用IC,设备太贵,只要求对产品痕量磷酸根离子进行测试,是否有针对性强相对IC便宜的设备推荐?谢谢
各位好,最近我们用万[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]做痕量硫酸根,不知为什么,一旦浓度低于50 ppb,就会在硫酸根位置出负峰。后来咨询了仪器公司的工程师,他们开始说是我们实验室的水不好,后来借了其它实验室的饿Millipore的水,还是不行,后来他们又告诉我们不要用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]做痕量硫酸根,而应该用电位滴定,可是当时购买的时候明明说可以做到0.5 ppb的啊。我们用的是MIC,据说这是最好的[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]了,为什么连硫酸盐都做不了啊?难道是我们实验室的水的问题吗?还是我们的水平问题?我们确实没有做过[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url],不过以前做过液相的,应该差不多吧?做痕量硫酸盐需要注意什么呢?求各位大侠指点迷津!
大家MS方法设置[font=Arial, sans-serif][size=13px]痕量离子检测勾不[/size][/font]
EDTA滴定痕量金属离子滴定痕量金属离子(0.1ppm-2ppm的Hg2+\Cu2+\Ni2+\Ca2+\Mg2+)用的EDTA的通常的浓度是多少?
各位老师,5975C工作站痕量离子检测(TID)这个你们开启吗?做农残检测需要用到这个功能吗?
对于电子产品、核电力等行业来说,水的纯度具有极其重要的地位。痕量离子都会使产品的纯度不达标而成为废品,或对电机表面产生腐蚀作用。离子色谱是快速、灵敏测定阴阳离子的好方法,已成为精细产品制造业必备的仪器,以直接进样的方式可以测定ug/L级的离子。经浓缩富集,可以测定至ng/L级。本文使用青岛普仁仪器有限公司生产的PIC-10型离子色谱仪(配有五极电导检测器)对纯水中痕量的F-和Cl-进行分析,优化了色谱条件,以直接进样的方式可灵敏的测定几个ug/LF-和Cl-,得到了较好的结果。 色谱条件: 离子色谱仪:PIC-10型,青岛普仁仪器有限公司(配有五极电导检测器) 色谱柱:Shodex 52 4E (4.0*250mm) 淋洗液:Na2CO3+NaOH 流速:0.7mL/min 检测器:抑制电导检测 进样体积:100uL 色谱柱及检测器温度:36℃ 色谱条件的选择与优化: 为灵敏的测定ug/L级离子,需使用高效的离子交换柱,本文使用Shodex 52 4E (4.0*250mm)色谱柱,该色谱柱对SO42-离子的塔板数可达14000/m,是测定痕量离子的首选。但该色谱柱推荐使用的淋洗液为3.6mMNa2CO3,其淋洗离子仅有负二价的CO32-;由于碳酸盐淋洗液在抑制电导检测中存在水负峰,对弱保留的F-定量产生一定的干扰,淋洗液中CO32-的浓度变化也不会对负一价的F-、Cl-的保留时间、信噪比产生明显的影响,因此降低淋洗液中CO32-的浓度并添加OH-作为负一价的洗脱离子。采用大体积直接进样的方式是测定痕量离子的常用方法,可省去浓缩富集的时间,但对于4mm内径的色谱柱而言,进样体积一般不超过200uL。通过不断尝试,笔者发现在100uL进样体积时,水负峰不干扰F-定量,且F-、Cl-能够达到较高的信噪比,因此使用100 uL进样体积。 实验前期准备 使用电阻率大于18.2兆欧的水清洗容量瓶3-5遍,并注满容量瓶,盖紧瓶盖,浸泡4小时。使用优级纯的试剂配制F-、Cl-溶液,ug/L级的F-、Cl-现用现配,并在配置后6小时内使用。 实验结果 首先测定18.2兆欧去离子水的空白,色谱图如下所示。http://www.qdpr.com/uploads/161121/2_110622_1.jpg 从色谱图中可以看出,去离子水在8.3min和11.5min检出两种物质,其中11.5min的色谱峰与Cl-保留时间一致,说明去离子水中存在痕量的Cl-(信噪比为4.6)。F-(保留时间为6.2min)并未检出。 使用容量瓶配制各种溶液之前,使用去离子水反复清洗并浸泡4小时。浸泡4小时后,测定溶液的空白值,其色谱图如下所示。http://www.qdpr.com/uploads/161121/2_110650_1.jpg 从色谱图中可以看出,去离子水在浸泡容量瓶4小时后,有三种物质被检出。此样品中Cl-的信噪比为5.4,F-同样未检出。 在测定了去离子水和容量瓶的空白后,将含有2.5ug/LF-、Cl-的溶液注入离子色谱仪,得到以下色谱图。http://www.qdpr.com/uploads/161121/2_110727_1.jpg 从色谱图中可以看出,F-、Cl-在色谱图中均明显检出,此外还有三种未知组分。F-、Cl-的信噪比分别为3.4和4.0(Cl-未扣除空白)。 将含有5.0ug/LF-、Cl-的溶液注入离子色谱仪,得到以下色谱图。http://img60.chem17.com/9/20161121/636153382766497363235.jpg 从色谱图中可以看出,F-、Cl-在色谱图中均明显检出,此外还有两种未知组分。F-、Cl-的信噪比分别为6.0和8.0(Cl-未扣除空白)。结语 使用国产的PIC离子色谱仪和五极电导检测器,能够灵敏的测定ug/LF-和Cl-。去离子水和容量瓶空白样品表明,F-在该色谱条件下不存在干扰,可灵敏、准确的测定。空白样品中存在痕量的Cl-,其信噪比大于三,因此对实际样品中几个ug/LCl-只能灵敏而不能准确的测定。使用更加纯净的水作为溶剂,当去离子水和容量瓶等空白样品中Cl-低于三倍的信噪比时方能准确测定痕量的Cl-。参考文献略作者:青岛普仁仪器有限公司 王存进
摘 要:随着分析科学的不断发展,常用的元素分析方法,如光谱技术AES ,AFS) 和质谱等已不能满足环境和生物样品中痕量、超痕量元素的赋存形态分析。以色谱联用技术为代表的元素形态分析测试技术(如:液相色谱- 原子光谱联用、色谱- 电感耦合等离子质谱联用、毛细管电泳- 电喷雾离子化质谱联用技术等) 已成为国内外研究的热点。本文扼要的介绍了近年来国内外在环境和生物样品中痕量、超痕量元素砷、硒、汞形态分析的色谱联用技术研究进展,并侧重于样品前处理方法、痕量或超痕量元素的形态分析技术。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=31681]色谱联用技术在环境和生物样品中痕量超痕量元素形态分析研究进展[/url]
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=103305][url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定饱和卤水中痕量碘离子[/url]
最近要测定次磷酸根(H2PO2-),离子色谱是用的最常用的KOH淋洗系统,不知道是否可以测定次磷酸根。我粗略测了一下,配制约1mg/L的溶液,在6min左右有一个巨大的色谱峰(F离子的保留时间也在这附近),稀释一倍后,峰也减少了约一倍,现在在考虑这个峰是不是次磷酸根。但存在两个问题:1. 次磷酸根的保留时间是否应该是比较小的,在cl之前,跟f的保留时间差不多。2.同浓度下(1mg/L),次磷酸根的峰高/峰面积远高于其他离子,估计有100倍左右了,所以觉得有点奇怪。
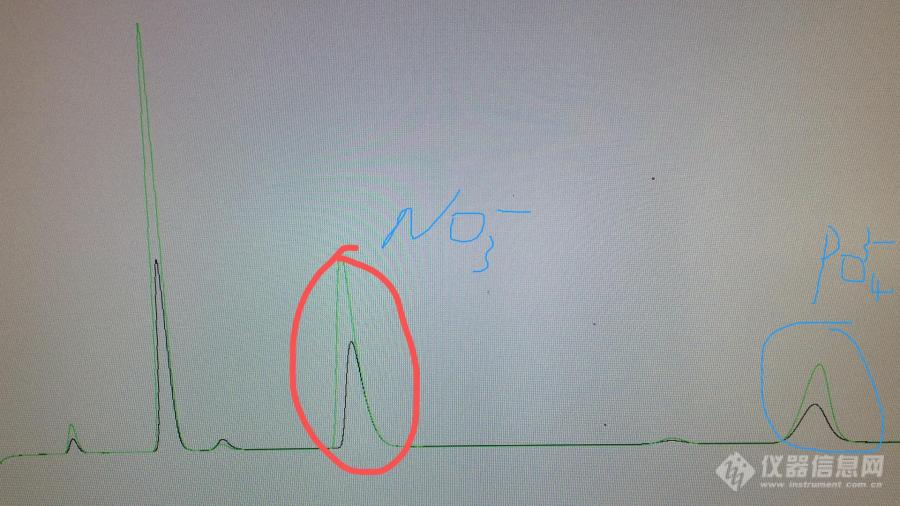
同事做氟离子、氯离子、硝酸根离子、磷酸根离子 ,氟离子和氯离子出峰时间差不多,硝酸根离子高浓度的时间会前移、磷酸根离子高浓度的会延时;请问这个是什么原因啊?(硝酸根离子浓度是1、20PPM;磷酸根离子浓度是0.5、10PPM)[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2019/04/201904031533437041_1228_3236392_3.jpg!w690x387.jpg[/img]
小弟所在的公司是生产IC厂和光伏厂用高纯化学试剂的,由于这两年太阳能光伏产业遭受打击,公司欲重点开发IC厂的客户。可是他们的要求好高啊:比如,浓硝酸中氯离子,硫酸根离子都要限制在0.5PPM之内。这个测试真让人头疼。不能上离子色谱,高浓度硝酸根离子严重影响测定。阶段采用,富集,提高氯离子和硫酸根离子的含量,然后再测浊度(氯转化为氯化银,硫酸根转化为硫酸钡。)可是,硫酸根的标线很难做出很好的线性,数据不敢报出去吧http://simg.instrument.com.cn/bbs/images/default/em09509.gifhttp://simg.instrument.com.cn/bbs/images/default/em09509.gif
[font=&]【题名】: 用pCl电极测定痕量氯离子的简便方法——两点定位比较法[/font][font=&]【全文链接】: https://www.cnki.com.cn/Article/CJFDTOTAL-HDDL198405003.htm[/font]
想测定液体中的磷酸根和亚磷酸根,离子色谱能不能测?如果能测的话选什么条件?谢谢?
看了不少资料用石墨炉原子吸收法测定土壤中痕量镉,本人按方法做了一遍,可以标样结果偏高很多,不知是什么原因。采用的是硝酸,氢氟酸和高氯酸溶样,磷酸二氢铵做基改,不知坛子里大神用何种方法,求指教~
我们用的ics1000[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]阴离子分析做的标线里有磷酸根离子但是做磷酸根离子
刚接触[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url],要扩项,做电厂水汽中痕量阴离子,现在就有氯离子做样,用的ICS-1100,AS19+AG19+抑制器,4mm的,配的20mMol氢氧化钾淋洗液,等度。样品浓度5,10,15,20ug/L。。请大神解答一下,为什么浓度越高,反而峰高峰面积越小了?然后是请教一下现有的配置能不能做痕量的阴离子,如果不能需要更换什么?[img]https://ng1.17img.cn/bbsfiles/images/2022/08/202208111604365543_5297_5501758_3.png[/img]
各位专家,我有一种高纯气体(6N)需测量其中痕量C、O(1ppm)离子,但不知其中含C、O的确切组分,如何定量?感谢!
大体积直接进样[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定饮用水中痕量溴酸盐史亚利1,2,蔡亚岐1,刘京生1,牟世芬1*,温美娟2(1.中国科学院生态环境研究中心环境化学与生态毒理学国家重点实验室,北京 100085;2.北京科技大学化学系,北京 100083)摘要:本文建立了一种无需样品前处理,直接大体积进样,电导检测饮用水中痕量溴酸盐的[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]新方法。分析柱为容量高、亲水性强的Dionex IonPac AS19阴离子交换柱,EG40在线产生KOH淋洗液,等浓度泵作梯度淋洗。该方法对溴酸盐的检出限为0.2µ g/L,在1~100µ g/L范围内具有良好的线性(r=0.9996)。将该方法用于北京市自来水和市售瓶装水样品中痕量溴酸盐的检测,实际样品的加标回收率在90%~106%之间,1µ g/L溴酸盐连续进样10次,相对标准偏差(RSD)为5.7%。关键词:溴酸盐,大体积直接进样,[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url],饮用水请到以下地址下载PDF全文阅览:http://www.instrument.com.cn/netshow/SH100244/paperDetail.asp?ID=12003[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=7547]相关附件[/url]
利用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定磷酸二氢锂固体中氯离子和硫酸根离子含量时,发现氯离子前面出现负峰,很容易影响氯离子的定量结果。在后面还会出现很大的一个峰,但不是在磷酸根出峰时间出的。样品的处理方式主要为称取一克固体样品溶解在100毫升水中,之后将样品溶液过氢柱后进样测定。求助一下,有做过类似的实验,或者出现过类似情况的吗?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/02/202202181331448878_8181_3920308_3.png[/img]
[align=center]痕量分析时的注意事项[/align] 在一些特殊行业中,需要测试一些超痕量的离子含量,因此,怎么检测含有超痕量离子的样品是非常重要的。超痕量分析的关键在于富集样品,可以通过富集柱或者大体积进样方式,与此同时,原本的系统污染也会随之同步增大,会对检测结果造成干扰,使得结果不可信。因此,如何减少系统污染,对于超痕量分析来说是重中之重。1 系统污染的来源及防控进行超痕量检测时,最大的难点在于系统的污染。污染物会进入试剂或样品中,对检测结果造成干扰(直接干扰与间接干扰如溶出与目标峰相邻的峰的离子)。这里把可能的系统污染列举下。①来自空气中的污染及其防控:自然界空气中含有各种气体、液体和固体颗粒物,如气溶胶和尘埃,这些污染物质通过各种渠道进入分析实验室。空气中的污染物也可能来源于实验室内的各种仪器、设备、试剂和分析人员(头发、皮肤屑、衣物和化妆品)。挥发性物质的污染:如氯化氢、氨、汞蒸气、挥发性有机物等。比如笔者到过一个实验室,其需要检测某样品中的氯离子含量,但这个实验室同时使用盐酸提取其它样品,这样氯离子检测结果就很可能偏高。为了减少空气污染对超痕量分析的影响,我们要充分利用洁净实验室、洁净通风柜、超净工作台、手套箱以及各种封闭装置。对于有条件的实验室,通风用的空气亦需过滤。工作时穿戴手套、工作帽、工作服和工作鞋,避免皮肤、头发和衣服上的微粒带进实验室并在空气中传播造成污染。其中手套必须不透皮肤油脂和汗水,不得用滑石粉润滑。PVC、聚乙烯、乳胶和丁睛手套都能很好的防止物理及化学污染。②来自设备设施等污染及其防控:直接与样品接触的容器或其它设备的表面可能因为溶出从而引起污染。玻璃或石英材质的表面存在一层很薄的活化层,使容器表面与溶液中的离子之间可能发生吸附、离子交换、渗透等复杂的物理化学变化。而且玻璃及石英不能作为碱及氟化物的容器。有些钠玻璃不适合用来做超痕量分析——因为其中的钠离子等组分会溶入样品及试剂中。聚四氟乙烯(PTFE)和聚丙烯(PP)等高分子聚合物材料的容器具有良好的耐化学腐蚀性,但不耐高温,且有透气性,不能长时间保存有挥发性的溶液。某些品牌的PP瓶也存在溶剂吸附与溶出现象,为了减少吸附及溶出影响,低浓度的溶液需要现用现配,超纯水也需要现场制取,不宜久存。③来自试剂的污染及其防控:主要是实验用水易受污染,导致结果出现偏差。这里推荐使用怡宝、娃哈哈、屈臣氏等品牌的纯净水经过超纯水机(需要勤换耗材)过滤后的超纯水作为实验用水,并且使用时要现场制水,因为空气中的二氧化碳会溶于水中并电离出碳酸根离子干扰分析。2 如何评估实验过程中的污染与损失评估实验过程中的污染与损失,进行空白实验是可行的方法。在相同条件下,同样品分析平行地进行“空白实验”,所得的“空白值”要从分析值中扣除。那么,我们是否可以通过扣除某次空白值的方法有效地将沾污等因素加以准确校正呢?其实是不能的。因为很多类型的污染并不能重现,例如空气污染的程度随时间和地点的不同而不同,由容器表面吸附及溶出造成的污染与容器的材质、品牌、所用的清洗方法以及样品分析和空白实验中溶液的组成的差异有很大的关系。另外,在进行样品分析和空白实验时,待测超痕量离子的损失可能同污染一起以不同的方式同时发生。在偶然情况下污染和损失可能会互相抵消。所以,扣除空白值的操作仅限于空白实验和样品实验同批进行时才有效。总之,对于痕量分析来说,只有从人机料法环各个方面都进行污染排除,才能将分析做好。
2019.3月做磷酸根(厂家带的标液),最低点做到了0.25mg/L,线性也很好。4月再做响应衰减1mg/L的没有峰了,最高点衰减好几倍。现在更是做不出来。问工程师说我们标准溶液有问题,按照标准上称量磷酸二氢钾1mg/L依然没风,网上看资料说柱效低了,这个别的离子没有问题,不解
在痕量,超痕量ICP分析中(纯度99.999%)对于环境,水质,溶剂(酸)有没有特殊的要求? 谁有这样的文献和相关资料, 麻烦发一份给我,谢谢!我的邮箱:gongmm@lsam.cn
岛津GC-2014C的仪器上,在SPL进样口的进样模式里面有不分流进样模式,不分流进样模式是针对于痕量化合物组分的分析,这个痕量有没有标准呢?多少个ppm才算是痕量呢?
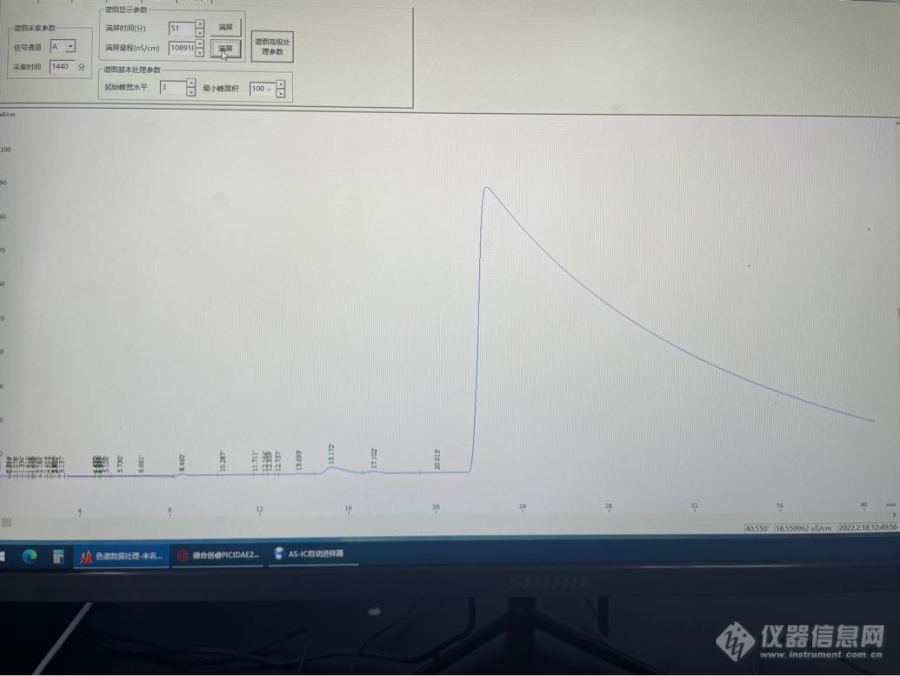
仪器型号:赛默飞 dionex ICS-600色谱柱:Thermo AS23(4×250mm)+AG23(4×50mm)淋洗液:0.8mmol/L碳酸氢钠+4.5mmol/L碳酸钠流速:1.0mL/min抑制器型号:Dionex? AERS? 500抑制器电流:30mA现象描述: 最近用此款[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱仪[/color][/url]以及上述色谱条件,检测某样品中磷酸根含量(预实验得出含量在40mg/L左右,保留时间在19.7min左右)。在方法学开发时,发现连续进样后,色谱图会出现明显的基线漂移,而且这种漂移会严重影响后面几次进样的色谱图,歪七扭八的。 尝试在样品之间插入2-3针纯水作为空白过渡一下,结果这些空白的色谱图,从磷酸根的保留时间开始,会出现一个很大的馒头峰,同样会严重影响后面几次进样色谱图的形状和基线。 考虑到预实验中样品的含量,这次做的方法学开发,线性范围选择了10、20、40、60、80、100mg/L这几个点。因为磷酸根浓度到了1mg/L,色谱峰的峰型很差,而且信噪比达不到3。 请问各位大神,基线漂移的问题有没有什么办法改善或解决?
火力发电厂[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定痕量阴离子--电力部颁标准方法(正式版)[~86829~]
请教各位大虾,有谁做过双氧水中痕量三价铁离子的含量分析?[em0808] [em0808] [em0808]
【题名】:磷酸根离子选择电极的研究与开发【全文链接】: https://cdmd.cnki.com.cn/Article/CDMD-10295-1016265033.htm
如题,离子色谱测磷酸根离子配的5,10,20,40,50ppm的浓度配了很多次都不成线性是怎么回事?其他离子都正常,求助各位老师!
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=73759]等离子体质谱法测定多目标地球化学调查样品中30种痕量元素[/url]
用磷酸二氢钾配置的磷酸根浓标,做了两次标线都不成线性。仪器是戴安ICS-900, 柱子好像是AS-9c,具体记不清了,碳酸钠淋洗液,磷酸根出峰在硫酸根之前。其他阴离子线性都很好,就磷酸根差。标曲浓度梯度是0.5-10mg/L。都有出峰,就是没线性。







 我要推广仪器
我要推广仪器
 下载APP
下载APP