做塑化剂时,低浓度的标准溶液分离效果不好,且出现了一个鬼峰,但高浓度的标准溶液分离效果很好,也没有鬼峰,是什么原因啊?
液相色谱中可否采用毛细管来提高分离效果?为什么?请书面的语言回答,谢谢!
用什么指标来判断高效液相色谱中两峰的分离效果那个评价指标是什么,怎么得出或怎么计算出,详细点
优化液相色谱(HPLC)的分离效果是确保分析结果准确性和重现性的关键步骤。以下是一些常用的策略来优化HPLC分离效果: 1. 选择合适的色谱柱: 根据待分离化合物的性质选择合适的色谱柱类型(如反相色谱柱、离子交换柱、亲和色谱柱等)。 色谱柱的尺寸(内径、长度)、填料粒度和类型都会影响分离效果。 2. 调整流动相组成: 改变流动相的组成,如溶剂比例、pH值、离子强度等,可以优化分离。 使用缓冲液可以帮助控制pH值,改善峰形和分离度。 3. 优化梯度洗脱程序: 设计合适的梯度洗脱程序,使各组分在色谱柱上逐渐分离。 避免过快的梯度变化,以免造成峰展宽或保留时间的不稳定。 4. 调整流速: 流速影响色谱柱的柱压、分离效率和保留时间。通常,降低流速可以提高分离度,但会增加分析时间。 5. 优化柱温: 控制色谱柱的温度可以改善分离度,减少峰展宽,并提高保留时间的重现性。 6. 改进样品制备: 确保样品纯净,避免杂质干扰。 使用适当的样品制备方法,如固相萃取、液-液萃取等,以富集目标化合物或去除干扰物质。 7. 使用合适的检测器: 根据分析物的性质选择合适的检测器(如紫外检测器、荧光检测器、电喷雾检测器等)。 以下是一些具体的优化步骤: 初步分离:首先使用等度洗脱或简单的梯度洗脱程序来观察样品的初步分离情况。 峰优化:针对特定峰,调整流动相的组成和梯度程序,改善峰形和分离度。 系统适应性:在确定最佳条件下,测试系统适应性,确保分离效果在不同时间、不同批次之间稳定。 方法验证:对优化后的方法进行验证,包括专属性、线性、范围、准确度、精密度和耐用性等参数。 通过上述步骤,可以逐步优化[url=https://insevent.instrument.com.cn/t/5p]液相色谱的分离效果,达到最佳的分析性能。
今天讨论到一个问题,同样一根色谱柱,装在GCMS与GC(FID检测器),分别分析同一个有机物样品,在相同的柱温、进样口及检测器温度的条件下,GCMS的分离效果要比GC的好吗?个人觉得这两者的分离效果是一样的,因为色谱柱及测试温度都一样,此时分离效果是有柱子本身决定的,难道载气也会有影响到分离效果(GC-MS一般用的是He,而GC-FID用的是N2)http://simg.instrument.com.cn/bbs/images/brow/emyc1010.gif期望高手们来解答一下,要是有理论材料说明的更好http://simg.instrument.com.cn/bbs/images/brow/em09505.gif
请问有没有这种可能?一直以为分流只是为了减少进入柱子的体积,可以避免柱子过载,但是发现好像分流后分离效果好了很多。原来紧紧挨着的峰现在保留时间差了1分钟还多。请问分流都有哪些优缺点啊?如何确定最好的分流比呢?
http://ng1.17img.cn/bbsfiles/images/2011/11/201111241556_332682_1835131_3.jpg 18PAH-TIChttp://ng1.17img.cn/bbsfiles/images/2011/11/201111241558_332684_1835131_3.jpg B(b,j,k)f & B(e,a)p看大家讨论18PAH比较多,下面也分享下我做的18PAH效果图。标准溶液浓度:200ng/ml实验过DB-5/HP-5,其中B(b,j)f很难得到很好的分离,用安捷伦DB-EUPAH分离效果图见上!DB-EUPAH对18PAH能有比较好的分离!
分离效果的好坏,取决于什么?
如果进行多种蛋白质的混合样品,在提高分离度上怎么才能得到提高呢?我现在用的是C8柱,流动相是乙腈和水,添加1%甲酸,梯度洗脱,但是效果不是很好,总有两三种蛋白分离不开,而且时间很长,一般要40分钟。我现在希望把分离时间缩短,几种蛋白尽量分开。大家有没有做过这方面的?
HP-5的色谱柱初始温度40,保持10min进样0.2ul,样品含有甲醇,乙腈,二氯甲烷,乙酸乙酯,丙酮五个峰在6min都出峰了,但都不能分开在不更换色谱柱的情况下,改变什么色谱条件可以改变分离效果?
[color=#000000]我现在在用凝胶色谱柱检测甘油一酯、二酯、三酯及游离脂肪酸的含量。流动相是四氢呋喃,检测是ELSD。在检测过程中,分离效果不稳定,同样的条件,有时候分离度可以达到大于1.5,有时候完全分不开。而且跑四氢呋喃空白,也会在目标峰的位置有信号响应,响应值也不小。[/color]
购买的毛细管柱KB-WAX 60m*0.32mm*0.25μm分析邻二甲苯的各个组分,用了不到两个月,分离效果就变得很差,分离不开,而且拖尾很严重,柱温100℃恒温,进样器280℃,检测器280℃,拿出色谱柱,发现色谱柱上有很多黑点,请问是什么原因?
气化室即进样口及内腔,属于进样系统的一部分,是气相色谱仪的重要组成部分。由于气相色谱柱所使用的温度一般不超过400℃,所以气化室温度低于400℃,经常根据化合物的需要设定为200-300℃左右。那么,气化室温度对样品的分离效果有什么影响呢?
请问一般讲氮气和氦气那个分离效果好?
请问:怎样才能装好色谱住,保证有较好的分离效果?
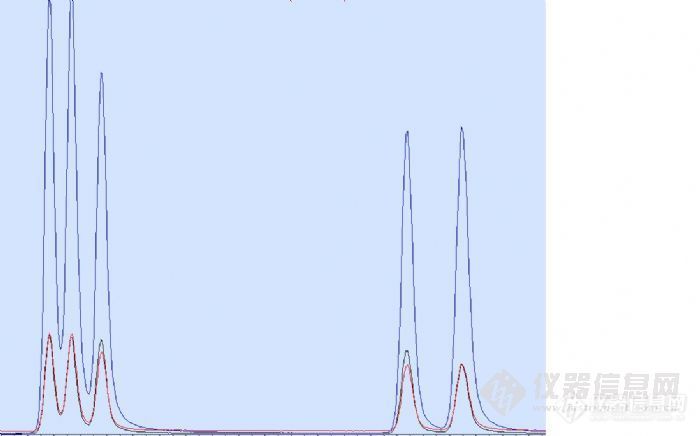
之前用6820分离三苯,用乙酸乙酯作溶剂,结果苯和溶剂峰分离的不好,请教各位大虾,改变色谱条件可以使效果好点吗,万分感谢?附上检测的图片。

1、进料温度浆液的温度,可以直接影响母液的粘度,通常来说,溶液温度越高,则粘度越低,固相上的液膜就越薄,细小粒子越容易沉降,毛细孔中所含液体越少,对于追求固相干燥度的离心机来说分离效果就会越来越好。http://ng1.17img.cn/bbsfiles/images/2015/09/201509291639_568537_3005330_3.jpg
色谱柱长对测定结果的影响有哪些?是否越长分离效果越好?
有没有人做过构象异构体气相分离的?什么色谱柱分离效果好一点
如果高效液相分离方法的是理论塔板数为2500~3500之间,是否意味着分离效果不好。
一般用于分离空气中的油的油分离器,可不可以用于分离氨气中的油,分离效果会不会有很大影响?
各位大侠, 我做利用C18柱子液相时突然想到,液相柱子对某个样品中不同组分的分离效果很好,那利用这根柱子的理化特性,有没有相似的填料如树脂、硅胶、氧化铝等,来实现组分分离,在生产上加以应用?谢谢!
之前和同事讨论分离效果,有人说C8的分离效果比C18的差,是这样吗
在做核苷酸分离实验,流动相是离子对缓冲盐溶液(四丁基硫酸氢铵+磷酸氢二钾):甲醇=1000:40,预混后的流动相pH值为3.43。 第一次用汉邦的C18柱做,分离效果很好。第二次做重复试验时,条件完全相同,柱子也相同,结果却完全分不开了。 后来又换了根德国某品牌的C18柱做,也是第一次做效果很好,第二次做,同样的条件同样的柱子,又完全分不开了。 每次用完后,都是先用5%的甲醇冲洗1小时后,梯度升到100%甲醇再冲洗至少两小时。 请教前辈,是什么原因导致实验无法重复?用离子对试剂做流动相时有什么要注意的吗?[em09512]
反相液相色谱中 调整流动相组分比例影响分离效果 是通过改变流动相比例改变流动相极性 来改变分离效果的吗
气象色谱分离效果不明显什么原因,峰分不开,有些组分出不来
各位同仁,大家好,请问HPLC柱子的直径对药物分离效果有影响吗?如出峰时间及峰形,谢谢
国产sephadex LH-20的分离效果和进口的比较,质量差别大吗?有谁比较过,请赐教,谢谢。
如题。一般情况下,为了提高分离度,会选择增加柱温来达到目的。那么,柱温提高后,就一定有利于分离效果吗?
因为近期实验室可能姚进行蛋白质组分的鉴定,可能姚用到HPLC,但是有人说,用HPLC鉴定&分离蛋白质的效果不好,详听听各位的看法!谢谢







 我要推广仪器
我要推广仪器
 下载APP
下载APP