量,μg;A2为试剂空白液中镉的质量,μg;m为试样质量或体积,g或mL。计算结果保留两位有效数字。⑥精密度 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的15%。
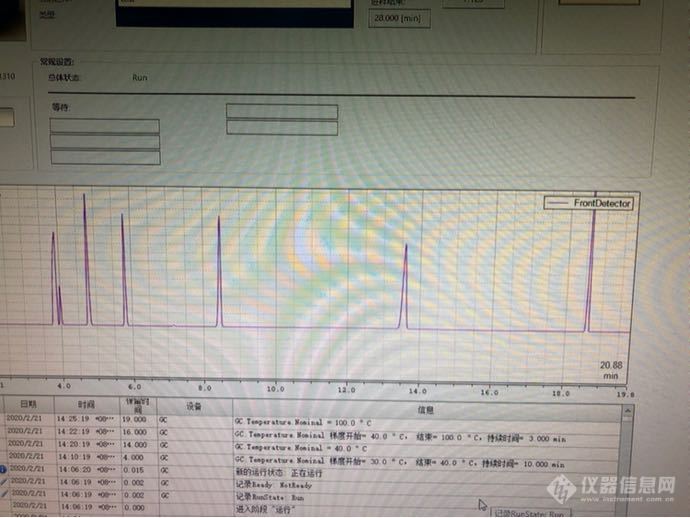
我买的坛墨的7种饱和脂肪酸酯类化合物混标!甲酸甲酯 乙酯 乙酸甲乙丙丁戊酯。现在问题来了。我甲酸乙酯和乙酸甲酯一直分不开 。下面是我条件:30 度保持4min 以1 度/min 升到40度 30度/min到100。后面都没问题 甲酸甲酯我温度低一点也能和cs2分开。可是甲酸乙酯和乙酸甲酯...分不开。那我怎么做呢?是不是要单独做一个乙酸甲酯曲线方法验证。柱子是 ffap 30 0.32 0.25[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/02/202002211425479329_4319_2990176_3.png[/img]
乙酸甲酯里面有微量醋酸,有什么好的检测方法吗
目前我有一个体系中含有甲醇,乙酸,乙酸甲酯,以及少量的硫酸,我想问怎么才能够准确的知道甲醇,乙酸,乙酸甲酯的量,另外气相在进样过程中如何避免甲醇和乙酸之间的反应
用磷酸二氢钾与乙腈测定葡萄酒中的脱氢乙酸,脱氢乙酸的出峰时间大概是好久呢?怎么测出来的曲线不是很好,都很质疑测出来的是不是脱氢乙酸。麻烦哪位做过的给点经验,我才刚刚接手这台仪器,很多都不懂。
如果同时想做苯,甲苯,乙酸乙酯,乙酸丁酯,丙酮,丁酮,三氯乙烯,二氯乙烷,环己酮的话应该悬着什么样的色谱柱,什么条件.......如果实在做不到一起的话,最合适的搭配,方法,条件是什么???.
用GB/T5750.10-2006的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]检测水中的二氯乙酸和三氯乙酸。萃取衍生过程和标准方法一样,用的是100ug/mL的混标,配制的标准曲线点两组分浓度一样。问题:1.为什么三氯乙酸的峰比二氯乙酸峰小呢?标准上三氯乙酸的峰比二氯乙酸的峰大很多,况且三氯乙酸多一个氯响应应该更大才对塞2.有做过这个实验的老师,麻烦问哈你们用的甲基叔丁基醚是哪个厂生产的啊,有没有卖色谱纯的厂家给推荐一个,还有内标1,2-DBP也是有没有色谱纯的?感觉我的基线比较毛燥。[img]https://ng1.17img.cn/bbsfiles/images/2022/07/202207081336599901_2592_2887366_3.png[/img]
大家好,我现在在做乙酰乙酸甲酯气相色谱检测,用同一根色谱柱进样,有时候时两个峰,有时候是一个峰到底是什么原因?乙酰乙酸甲酯有互变异构体,在运行过程中有出现两个峰的可能。 现在能找到乙酰乙酸甲酯和乙酰乙酸乙酯的国标。乙酰乙酸甲酯的国标中是一各峰,而乙酰乙酸乙酯是两个峰,这两个东西的性质是相似的,为什么会出现不同的峰形呢?
求助三氯吡氧乙酸、2,4-二氯苯氧乙酸正丁基酯(2,4-D丁酯)的检测方法
生活饮用水消毒副产物二氯乙酸,三氯乙酸不成线性,空白出峰
水质106项中有二氯乙酸和三氯乙酸,其方法原理是在硫酸和甲醇的条件下将酸酯化为酯,将其沸点降低再进行检测虽然衍生步骤也只是稍微有点麻烦但我有个问题不大明白,想请教下各位二氯乙酸和三氯乙酸沸点其实也不算太高吧,也就不到200度,比起百菌清溴氰菊酯什么的要低,而且气相主要也是靠卤乙酸中的氯来进行响应吧那为什么不能直接做?是因为他们属于中强酸怕坏了柱子么?P.s 顺道问问大家关于5个9的高纯氮一般用到几就不能用了啊...上次没注意用到了0结果吧柱子给毁了,割了1m都没大见效ECD也烘了好久...
我一向认为乙酸乙酯卡尔费休滴定用普通试剂就可以了,有个羰基不等于醛酮.RdH的应用手册上也说直接滴定就可以了。但是梅特勒和万通的人都说要用醛酮试剂,有道理吗?
工作场所空气中37种挥发性有机物的测定(GBZ/T 300.59-2017)方法验证,方法上用的是二硫化碳中37种VOCs和VOC专用柱,上面给的谱图显示能分开,没看到二硫化碳的溶剂峰;但实际上我选用同样的标液,DB-624UI(30m×025mm×1.40um)的柱子,升温程序35℃保持10min,5℃/min速率升到80℃保持5min后再以10℃/min升到240℃,分流比为10时,根本找不到乙酸甲酯的峰,而且二硫化碳的峰特别大,丙酮峰还在二硫化碳溶剂峰的前面;所以想请教各位老师有没有人做过这个方法验证,以及是否碰到这样的问题,有什么好的建议吗(仪器是PE的GCMS,clarus SQ 8T的)

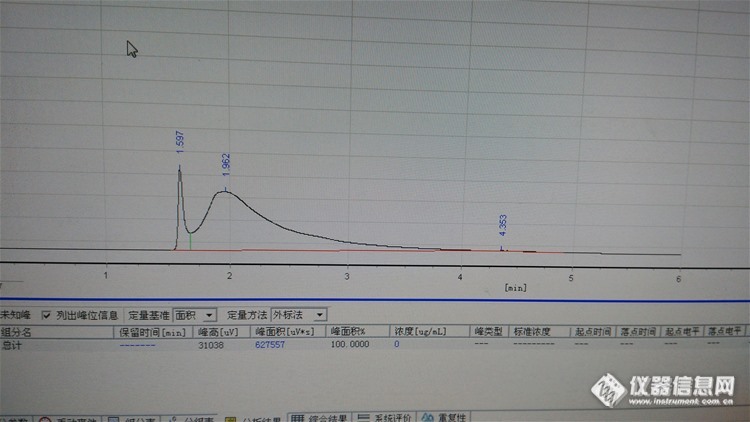
帮我看下乙酸乙酯的标线做的怎么样?1分钟多的是二硫化碳,我用的乙酸乙酯是分析纯的,我推算是1.6分钟那个是乙酸乙酯,其他是杂质峰,老师们觉得呢[img=,690,949]https://ng1.17img.cn/bbsfiles/images/2019/07/201907051351244896_5205_1791505_3.jpg!w690x949.jpg[/img]
测定乙酸乙酯残留,溶剂为二甲基亚砜,测定精密度,发现有一些溶剂峰没有出现,而有的溶剂峰却出现了。请教大家是怎么回事,应该主溶剂的峰都是应该出现的啊,而且很大。溶剂峰和乙酸乙酯峰分离度很好。乙酸乙酯出峰在10分钟,二甲基亚砜出峰在17分钟。
各位老师:HPLC色谱条件下进样乙酸乙酯,分别在磷酸二氢钾(ph3.0)和甲酸流动相体系下乙酸乙酯的响应值相差较大,是否有同样的问题出现?







 我要推广仪器
我要推广仪器
 下载APP
下载APP