最近在做一个黄酮苷元的尿样分析项目,现在遇到的问题是在“空白(也就是未服药的人的尿样)”样品中也会有色谱峰出现。我用的是选择(二级)离子扫描模式(SRM),换了不同的柱子、流动相、正负离子扫描以及前处理方式(液液萃取、沉蛋白、SPE)结果都不行,郁闷死了,已排除样品污染,现在越来越怀疑“空白”样品中也含有目标成分,请教各位DX有没有什么好方法。
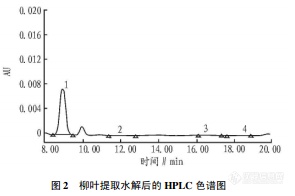
【作者】 刘卫; 邓淑凤; 刘景东; 秦葵; 孙晓丽;【机构】 第四军医大学白求恩军医学院;【摘要】 测定柳叶中黄酮醇苷元成分槲皮素的含量。于4月中旬采摘柳叶嫩叶,用无水甲醇连续提取,将提取液用盐酸水解后采用高效液相色谱法测定槲皮素的含量。色谱条件为:色谱柱为Diamonsil C18柱(150 mm×4.6 mm,5μm),流动相为浓度0.07%磷酸-无水甲醇(50∶50,V/V);流速为1.0 ml/min;检测波长为260 nm;进样量为10μl;灵敏度为0.000 1 AUFS;柱温为室温(20℃)。柳叶嫩叶中黄酮醇苷元以槲皮素为主,含量在0.36%~0.74%。柳叶中槲皮素含量测定方法的建立,为有效利用现有柳资源提供理论依据。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207241340_379384_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207241341_379385_2379123_3.jpg

大家好,我是新手,最近想要用气质检测一个黄酮苷元组分,我查了些资料,有报道表明HP-5-MS(长30m,膜厚0.25um,内径0.25mm)可用于分析BSTFA+1%TMCS衍生化的黄酮苷元(见附件),我们学校的气质柱为Rtx-5MS(长30m,膜厚0.25um,内径0.25mm),似乎HP-5-MS和Rtx-5MS都是非极性的?所以我就用Rtx-5MS进行了分析,结果如下:黄酮苷元不经过衍生化直接进样1ul,GC/MS只出现一个黄酮苷元峰(之前我HPLC-DAD分析过有15-20左右黄酮苷元);经过衍生化后进样1ul分析,一个峰都没有;非常郁闷,不知道啥原因?是柱子不适合分析黄酮苷元?还是其他参数条件设置有问题?希望各位高手多多指教!http://ng1.17img.cn/bbsfiles/images/2011/06/201106010914_297216_1736519_3.jpg
有高手做过黄酮醇苷的检测嘛??我现在要做,难做吗??急........我要用液相测定的是银杏提取液中的黄酮总量,包括槲皮素,异鼠李素,山奈素.....因为课题比较急,希望高手伸出援手,谢谢.......
请问下高手们,槐米中芦丁(芦丁又称芸香苷,属于黄酮苷)的提取时,可以选择碱溶酸沉的方法来提取,有点困惑的是苷的极性很大可以溶于水,而且苷不是在酸水中可以水解的吗,为什么可以用碱溶酸沉的方法来提取??
分析一中药山楂叶中金丝桃苷,出峰总是不好,总有一成分分不开,分离度不好的时候直接就叠成一个峰。因为山楂叶中还含有槲皮素(金丝桃苷的苷元也是槲皮素),请问分不开的这个峰是不是可能是槲皮素,怎么来检验呢?
各位老师,这是一黄酮苷类的化合物,母核是黄酮二糖苷,现在多出一组信号,无法拼出合理结构,δCδHDept170.6-C季碳107.54.16(s)-CH83.7-C季碳74.84.25(q)-CH60.24.05(q)-CH240.31.98(m)-CH222.41.20(t)-CH314.01.12(d)-CH3根据QC和BC,我们推出以下结构,通过chemdraw模拟了一下碳氢数据,也基本符合。但是感觉是不是不太符合生源关系。有那位大侠能给出一些建议。样品纯度不是很高,大概92%左右,高分辨扣除母核外,这个集团的分子式应为C8H14O5,已经加了一个氢。谱图都是纸版的,都没有扫描,麻烦大家先帮着看一下,给出一些可能的结构及推测,有见过类似集团的就更好了,谢谢了。
[color=#333333]甘草是一种药食同源的草本植物,广泛用于中药处方与食品工业中。现今使用的甘草主要为其根与根状茎,而地上部分却作为畜牧饲料或燃料低值化处理。目前关于甘草根及根状茎的成分及生理活性研究已相当充分,而对甘草地上部分却研究较少。本论文通过对比分析光果甘草(Glycyrrhiza glabra L.)叶与根中物质组成和生理活性,确定光果甘草叶的研究价值 通过色谱分离和光谱技术分离并鉴定了光果甘草叶中的黄酮并对其活性进行评价 优化了光果甘草叶黄酮的测定和提取方法,并通过大孔树脂对光果甘草叶黄酮进行富集研究 研究了光果甘草叶黄酮对猪肉及其制品储藏过程中油脂氧化和蛋白氧化的抑制作用,以期为甘草叶的高值利用提供理论指导。[/color]
我是新人,最近在实验中遇到了些麻烦,还要大家帮助一下啦麦冬中主要成分为多糖,皂苷,黄酮具体怎样用薄层方法鉴别这三种物质具体用什么展开剂,比例多少,显色剂是什么先谢谢大家了
[align=center][b]HPLC法测定黄芪水提物中的毛蕊异黄酮苷含量[/b][/align][b]黄芪中的主要有效成分除皂苷外就是异黄酮类化合物。异黄酮类成分具有调节免疫、抗肿瘤、抗突变、抗氧化、抗炎、抗突变抗辐射、抗心肌缺血、抗心律失常、抗病毒、抗细胞凋亡、保肝、防止动脉粥样硬化等作用[sup][/sup],其代表成分就是毛蕊异黄酮 7-O-β-D 吡喃葡萄糖苷(即毛蕊异黄酮苷)。2010版药典才开始将毛蕊异黄酮苷收录为黄芪中有效成分含量测定项。本章实验借鉴药典中的测定方法,对不同工艺条件下获得的黄芪水提物中的毛蕊异黄酮苷含量进行考察,以期为优化芪龙胶囊和黄芪配方颗粒中黄芪的提取工艺参数提供科学基础和理论依据。[b]1 材料和仪器1.1 样品 [/b] 收集9组黄芪水提取物样品,黄芪饮片为济南济成堂中药饮片有限公司提供(批号18033101)。[b]1.2 试剂 [/b]毛蕊异黄酮苷对照品(成都瑞芬思生物科技有限公司批号M-020-170926),乙腈为色谱纯(天津市科密欧化学试剂有限公司);甲酸(天津市科密欧化学试剂有限公司);超纯水。[b]1.3 仪器 [/b]液相色谱系统,包括日本岛津公司LC-20AT型液相色谱仪,LC-20AT岛津输液泵,CTO-20A柱温箱,SIL-20A自动进样器,SPD-20A紫外-可见光检测器;超声波清洗机KS-300E(宁波科生仪器厂);电子天平MS205DU(梅特勒/瑞士)。[b]2 方法学考察2.1 色谱条件及系统适应性试验[/b]DiamonsiL(钻石)C18柱(250*4.6 mm,5 mm)。以乙腈为流动相A,0.2%甲酸溶液为流动相B,梯度洗脱,A相:0→20 min A为20→40%;20→30 min A保持40%;30→40 min A保持20%。检测波长260 nm;流速为1 mL/min;柱温35 ℃进样量为10 μL。毛蕊异黄酮苷对照品溶液以及黄芪水提物样品色谱图见图4-1与图4-2。[/b][align=center][img=,690,365]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131706005051_4964_3237657_3.png!w690x365.jpg[/img][/align][align=center]图4-1 毛蕊异黄酮苷对照品色谱图[/align][align=center][img=,690,384]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131706118782_6652_3237657_3.png!w690x384.jpg[/img][/align][align=center]图4-2 黄芪水提物样品色谱图[/align][b]2.2 供试品溶液的制备[/b]精密称定1/50重量的黄芪水提物样品(折合黄芪药材2 g)置于锥形瓶中,精密加入50 mL甲醇,超声30 min,过滤,将滤液放至水浴锅上蒸干,残渣用甲醇溶解,并定容至25 mL,用0.22 μm的微孔滤膜过滤,即得。[b]2.3 对照品储备溶液的制备[/b]精密称取黄芪甲苷标品5.10mg至10 mL容量瓶中,用甲醇溶解后定容,摇匀,即得浓度为0.51 mg/mL的对照品储备溶液。[b]3 结果3.1 线性关系考察[/b]精密吸取 2. 3 项下对照品储备溶液 4,2,1,0.5,0.25 mL至10 mL容量瓶中,用甲醇定容,得到浓度为204.00,102.00,51.00,25.50,12.75 μg/mL的对照品溶液。按“2.3”项下色谱条件分别进样10 μL,利用自动积分功能测定峰面积积分值,并以峰面积积分值与浓度进行线性回归。如图3,得回归方程为:Y =25445X + 44225(r[sup]2[/sup]= 0.9994)提示毛蕊异黄酮苷在12.75~204.00 μg/mL范围内线性关系良好。毛蕊异黄酮苷对照品标准曲线如图4-3所示。[align=center][img=,690,406]https://ng1.17img.cn/bbsfiles/images/2019/08/201908131706302801_395_3237657_3.png!w690x406.jpg[/img][/align][align=center]图4-3 毛蕊异黄酮苷标准曲线[/align][b]3.2 精密度实验[/b]按2. 2 项下方法制备供试品溶液,精密吸取供试品溶液10 μL,重复进样 6 次,记录色谱图峰面积,测定毛蕊异黄酮苷含量,计算得相对标准偏差RSD为1.6% ,提示该方法精密度良好。[b]3.3 重复性实验[/b]取同一黄芪样品 6份,按“2. 2”项下方法制备供试品溶液,在拟定分析条件下,精密吸取供试品溶液10 μL,测定毛蕊异黄酮苷含量,计算得RSD为1.1%,提示该方法重复性良好。[b]3.4 稳定性实验 [/b]按2. 2 项下方法制备供试品溶液,分别在0 h、3 h、6 h、9 h、24 h、48 h后,准确吸取 10 μL 进样分析,测定毛蕊异黄酮苷含量,计算得 RSD 为 0.6%,提示黄芪供试品溶液在48h内稳定性良好。[b]3.5 加样回收率实验[/b]精密称取 6 份毛蕊异黄酮苷含量已知的黄芪水提物样品,每份折合黄芪药材 0. 5 g,分别准确加入样品中毛蕊异黄酮苷含量的50%,50%,100%,100%,150%,150%重量的毛蕊异黄酮苷标品,按2. 2 项下方法制备供试品溶液,准确吸取 10 μL 进样分析,测定毛蕊异黄酮苷含量,计算回收率,结果见表4-1。方法平均回收率为108.48%,表明该方法具有较好的回收率。[align=center]表4-1 毛蕊异黄酮苷加样回收率测定结果[/align] [table][tr][td] [align=center]样号[/align] [/td][td] [align=center]样品中的量/mg[/align] [/td][td] [align=center]加入量[/align] [align=center]/mg[/align] [/td][td] [align=center]测得量/mg[/align] [/td][td] [align=center]回收率[/align] [align=center]/%[/align] [/td][td] [align=center]平均值/%[/align] [/td][td] [align=center]RSD[/align] [align=center]/%[/align] [/td][/tr][tr][td] [align=center]1[/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]0.52 [/align] [/td][td] [align=center]1.64 [/align] [/td][td] [align=center]111.54 [/align] [/td][td=1,6] [align=center]108.85 [/align] [/td][td=1,6] [align=center]2.9 [/align] [/td][/tr][tr][td] [align=center]2[/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]0.52 [/align] [/td][td] [align=center]1.64 [/align] [/td][td] [align=center]112.08 [/align] [/td][/tr][tr][td] [align=center]3[/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]2.22 [/align] [/td][td] [align=center]109.20 [/align] [/td][/tr][tr][td] [align=center]4[/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]2.24 [/align] [/td][td] [align=center]111.02 [/align] [/td][/tr][tr][td] [align=center]5[/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]1.59 [/align] [/td][td] [align=center]2.72 [/align] [/td][td] [align=center]104.57 [/align] [/td][/tr][tr][td] [align=center]6[/align] [/td][td] [align=center]1.06 [/align] [/td][td] [align=center]1.59 [/align] [/td][td] [align=center]2.72 [/align] [/td][td] [align=center]104.67 [/align] [/td][/tr][/table][b]3.6 毛蕊异黄酮苷的含量测定结果[/b]取9组黄芪水提物按照“2.2”项下操作,制备供试品溶液,准确吸取 10 μL 进样分析,测定毛蕊异黄酮苷的含量。结果见表2。[align=center]表4-2 9组黄芪水提物中毛蕊异黄酮苷含量测定结果[/align] [table][tr][td] [align=center]批号[/align] [/td][td] [align=center]峰面积[/align] [/td][td] [align=center]含量(mg/g)[/align] [/td][/tr][tr][td] [align=center]1[/align] [/td][td] [align=center]1123989.50 [/align] [/td][td] [align=center]0.53 [/align] [/td][/tr][tr][td] [align=center]2[/align] [/td][td] [align=center]2707047.50 [/align] [/td][td] [align=center]1.31 [/align] [/td][/tr][tr][td] [align=center]3[/align] [/td][td] [align=center]1947171.50 [/align] [/td][td] [align=center]0.93 [/align] [/td][/tr][tr][td] [align=center]4[/align] [/td][td] [align=center]3573103.75 [/align] [/td][td] [align=center]1.73 [/align] [/td][/tr][tr][td] [align=center]5[/align] [/td][td] [align=center]1824562.00 [/align] [/td][td] [align=center]0.87 [/align] [/td][/tr][tr][td] [align=center]6[/align] [/td][td] [align=center]2767419.00 [/align] [/td][td] [align=center]1.34 [/align] [/td][/tr][tr][td] [align=center]7[/align] [/td][td] [align=center]2077551.50 [/align] [/td][td] [align=center]1.00 [/align] [/td][/tr][tr][td] [align=center]8[/align] [/td][td] [align=center]1677225.50 [/align] [/td][td] [align=center]0.80 [/align] [/td][/tr][tr][td] [align=center]9[/align] [/td][td] [align=center]1725416.00 [/align] [/td][td] [align=center]0.83 [/align] [/td][/tr][/table][b]3.7 毛蕊异黄酮苷转移率测定正交试验结果[/b]水提的毛蕊异黄酮苷转移率考察正交试验与水提的出膏率考察正交试验设计相同,即以水作为提取溶剂,把影响药材提取效果的用水量(A)、提取时间(B)、提取次数(C)确定为考察因素,以上三个考查因素各分3个水平考察,见表4-3。[align=center]表4-3水提实验因素水平表[/align] [table][tr][td=1,2] [align=center]水平[/align] [/td][td=3,1] [align=center]因素[/align] [/td][/tr][tr][td] [align=center]A(用水量/倍)[/align] [/td][td] [align=center]B(提取时间/h)[/align] [/td][td] [align=center]C(提取次数/次)[/align] [/td][/tr][tr][td] [align=center]1[/align] [/td][td] [align=center]4[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]1[/align] [/td][/tr][tr][td] [align=center]2[/align] [/td][td] [align=center]8[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]2[/align] [/td][/tr][tr][td] [align=center]3[/align] [/td][td] [align=center]10[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]3[/align] [/td][/tr][/table][b] [/b]毛蕊异黄酮苷转移率=各实验组黄芪提取物中毛蕊异黄酮苷含量/原黄芪药材中毛蕊异黄酮苷含量×100%。原药材中毛蕊异黄酮苷的含量按照药典的方法测得的结果为0.1425mg/g。跟据实验数据,得到水提实验中设定的不同工艺条件下的毛蕊异黄酮苷的转移率,其中因素D为误差项,作直观分析表和方差分析表,见表4-4,4-5。[align=center]表4-4 毛蕊异黄酮苷转移率考察L[sub]9[/sub](3[sup]4[/sup])正交试验表[/align] [table][tr][td] [align=center]批号[/align] [/td][td] [align=center]A[/align] [/td][td] [align=center]B[/align] [/td][td] [align=center]C[/align] [/td][td] [align=center]D[/align] [/td][td] [align=center]毛蕊异黄酮苷[/align] [align=center]转移率/%[/align] [/td][/tr][tr][td] [align=center]1[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]37.21[/align] [/td][/tr][tr][td] [align=center]2[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]91.77[/align] [/td][/tr][tr][td] [align=center]3[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]65.58[/align] [/td][/tr][tr][td] [align=center]4[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]121.62[/align] [/td][/tr][tr][td] [align=center]5[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]61.36[/align] [/td][/tr][tr][td] [align=center]6[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]93.85[/align] [/td][/tr][tr][td] [align=center]7[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]70.08[/align] [/td][/tr][tr][td] [align=center]8[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]56.28[/align] [/td][/tr][tr][td] [align=center]9[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]3[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]57.94[/align] [/td][/tr][tr][td] [align=center]K1[/align] [/td][td] [align=center]194.56[/align] [/td][td] [align=center]228.91[/align] [/td][td] [align=center]187.34[/align] [/td][td] [align=center]156.51[/align] [/td][td] [align=center] [/align] [/td][/tr][tr][td] [align=center]K2[/align] [/td][td] [align=center]276.83[/align] [/td][td] [align=center]209.41[/align] [/td][td] [align=center]271.33[/align] [/td][td] [align=center]255.70[/align] [/td][td] [align=center] [/align] [/td][/tr][tr][td] [align=center]K3[/align] [/td][td] [align=center]184.30[/align] [/td][td] [align=center]217.37[/align] [/td][td] [align=center]197.02[/align] [/td][td] [align=center]243.48[/align] [/td][td] [align=center] [/align] [/td][/tr][tr][td] [align=center]优水平[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]1[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center] [/align] [/td][/tr][tr][td] [align=center]R[/align] [/td][td] [align=center]92.53[/align] [/td][td] [align=center]19.50[/align] [/td][td] [align=center]83.99[/align] [/td][td] [align=center]99.19[/align] [/td][td] [align=center] [/align] [/td][/tr][/table][b] [/b][align=center]表4-5 毛蕊异黄酮苷转移率考察方差分析表[/align] [table][tr][td] [align=center]方差来源[/align] [/td][td] [align=center]离差平方和[/align] [/td][td] [align=center]自由度[/align] [/td][td] [align=center]F[/align] [/td][td] [align=center]显著性[/align] [/td][/tr][tr][td] [align=center]A[/align] [/td][td] [align=center]1715.05[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]1.50[/align] [/td][td] [align=center]-[/align] [/td][/tr][tr][td] [align=center]B[/align] [/td][td] [align=center]64.09[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]0.04[/align] [/td][td] [align=center]-[/align] [/td][/tr][tr][td] [align=center]C[/align] [/td][td] [align=center]1407.78[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]1.13[/align] [/td][td] [align=center]-[/align] [/td][/tr][tr][td] [align=center]方差来源[/align] [/td][td] [align=center]离差平方和[/align] [/td][td] [align=center]自由度[/align] [/td][td] [align=center]F[/align] [/td][td] [align=center]显著性[/align] [/td][/tr][tr][td] [align=center]D [/align] [/td][td] [align=center]1950.20[/align] [/td][td] [align=center]2[/align] [/td][td] [align=center]-[/align] [/td][td] [align=center]-[/align] [/td][/tr][/table]注:F[sub]0.1[/sub](2,2)=9,F[sub]0.05[/sub](2,2)=19,*为有显著性,-为无显著性。从正交试验结果可知:水提实验中,各因素对毛蕊异黄酮苷转移率的影响大小顺序为:A(用水量)C(提取次数)B(提取时间);每个因素3水平之间的趋势为A[sub]2[/sub]A[sub]1[/sub]A[sub]3[/sub],B[sub]1[/sub]B[sub]3[/sub]B[sub]2[/sub],C[sub]2[/sub]C[sub]3[/sub]C[sub]1[/sub],直观分析得提取工艺为A[sub]2[/sub]B[sub]1[/sub]C[sub]2[/sub],即加水8倍量,提取2次,每次1h。表4-5的方差分析结果表明: A、B、C三因素的对毛蕊异黄酮的转移率影响都无统计学差异(PC(提取次数)B(提取时间);每个因素3水平之间的趋势为A[sub]2[/sub]A[sub]3[/sub]A[sub]1[/sub],B[sub]1[/sub]B[sub]3[/sub]B[sub]2[/sub],C[sub]2[/sub]C[sub]3[/sub]C[sub]1[/sub],直观分析得提取工艺为A[sub]2[/sub]B[sub]1[/sub]C2,即加水8倍量,提取2次,每次1h。表4-7的方差分析结果表明:A、B、C三因素对综合评分的影响都无统计学差异(P0.05)。[b]4 讨论[/b]本章实验借鉴药典中测定黄芪药材中毛蕊异黄酮苷含量的方法,利用紫外-可见光检测器的高效液相色谱仪测定黄芪水提物中毛蕊异黄酮苷的含量。与药典中方法测得的结果相比,本实验测得的结果除色谱峰分离度稍差外,其他方法学考察指标显示良好。从毛蕊异黄酮苷转移率测定正交试验结果来看,第4组实验毛蕊异黄酮苷转移率最高,这也是正交结果分析中最佳提取工艺,即加水8倍量,提取2次,每次1小时。本结果与上一章实验中第四组黄芪水提物中黄芪甲苷的转移率最高结果一致,但其他组别毛蕊异黄酮苷的转移率高低顺序与上一章黄芪甲苷的转移率高低顺序并不一致,这说明毛蕊异黄酮苷和黄芪甲苷对提取工艺的要求并不完全一致。同一种原药材,加工成不同功效的药物,那么发挥药效的物质也有可能不同,因此相应的提取工艺也是需要根据药效物质适时调整的。另外,用水量在设置的三个因素中对毛蕊异黄酮苷的转移率影响最大,但仍无统计学差异(P0.05),说明用水量、提取次数、提取时间三种工艺的改变对黄芪水提物中毛蕊异黄酮苷的含量无显著性影响。从综合评分计算正交试验结果来看,第4组实验综合评分最高,这也是正交结果分析中最佳提取工艺,即加水8倍量,提取2次,每次1小时。用水量在设置的三个因素中对综合评分影响最大,但仍无统计学差异(P0.05),说明用水量、提取次数、提取时间三种工艺的改变对黄芪水提工艺的综合评分无显著性影响。综合出膏率、黄芪甲苷和毛蕊异黄酮苷的含量得出综合评分来优选黄芪水提的最佳提取工艺,能够从化学成分的角度来客观全面地评价和研究黄芪水提的关键环节,这也为芪龙胶囊和黄芪配方颗粒水提环节工艺的优化提供借鉴和指导。[align=center] [/align][align=center]参考文献[/align] 陈建真,吕圭源, 叶磊, 等.黄芪黄酮的化学成分与药理作用研究进展. 医药导报, 2009, 28(10): 1314-1316. 赵四清,周日宝, 陈胜璜, 等.不同的产地加工方法对中药材金樱子质量的影响. 湖南中医学院学报, 2005, 25(3): 21-22.
查了有关甘草黄酮的文献资料,用紫外分光光度计,500nm处检测,溶剂用80%乙醇。
花青苷和黄酮类(多酚类化合物Anthocyans and flavonoids)分子结构特点:含有苯并吡喃环,它们是植物组织中的水溶性色素的主要成分,具有各种色泽。常见有三种类型:花青苷素、黄酮类和儿茶素,均属于多酚化合物类,大量存在于自然界中。1.花青苷的色泽——红色(1)花青苷在酸性条件下呈红色,在中性条件下呈无色,在碱性条件下呈蓝色。(2)金属离子sn2+、Fe2+、Cu2+、Al3+与花青苷结合使花青苷呈蓝色。(3)亚硫酸盐类可以对花青苷进行漂白使之褪色,通过加热或酸化处理可去掉亚硫酸,使花青苷再生,重新恢复原来的红色。2.无色花青苷(Leucoanthicyanins)无色花青苷的结构与花青苷相似,以三聚体以上存在。自然界中无色花青苷存在于苹果、梨、葡萄和山楂等水果之中。它们与涩味有关,在无机酸中加热可以转化为花青苷,可参加酶促褐变反应。既可赋予食品(如酒、茶、香蕉、巧克力、越橘)以特殊的风味,也可影响食品的色泽,如使罐头果肉变红、变褐,在啤酒或其他酒中形成混浊物。3.黄酮类黄酮是一种多种多样,广泛存在着的呈无色至黄色的色素,其结构上与花青苷类不同之处在于它具有的是苯并吡喃酮结构。黄酮类的热稳定性比花青苷类好,热加工对它们的破坏不大。一些金属离子形成深色化合物,往往会造成食品的异常色泽。4.单宁(Tannin)来源:单宁存在于柿子、茶叶、咖啡、石榴等植物组织中,在未成熟时含量尤为多,它们的结构较为复杂,多是高分子多元酚类的衍生物,水解后可生成葡萄糖、没食子酸或其他多酚酸(鞣酸)。性质:①单宁与食品的涩味有关,能参加酶促褐变反应;②与Fe3+形成黑色物质;③与蛋白质形成不溶性沉淀可以用来对果汁的澄清;④含单宁高的植物可以作为制革工业中的植物性鞣质原料。
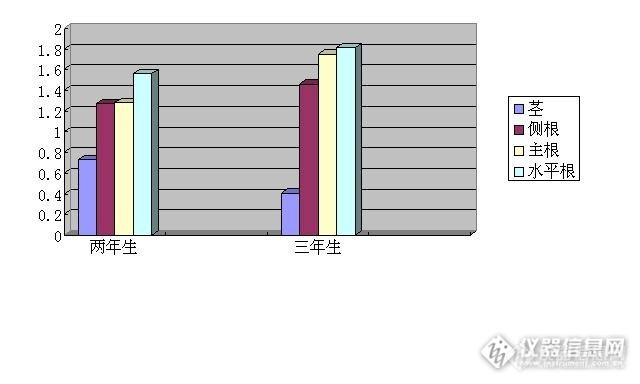
本实验分别提取两年生、三年生无腺毛甘草主根、侧根、水平根及茎中的黄酮,并测定其含量,初步找出消长规律。以甲醇为提取溶剂,索氏提取法提取黄酮,分光光度法测定总黄酮含量。结果表明两年生无腺毛中主根、侧根、水平根及茎中的黄酮含量分别为1.28%、1.28%、1.56%、0.73%;三年生无腺毛中主根、侧根、水平根及茎中的黄酮含量分别为1.75%、1.46%、1.81%、0.41%。随着年限的增大,无腺毛甘草中根部的黄酮含量逐渐增大。同龄期无腺毛甘草中黄酮含量:水平根最高,主根和侧根次之,茎最小。 无腺毛甘草 黄酮 含量测定 消长规律1.前 言无腺毛甘草(Glycyrrhiza eglandulosa X.Y.Li)是石河子大学李学禹教授在新疆发现、1993年命名的新种,也是我国的特有种。它生长的环境条件比乌拉尔甘草(G. uralensis)更恶劣,因而更加耐干旱、耐盐碱,而且是很适合于低产地、弃耕地等恶劣环境条件下生长的抗逆性极强的甘草物种。这个种在形态分类系统学、细胞学、生态学都有研究,根据细胞染色体组型分析与形态数值分析,都证实它与乌拉尔甘草、光果甘草亲缘关系较近,但在化学成分方面未进行系统研究。根据资料报导,产于新疆的胀果甘草(G.inflata)、黄甘草(G.eurycarpa)及云南的云南甘草(G.yunnanensis)都发现过大量的具有生理活性的新的化学成分。由此可知:一个形态性状特异、分布于逆境条件下能正常生长的新物种,肯定具有抗逆性较强的基因所决定的代谢产物,肯定有特殊性。为此,我们拟从应用开发的角度,提取无腺毛甘草中黄酮类化合物,对其进行含量测定,并进一步研究其在不同龄期、不同部位黄酮类化合物的消长规律,为进一步研究其化学组成和结构打下基础,并且为退耕还草大面积栽培无腺毛甘草提供科学依据。2.实验部分2.1实验仪器、样品与试剂2.1.1实验仪器:紫外可见分光光度计(上海棱光技术有限公司),索氏提取器(自治区化玻站提供),烘干箱(湖北省黄石医疗器材厂),电光分析天平(上海棱光技术有限公司),精密PH计(上海雷磁仪器厂)。2.1.2实验样品:两年生、三年生的无腺毛甘草(石河子大学甘草栽培基地李学禹教授提供)。2.1.3实验试剂:柚皮苷对照品(中国药品生物制品检定所),10%的氢氧化钾溶液,甲醇(AR)。2.2黄酮类化合物的提取分别称取3份两年生无腺毛甘草的主根粉碎后放入索氏提取器中,加入甲醇回流2h,冷却后分别将回流液转移至容量瓶中,提取两次,提取液合并,用甲醇定容。用同样的方法分别提取两年生无腺毛甘草的水平根、侧根和茎以及三年生无腺毛甘草中主根、侧根、水平根、茎中的黄酮。2.3黄酮类化合物的含量测定2.3.1对照品溶液的制备准确称取柚皮苷对照品适量,用甲醇溶解并定容于10mL容量瓶中,制得浓度约为1.0mg.mL-1的对照品溶液。2.3.2最大吸收峰的确定精确吸取柚皮苷对照品溶液0.5ml,加入5ml甲醇,再加入10%KOH溶液2.5ml,室温放置5min,用甲醇稀释至50ml[
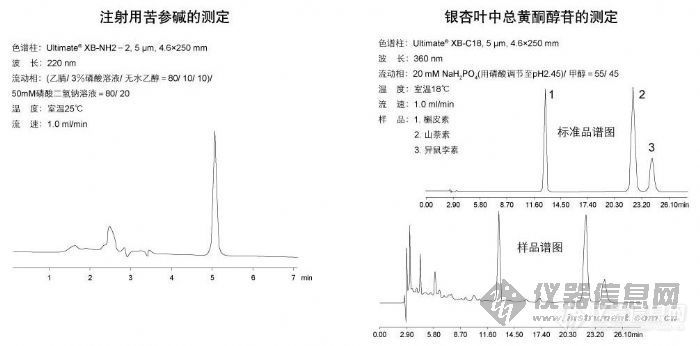
注射用苦参碱的测定和银杏叶中总黄酮醇苷的测定http://ng1.17img.cn/bbsfiles/images/2009/10/200910161352_175975_1896702_3.jpg
各位老师,帮忙想个办法能从黄芪蜜中分离出来毛蕊异黄酮葡萄糖苷
检测中药中的多糖,皂苷,黄酮,木脂素用什么液相色谱柱?
黄芪净药材—毛蕊异黄酮葡萄糖苷梯度设置请教一下怎么设置的 为什么我的不出峰 有没有做过的 请教请教[img]https://ng1.17img.cn/bbsfiles/images/2021/06/202106032032470096_8789_4180826_3.png[/img]

黄酮类化合物在自然界分布非常广泛,是一类非常重要的天然有机化合物。传统意义上黄酮类化合物主要是指基本母核为2-苯基色原酮类化合物,现在泛指两个具有酚羟基的苯环(A-与B-环)通过中央三碳原子相互连接而成的一系列化合物。根据黄酮类化合物结构特点,可分为黄酮、黄酮醇、二氢黄酮、二氢黄酮醇、黄烷醇、查耳酮、异黄酮、双黄酮、花色素等种类。黄酮类化合物具有多方面的生物活性,如葛根总黄酮及葛根素(puerarin)、银杏叶总黄酮等具有扩张冠状血管作用,临床用于治疗冠心病;水飞蓟素(silymarin)、异水飞蓟素( silydianin)及次水飞蓟素(slychristin)等有肝脏保护作用,临床上用于治疗急、慢性肝炎、肝硬化及多种中毒性肝损伤等;木犀草素(luteolin)、黄芩苷( baicalin)、黄芩素(baicalein)以及槲皮素等具有抗菌、抗病毒作用;牡荆素( vitexin)、桑色素、儿茶素等具有抗肿瘤作用等。游离黄酮类化合物一般难溶或不溶于水,易溶于甲醇、乙酸乙酯、氯仿、乙醚等有机溶剂及稀碱水溶液中。黄酮苷一般易溶于水、甲醇、乙醇等强极性溶剂中。花色苷及其苷元以离子形式存在具有盐的通性,亲水性较强,水溶度较大。黄酮化合物单体的分离主要依靠各种色谱方法来实现,除经典的柱色谱法和薄层色谱法、HPLC外,近年来HSCCC已经得到广泛的应用。对于多数极性较弱的黄酮苷元,在进行HSCCC分离实验时,通常可以选用氯仿-甲醇-水的溶剂系统,而氯仿-甲醇-水(4:3:2或5:3:2)则是最常用的溶剂系统。根据被分离样品的具体情况,在上述溶剂系统的基础上,对组成诸元的比例进行适当的调整,就能获得良好的分离效果。还有些苷元也可采用正己烷(石油醚)-乙酸乙酯-甲醇-水的溶剂系统,通过调整溶剂的组成比例来实现有效分离。对于极性较强的黄酮糖苷类成分的HSCCC分离,通常使用的是乙酸乙酯–水为基本结构的溶剂系统,可以通过添加正丁醇、甲醇、乙醇、乙酸来调节溶剂系统的极性。分离这类化合物的典型性溶剂系统有:氯仿-[color
寒冷的冬天,整个人都是懒洋洋的,尽管有很多试验经历、色谱柱使用体会、很多的原创素材,都懒的起笔。2013年马上来临,趁着2012年的第五届原创大赛,赶快记录一篇原创,分享自己更多的使用体会与试验经历给大家探讨。这次检测的是大豆异黄酮,说起这个,估计很少人接触,而我已经跟它已经认识了好几年。异黄酮是一类物质,有苷和苷元之分,可是接触了很久都不知道苷和苷元有什么区别,为什么这个叫苷,另外一个就叫苷元。而且保健品中有很多叫甙,我也搞不清楚甙和苷有什么区别。大豆异黄酮的测定,目前存在的标准方法:GB/T 23788-2009保健食品中大豆异黄酮的测定方法 高效液相色谱法NY/T 1252-2006大豆异黄酮http://ng1.17img.cn/bbsfiles/images/2012/12/201212281607_416709_1608710_3.jpg实验过程:色谱柱:Topsil 液相色谱柱(C18,5um,4.6*250mm)检测波长:260nm流动相:乙腈+0.1%磷酸水溶液,梯度洗脱流速:1.0mL/min进样量:20ul先晒晒标准上的图谱http://ng1.17img.cn/bbsfiles/images/2012/12/201212281611_416712_1608710_3.jpg以下是我测定的色谱图,根据出峰顺序依次是大豆苷、染料木苷、大豆素、染料木素。标准色谱图http://ng1.17img.cn/bbsfiles/images/2012/12/201212281614_416716_1608710_3.jpg保健食品样品色谱图http://ng1.17img.cn/bbsfiles/images/2012/12/201212281615_416718_1608710_3.jpg总结:1、总体来说,4个组分的保留时间和色谱峰都能与标准对应重现2、色谱峰的理论塔板数都很好,这根柱子已经用了一段时间,具体测了多少样品就不清楚了3、以前检测测的是大豆素和染料木素,同时测定4个还是第一次,效果还算满意你测试过大豆异黄酮吗?有经验赶快讨论吧
有谁知道食品中总黄酮的测定方法、及保健食品中人参皂苷的测定方法!谢谢!十万火急!!!
植物性食物是黄酮类物质的优秀来源,尤其是洋葱、欧芹、紫甘蓝、浆果、柑橘、茶叶、豆类及豆制品中,黄酮类物质含量很丰富,大家可适当多吃。物是黄酮类物质的优秀来源,尤其是洋葱、欧芹、紫甘蓝、浆果、柑橘、茶叶、豆类及豆制品中,黄酮类物质含量很丰富,大家可适当多吃。
检测发酵液中的有机酸,黄酮,皂苷、木脂素、葡萄糖、多糖用什么色谱柱?发酵液的PH范围在2-5之间
目前,[b]我国是植物提取物的第一原料供应大国,也是植物提取物应用大国[/b],据中国海关数据显示,2019年,我国植物提取物行业出口额达23.72亿美元(美国是最大的进口市场),进口额达8.49亿美元(美国、印尼和印度是前三进口市场)。在全球“禁抗、限抗”大背景下,国内外对可饲用植物提取物的需求日益增长,对于其产品和相应检测标准的需求也日益强烈。因为[b]没有统一的相关标准[/b],这就严重影响了其生产效率以及资源浪费,对从事可饲用植物提取物的生产、加工以及进出口贸易的相关企业造成了极大的困扰。因此必须尽快制定颁布并实施可饲用植物提取物的相关标准并实现标准的国际化,确保在国际贸易中有据可依,提高我国可饲用天然植物提取物在国际上的竞争力。[b]2024年3月15日,国家标准《饲料添加剂淫羊藿提取物中黄酮醇苷的测定 高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法》 正式发布[/b]。该标准由TC76(全国饲料工业标准化技术委员会)归口 ,主管部门为国家标准化管理委员会。主要起草单位为中国农业科学院北京畜牧兽医研究所 、中国医学科学院药用植物研究所 、天津博菲德科技有限公司 、湖南农业大学 、北京爱绿生物科技有限公司 、中国农业科学院饲料研究所。[来源:仪器信息网] 未经授权不得转载[align=right][/align]
维权声明:本文为xiaodu2007693原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。黄酮类化合物的分离体会 【黄酮类化合物简介】 黄酮类化合物(flavonoids)是一类存在于自然界的、具有2-苯基色原酮(flavone)结构的化合物。它们分子中有一个酮式羰基,第一位上的氧原子具碱性,能与强酸成盐,其羟基衍生物多具黄色,故又称黄碱素或黄酮。黄酮类化合物在植物体中通常与糖结合成苷类,小部分以游离态(苷元)的形式存在。绝大多数植物体内都含有黄酮类化合物,它在植物的生长、发育、开花、结果以及抗菌防病等方起着重要的作用。【本人研究内容】 我做的是某中药的化学成分研究,该中药中主要为黄酮类成分,遵循传统用药原则,我采取水煎煮,之后水提液浓缩至一定体积,过大孔吸附树脂,乙醇水梯度洗脱,梯度:水、20%乙醇、40%乙醇、60%乙醇、80%乙醇。每个梯度做为一个流分,再进行细分。下一步主要是运用聚酰胺柱色谱,中低压ODS,Sephadex LH20进一步分离,最后制备液相得到单体化合物。【经验分享】 主要和大家分享我使用聚酰胺柱的经验,我一般是聚酰胺薄膜结合聚酰胺柱,类似于硅胶板结合硅胶柱。所用的聚酰胺为30-60目,聚酰胺薄膜一盒50片,规格为8cm x 8cm,70块钱左右,可以根据需要剪成条状使用。聚酰胺薄膜常用的溶剂系统说明书上有,我常用甲醇-水,因为黄酮类化合物有一定的酸性,展开剂要加点酸,甲酸乙酸都可以,抑制其解离,保持游离状态的话,样品成点性好,否则拖尾很严重。在此需要指出的是,聚酰胺可以用反相溶剂系统,如甲醇水;也可以用正相溶剂系统,如二氯甲烷甲醇。要根据自己样品的极性来定。以我的80%乙醇洗脱物为例,进行详细的叙述。此部分极性较小。我用二氯甲烷溶解,拌样,挥干。洗脱系统为甲醇水,聚酰胺柱起始用水装柱,上样,上面要放一块棉花,然后用玻璃塞子压住(聚酰胺比较轻,防止加溶剂时冲起来),甲醇水梯度洗脱。之前点板显示,50%甲醇水刚刚展开一点,因此起始用50%甲醇水洗脱,然后梯度设为55%、60%、65%、70%、75%、80%、85%、90%、最后甲醇冲柱。过程中每100mL一接,蒸干转移至小瓶。最后用一大块聚酰胺对得到的流分进行合并。得到大概9-10个流分,HPLC-DAD分析,找到合适的液相色谱条件之后,用制备液相进行制备。【部分聚酰胺薄层谱图展示】http://ng1.17img.cn/bbsfiles/images/2010/09/201009211940_245940_2160661_3.jpg http://ng1.17img.cn/bbsfiles/images/2010/09/201009211940_245941_2160661_3.jpg左图:展开剂:(甲醇:水90:10)+2滴乙酸; 右图:展开剂:(甲醇:水80:10)+2滴乙酸,最右边的点为总样显色剂为:三氯化铁乙醇溶液【体会和教训】 黄酮类化合物用聚酰胺分离效果非常好,但是死吸附较严重,样品比较多时可以使用,少就算了。凝胶Sephadex LH20效果也非常好,建议多使用。黄酮类成分溶解性比较折磨人,但提醒一点在进行制备液相时,样品一定要用流动相溶解,或接近流动相,多加点总会溶解的,然后用0.45μm滤膜过滤。不用流动相溶解很容易把柱子弄裂分。原因在于,进样之后,样品析出,堵了柱塞板,导致局部流速过快,冲塌固定相。我就把老师的预柱整裂分了,多亏了预柱啊,保护了柱子,要不然老师肯定疯了,但是后来寄出去修预柱,别人检测又很神奇地自动好了,这有点解释不了,呵呵,这事也不是常能碰到的,最好按规矩来,否则耽误别人实验不太好。先就写这么多,实验也不是那么成功,但拿出来和大家交流,不对的地方请批评指正。【样品导致预柱裂分的后果-色谱图展示】http://ng1.17img.cn/bbsfiles/images/2010/09/201009212004_245944_2160661_3.jpg http://ng1.17img.cn/bbsfiles/images/2010/09/201009212007_245946_2160661_3.jpg左图为:样品制备时色谱图,当时已裂分右图为:预柱用纯品检测色谱图,连续进样2针,可见预柱裂分严重。
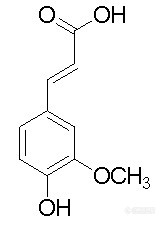
高效液相可同时测定植酸、黄酮和阿魏酸吗?检测波长该如何选择?流动相呢?阿魏酸:有顺式和反式两种,顺式为黄色油状物,反式为正方形结晶或纤维结晶,溶点为174℃,溶于热水,乙醇和乙酸乙酯,稍溶于乙醚,难溶于苯和石油醚。[img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912231143_191530_1638724_3.jpg[/img]黄酮:天然黄酮类化合物多以苷类形式存在 ,并且由于糖的种类、数量、联接位置及联接方式不同可以组成各种各样黄酮苷类。黄酮苷一般易溶于水、乙醇、甲醇等极性强的溶剂中;但难溶于或不溶于苯、氯仿等有机溶剂中。糖链越长则水溶度越大。黄酮类化合物因分子中多具有酚羟基,故显酸性。酸性强弱因酚羟基数目、位置而异。[img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912231143_191531_1638724_3.jpg[/img]植酸:淡黄色浆状液体,易溶于水、乙醇、丙酮,不溶于无水乙醚、苯、已烷、氯仿。[img]http://ng1.17img.cn/bbsfiles/images/2009/12/200912231144_191532_1638724_3.jpg[/img]
化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检测方法1 适用范围 本方法规定了用高效液相色谱法定性检测化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的方法。 本方法适用于化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的定性测定。2 方法提要 样品在经过提取后,经高效液相色谱仪分离,二极管阵列检测器检测,经与平行操作的补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮对照品及补骨脂对照药材比较,以保留时间和紫外光谱图定性,鉴别补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的存在。本方法对补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检出限和取样品0.5 g时的检出浓度见表1。 表1 4种补骨脂特征成分的检出限和检出浓度化合物检出限(ng)检出浓度(μg/g)补骨脂素0.30.6异补骨脂素0.30.6新补骨脂异黄酮0.30.6补骨脂二氢黄酮0.30.63 试剂和材料 除另有规定外,所用试剂均为分析纯,水为实验室用一级水。3.1 乙腈,色谱纯。3.2 补骨脂素,纯度≥99%。3.3 异补骨脂素,纯度≥99%。3.4 新补骨脂异黄酮,纯度≥98%。3.5 补骨脂二氢黄酮,纯度≥99%。3.6 补骨脂,中国食品药品检定研究院,供鉴别用。3.7 补骨脂特征性成分混合标准溶液(=0.1 μg/mL):分别称取补骨脂素(3.2)、异补骨脂素(3.3)、新补骨脂异黄酮(3.4)、补骨脂二氢黄酮(3.5)对照品各5 mg(精确到0.1 mg),置500 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,配制成质量浓度各为10 μg/mL的标准溶液。精密量取各标准溶液0.1 mL置10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得0.1 μg/mL的混合标准溶液。3.8 补骨脂标准储备溶液:取补骨脂对照药材0.2 g,置50 mL三角瓶中,加30 mL 70%乙醇回流提取1h,滤过,滤液置100 mL量瓶中,加70%乙醇稀释至刻度,摇匀,即得。4 仪器4.1 高效液相色谱仪:具二极管阵列检测器。4.2 分析天平:感量为0.1 mg。4.3 移液器。4.4 涡旋振荡器。4.5 超声波清洗仪(功率不低于200W)。4.6 高速离心机:转速不小于10000 r/min。
黄酮总量检测和单个黄酮分析。黄酮总量分析依据到底是依据芦丁还是其他黄酮参加显色反应,测试波长410还是中510nm。而单个黄酮测试,必须用响应黄酮作标曲还是其他也可以参考。谢谢!
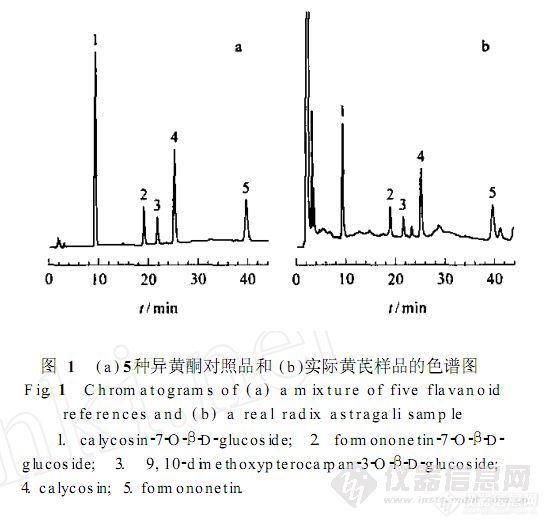
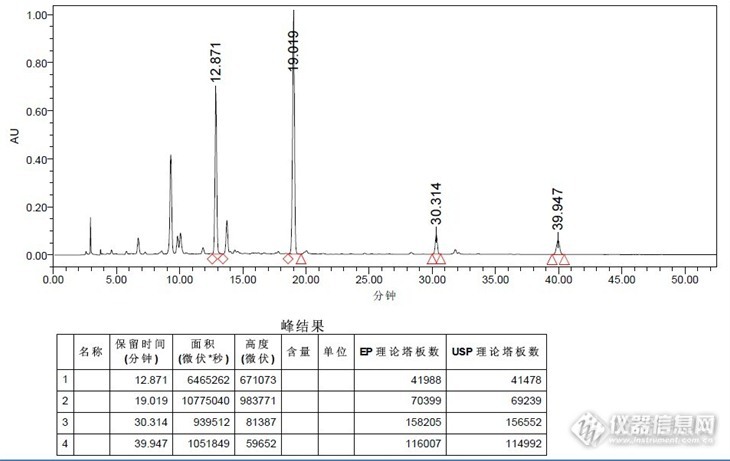
【作者】 王晓辉; 刘涛; 李清; 陈晓辉; 毕开顺;【Author】 WANG Xiaohui,LIU Tao,LI Qing,CHEN Xiaohui,BI Kaishun(School of Pharmacy,Shenyang Pharmaceutical University,Shenyang 110016,China)【机构】 沈阳药科大学药学院; 沈阳药科大学药学院 辽宁沈阳110016; 辽宁沈阳110016;【摘要】 对蒙古黄芪中5种异黄酮类成分的含量进行了反相高效液相色谱法测定。色谱柱为D iam ons il C18柱,流动相为乙腈-水系统,梯度洗脱,检测波长230nm,柱温35℃。毛蕊异黄酮-7-O-β-D-葡萄糖苷在20.12~201.2m g/L、芒丙花素-7-O-β-D-葡萄糖苷在4.62~46.2m g/L、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷在4.86~48.6m g/L、毛蕊异黄酮在9.24~92.4m g/L、芒丙花素在6.92~69.2m g/L时峰面积与浓度呈良好的线性关系,相关系数分别为0.999 2,0.999 7,0.999 7,0.999 5和0.999 5。5种成分的加样回收率均高于94%,相对标准偏差(RSD)小于3.2%(n=9)。该法简便快速,重复性良好,结果准确可靠,可用于黄芪药材中5种主要异黄酮类成分的含量测定。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301613_380608_2379123_3.jpg
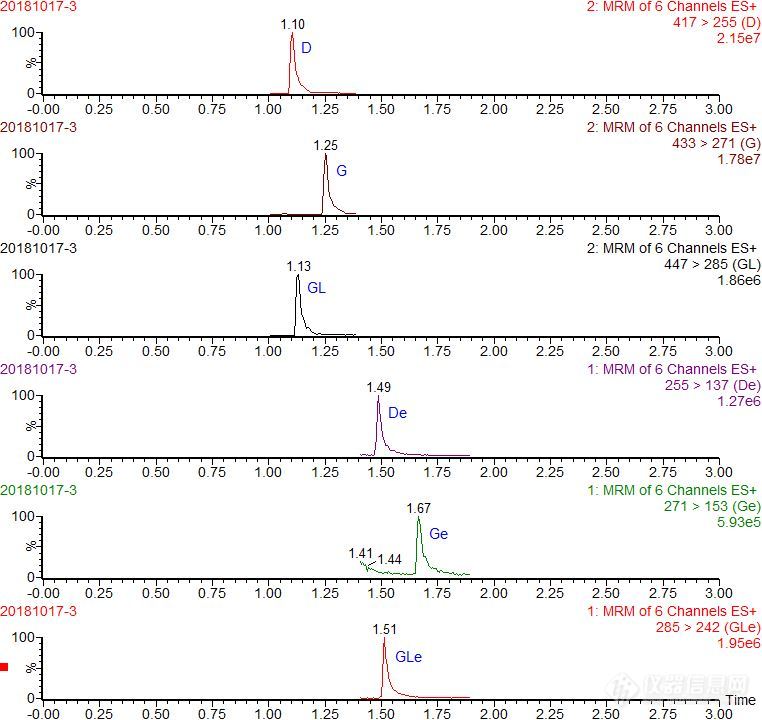
[align=center][b]超高效液相色谱-串联质谱法测定大豆中大豆异黄酮的含量[/b][/align][align=center]户江涛[/align][align=center](农业农村部豆类产品质量安全风险评估实验室(佳木斯),黑龙江省农垦科学院测试化验中心,黑龙江佳木斯 154007 )[/align]摘要:采用超高效液相色谱-串联质谱法建立了检测大豆中大豆异黄酮含量的分析方法。试样经90%甲醇水提取后,6种大豆异黄酮在C[sub]18[/sub]色谱柱上以0.1%甲酸水溶液和乙腈为流动相,进行液相色谱分离;质谱检测采用电喷雾正离子化模式和多反应监测模式(MRM)。结果表明,6种大豆异黄酮分别在0.01~0.5 mg/L(D、GL、G)和0.002~0.1 mg/L(De、GLe、Ge)范围内线性关系良好,相关系数(R)为0.9993~0.9998,定量限(LOQ)为0.0001 g/kg。在大豆空白样品添加浓度分别为0.01、0.05、0.2 g/kg(De、GLe、Ge)和0.2、1、2 g/kg(D、GL、G),6种大豆异黄酮的平均回收率为86.6%~96.2%,相对标准偏差(RSD)为1.07%~5.93%(n=6)。本方法简便、灵敏、抗干扰,适用于大豆中大豆异黄酮含量检测。关键词:超高效液相色谱-串联质谱;大豆;大豆异黄酮[align=center]Determination of soybeanisoflavone in soybean by ultra performance liquid chromatography-tandem massspectrometry[/align][align=center]HU Jiangtao[/align][align=center]([i]Laboratory of Qualityand Safety Risk Assessment for Soybean products, Ministry of Agriculture andRural Affairs, Testing and Analysis Center of Heilongjiang Academy of LandReclamation Sciences, Jiamusi 154007,China[/i])[/align][b]Abstract:[/b]A methodwasdeveloped for the determination of soybeanisoflavone in soybean by ultra performance liquid chromatography-tandem massspectrometry(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS). The samples were extracted by 90% methanol-water, then 6 soybean isoflavones were separated on aWaters BEH C[sub]18[/sub] column with gradient elution with the mobile phase of0.1% formic acid and acetonitrile, and finally detected by positive eletrosprayionization-mass spectrometry(ESI[sup]+[/sup]-MS/MS) in multiple reactionmonitoring(MRM) mode. The results showed the linearities of 6 soybean isoflavones were good in the concentrationrange of 0.01~0.5 mg/L(D、GL、G)and 0.002~0.1 mg/L(De、GLe、Ge), the correlation coefficients were 0.9993~0.9998. The limitof quantification(LOQ) of soybean isoflavone was 0.0001 g/kg. At the spiked levels of 0.01、0.05、0.2 g/kg(De、GLe、Ge)and 0.2、1、2 g/kg(D、GL、G) in the blank soybean samples, the mean recovery of soybeanisoflavone was 86.6%~96.2%, andthe relative standard deviation(RSD) was 1.07%~5.93%(n=6).This method is simple,sensitive, anti-jamming and suitable for simultaneous determination of soybean isoflavone in soybean.[b]Key words: [/b]ultra performance liquid chromatography-tandem massspectrometry (UPLC-MS/MS) soybean soybean isoflavone大豆异黄酮(soybean isoflavone)是一族化合物的统称,是大豆植物体内的一种次生代谢产物,是大豆主要活性成分之一,其母核为3-苯并吡喃酮,主要包括大豆苷、大豆黄苷、染料木苷及其相应苷元[sup][/sup]。研究表明,大豆异黄酮除具有天然抗氧化作用外[sup][/sup],还具有降低胆固醇含量、预防多种癌症及改善妇女更年期综合征等多方面生物功效[sup][/sup]。大豆异黄酮主要存在于大豆籽实中,其总含量约为0.4~5 g/kg,其中大豆苷、大豆黄苷和染料木苷这三种含量约占总量的97%~98%,而其对应的苷元含量仅占2%~3%左右[sup][/sup]。目前,大豆异黄酮的检测方法主要有高效液相色谱法(HPLC)[sup][/sup]、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[sup][/sup]、紫外分光光度法[sup] [/sup]、质谱法(HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS[sup][/sup])等。紫外分光光度法[sup] [/sup]只能测定大豆异黄酮总量,且灵敏度不高;[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[sup][/sup]需要对异黄酮进行衍生,前处理复杂;目前,大豆异黄酮检测现有的国家标准GB/T 26625-2011[sup] [/sup]采用的是高效液相色谱法(HPLC),在实际检测过程中发现,由于紫外检测器灵敏度不高,存在个别样品中异黄酮相应苷元检测不到的情况;同时大豆提取液中含有蛋白、脂肪等杂质影响色谱柱柱效,以至于不能满足分离度要求,严重干扰低含量组分峰面积积分定量。而[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法质谱检测器灵敏度高,通过选定大豆异黄酮的特征离子,能有效去除上述杂质干扰,定量更加准确可靠。目前,国内外采用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法检测大豆中大豆异黄酮含量的文献很少[sup][/sup]。本文对大豆中大豆异黄酮检测的前处理方法借鉴GB/T 26625-2011[sup][/sup],提取液改用UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS测定。该方法前处理过程简便、灵敏度高、分析时间短、抗干扰能力强,适用于大批量大豆样品中大豆异黄酮含量的检测。[b]1 实验部分[/b]1.1 材料与试剂大豆苷(daidzin,记为D,以下同)、大豆黄苷(glycitin,GL)、染料木苷(genistin, G)、大豆素(daidzein,De)、大豆黄素(glycitein, GLe)、染料木素(genistein,Ge)(纯度≥99%,Dr.Ehrenstorfer公司);甲醇、乙腈、甲酸(色谱纯,Fisher公司);实验用水为Millipore纯水仪制备。1.2 仪器与设备Acquity UPLC型超高效液相色谱仪(Waters公司);XEVO TQ-S三重四级杆质谱仪(Waters公司);KQ-500DE型超声波仪(昆山市超声仪器有限公司);涡旋混合器(IKA公司);CR21GⅢ型高速离心机(HITACHI公司)。1.3 大豆异黄酮标准储备液的配置分别称取适量的D、GL、G、De、GLe、Ge标准品,用甲醇配置成质量浓度为1mg/mL标准储备液,于-18℃冰箱保存(有效期6个月),待用;使用时用10%甲醇水逐级稀释成所需浓度的混合标准工作液,现用现配。1.4 样品前处理提取:称取粉碎均与后的试样1.0g(精确到0.01g)于50mL聚乙烯离心管中,加入10.0 mL90%甲醇水,涡旋混合30 s后置于60℃超声波清洗器中提取30 min,在离心机中以15000 r/min离心5 min,将上清液转移至100 mL容量瓶中,残渣再加入10.0 mL90%甲醇水溶液按上述步骤提取后,合并两次上清液于100 mL容量瓶中,用10%甲醇水溶液定容至刻度,摇匀。a)De、GLe、Ge的测定:取1 mL过0.22um有机系微孔滤膜,供UPLC/MS/MS分析测定;b)D、GL、G的测定:由于D、GL、G含量较高,需要将a)中过完滤膜的待测液用10%甲醇水稀释50倍后,供UPLC/MS/MS分析测定。1.5 液相色谱及质谱条件液相色谱:色谱柱:Waters BEH C[sub]18[/sub](1.7 μm,50mm×2.1mm);柱温:30℃;流速:0.5 mL/min;进样量:1μL;流动相A:乙腈;流动相B:0.1%的甲酸水溶液。梯度洗脱程序:0~0.5min,10% A;0.5~3. 0 min,10%~100% A;3. 0 ~4. 0 min,100%A,4 ~4.1.1min,100% A~10% A,4.1 ~5.0min 10% A。质谱:离子源:电喷雾离子源( ESI [sup]+[/sup] ) ;扫描方式:正离子扫描;检测方式:多反应监测( MRM);毛细管电压:3.2 kv;离子源温度:150℃;去溶剂气温度:500℃;去溶剂气流量:1000 L /h;定性、定量离子对及碰撞能量见表1。[align=center]表1大豆异黄酮的质谱参数[/align][align=center]Table 1 MRM parameters of soybean isoflavone[/align] [table][tr][td] [align=center]Analyte[/align] [/td][td] [align=center]Cone/V[/align] [/td][td] [align=center]Parent ion/(m/z)[/align] [/td][td] [align=center]Daughter ion/(m/z)[/align] [/td][td] [align=center]Collision energy/V[/align] [/td][/tr][tr][td] [align=center]D[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]417[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]255﹡[/align] 137[/td][td] [align=center]27[/align] [align=center]18[/align] [/td][/tr][tr][td] [align=center]G[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]433[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]271﹡[/align] 153[/td][td] [align=center]21[/align] [align=center]50[/align] [/td][/tr][tr][td] [align=center]GL[/align] [align=center] [/align] [/td][td] [align=center]30[/align] [align=center] [/align] [/td][td] [align=center]447[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]285﹡[/align] 270[/td][td] [align=center]25[/align] [align=center]46[/align] [/td][/tr][tr][td] [align=center]De[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]255[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]137﹡[/align] 181[/td][td] [align=center]30[/align] [align=center]26[/align] [/td][/tr][tr][td] [align=center]Ge[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]271[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]153﹡[/align] 215[/td][td] [align=center]30[/align] [align=center]25[/align] [/td][/tr][tr][td] [align=center]GLe[/align] [align=center] [/align] [/td][td] [align=center]25[/align] [align=center] [/align] [/td][td] [align=center]285[/align] [align=center][sup] [/sup][/align] [/td][td] [align=center]242﹡[/align] 168[/td][td] [align=center]27[/align] [align=center]35[/align] [/td][/tr][/table]﹡quantitativeion[b]2 结果与讨论[/b]2.1 色谱及质谱条件的优化流动相的选择:对比了酸性体系(0.1%甲酸水溶液)与非酸性体系(纯水、乙酸铵溶液)分别与甲醇、乙腈的流动相体系组合,结果发现目标物在酸性体系中比非酸性体系响应更高、峰形更好;同时大豆提取液中含有蛋白、脂肪等杂质可能会残留在色谱柱上,影响色谱柱的使用寿命,而乙腈比甲醇体系洗脱能力更强,可以有效去这些杂质。综合考虑目标物信号强度、色谱分离效果以及除杂等因素,本研究采用0.1%甲酸水溶液+乙腈流动相体系。质谱的选择:根据6种大豆异黄酮的分子量,用10%甲醇水配置1.0 mg/L 大豆异黄酮标准溶液直接注射到质谱中,在正离子模式下分别对各种组分进行母离子及对应子离子全扫描,最终确定的质谱条件见表1。2.2 质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)与色谱法(HPLC)的比较国家标准《GB/T 26625-2011粮油检验大豆异黄酮含量测定高效液相色谱法》[sup][/sup]中规定的大豆异黄酮检测方法为HPLC法。对同一大豆样品分别采用本文UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法(MRM色谱图见图1、2)和GB/T 26625 HPLC法检测,结果表明这两种方法测定的大豆异黄酮总含量值基本一致。由于De、GLe、Ge这三种苷元在大豆中含量很低,用HPLC法检测时,紫外检测器灵敏度不高,存在个别样品中上述三种组分检测缺失的情况;同时在实际大批量样品检测中发现,随着进样次数的增加,色谱柱柱效下降,大豆提取液中存在的蛋白、脂肪等杂质对含量低的目标物峰干扰越来越大,定量困难。研究发现,同浓度的大豆异黄酮在质谱检测器上的响应值要远远超过紫外检测器,同时质谱法可以通过选定大豆异黄酮的特征离子,有效地去除杂质的干扰,其目标物分离度不受色谱柱进样次数增加的影响,定量更加准确可靠。[align=center][img=,690,651]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050912587968_4111_3299836_3.jpg!w690x651.jpg[/img][/align][align=center]图1 大豆异黄酮标准溶液(0.01mg/L)MRM色谱图[/align][align=center]Fig.1 MRM chromatograms of soybean isoflavone standard solution at 0.01 mg/L[/align][align=center][/align][align=center][img=,690,653]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050913342201_5843_3299836_3.jpg!w690x653.jpg[/img][/align][align=center]图2 大豆样品中大豆异黄酮MRM色谱图[/align][align=center]Fig.2 MRM chromatograms of soybean isoflavone in soybean[/align]2.3线性范围和定量限吸取不同体积的大豆异黄酮标准储备液(1.3),用10%甲醇水分别配置0.002、0.005、0.01、0.05、0.1(De、GLe、Ge)和0.01、0.05、0.1、0.2、0.5(D、GL、G)的大豆异黄酮上机混合标准溶液,以各自定量离子的峰面积为Y对应质量浓度X(mg/L)做标准曲线,得到的线性方程和相关系数见表2。结果表明,大豆异黄酮标准溶液在各自浓度范围内线性良好,相关系数R为0.9993~0.9999。以10倍信噪比(S/N)计算,大豆异黄酮上机液最低定量浓度为0.001 mg/L,通过公式(1)计算得到大豆中大豆异黄酮含量,最终确定本方法大豆异黄酮的定量限(LOQ)为0.0001 g/kg。糠氨酸质量分数计算公式:[align=center][img=,207,87]https://ng1.17img.cn/bbsfiles/images/2019/08/201908050915166414_5621_3299836_3.jpg!w207x87.jpg[/img] ………………(1)[/align] 式中:X为试样中大豆异黄酮含量,以g/kg计;C为大豆异黄酮上机浓度(mg/L);V为定容体积(V=100)。表2 大豆异黄酮标准溶液的线性方程和相关系数[align=center]Table 2 Linear equation and correlation of soybean isoflavone in 10% methanol-water standard solutions[/align] [table][tr][td] [align=center]Analyte[/align] [/td][td] [align=center]Linear range/(mg/L)[/align] [/td][td] [align=center]Linear equation[/align] [/td][td] [align=center]R[/align] [/td][td] [align=center] [/align] [/td][/tr][tr][td] [align=center]D[/align] [align=center]GL[/align] [align=center]G[/align] [align=center]De[/align] [align=center]GLe[/align] [align=center]Ge[/align] [/td][td] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.01~0.5[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [align=center][sup]0.002~0.1[/sup][/align] [/td][td] [align=center]Y=2393.6x+479.38[/align] Y=1885x+139.66 [align=center]Y=1470.9x+187.97[/align] [align=center]Y=4287.9x+442.79[/align] [align=center]Y=3521.7x-103.62[/align] [align=center]Y=1993x+122.79[/align] [/td][td] [align=center]0.9995[/align] [align=center]0.9999[/align] [align=center]0.9993[/align] [align=center]0.9998[/align] [align=center]0.9997[/align] [align=center]0.9998[/align] [/td][td] [/td][/tr][/table]2.4回收率和精密度大豆中De、GLe、Ge含量较低,而D、GL、G含量较高,故本方法准确度实验分为高低浓度梯度组进行加标。称取大豆试样1.00 g,分别添加0.01、0.05、0.2 g/kg(De、GLe、Ge)和0.2、1、2 g/kg(D、GL、G),每个水平重复6次,同时做该大豆的空白本底实验。按照1.4前处理方法处理后上机检测,计算回收率(扣除空白),结果表明:不同添加浓度下,De、GLe、Ge的平均回收率为91.7%~96.2%,相对标准偏差(RSD,n=6)为2.78%~5.93%;D、GL、G的平均回收率为86.6%~93.8%,相对标准偏差(RSD,n=6)为1.07%~3.77%。[b]3 结语[/b]本文建立了超高效液相色谱-串联质谱法(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)测定大豆中大豆异黄酮含量的分析方法。该方法灵敏度高,线性范围宽,能同时覆盖大豆中多梯度浓度大豆异黄酮组分含量的测定。同时该方法具有较高的准确度和精密度,前处理步骤简单,分析速度快,可有效避免由于色谱柱柱效下降对最终检测结果的影响,特别适合大批量样品的检测。田娟娟, 宋宏哲, 张飞, 等. 水剂法纯化大豆异黄酮的研究. 大豆通报, 2005, 6:19-22. Hagen M K, Ludke A, Araujo A S, et al.Antioxidant characterization of soy derived products in vitro and the effect ofa soy diet on peripheral markers of oxidative stress in a heart disease model .Canadian Journal of Physiology and Pharmacology, 2012,90(8):1095-1103. 徐春华, 张治广, 谢明杰, 等. 大豆异黄酮的抗氧化和抗肿瘤活性研究研究 . 大豆科学, 2010, 29(5): 870-873. 李俏俏, 王清路, 薛金艳, 等. 大豆异黄酮对绝经女性血清中脂类物质的影响的研究 . 大豆科学, 2009, 28(1):172-174. 胡润芳, 张玉梅, 陈宇华, 等. 大豆异黄酮含量的初步研究. 东南园艺, 2017, 6:9-11. 刘琴, 朱媛媛, 白兴梁. 不同种类大豆中大豆异黄酮含量及抗氧化性比较. 北京工商大学学报(自然科学版), 2012, 30(6): 45-51. 袁凤杰, 姜莹, 董德坤, 等. 中国大豆核心种质异黄酮含量分析.中国粮油学报, 2011, 26(2):5-8. Tepavcevic V, Atanackovic M,Miladinovic J,et al. Isoflavone composition,total polyphenolic content,and antioxidant activity in soybeans of different origin. MedFood,2010,13(3):657-664 GB/T 26625-2011《粮油检验大豆异黄酮含量测定高效液相色谱法》. Liggins J,Bluck J C. Deidzein and genistein content of fruits and nuts. Journal ofNutritional Biochemistry,2000,11(6):326-331. 鞠兴荣, 袁建, 汪海峰. 三波长紫外分光光度法测定大豆异黄酮含量的研究. 食品科学, 2001, 22(5):46-48.
实验原理黄酮类化合物是植物的重要次生代谢产物,也是一些保健品和中药材的有效成分之一。黄酮类化合物的定量方法常用的有HPLC法和分光光度法,在实际生产和科研过程中,对于黄酮单体的定量常采用HPLC法,而对总黄酮的测定,考虑到方法的简便、快捷以及可行性,多采用在碱性介质中加铝盐显色的分光光度法。在碱性条件下黄酮类化合物与铝盐形成络合物、在500nm波长处有最大吸收峰。标准品选用芦丁。试剂和器材一、试剂芦丁标准品。5%NaNO2;10%A1(NO3)3;5%NaOH;70%乙醇。二、材料新鲜银杏叶。三、器材容量瓶10ml(×7),25ml(×1),100ml(×2);吸管 0.5ml(×2),1ml(×2),2ml(×1),5ml(×1);分光光度计。操作方法一、制作标准曲线精密称取芦丁标准品5mg,用70%乙醇溶解,定容于25mL容量瓶中,摇匀,得0.2mg/mL的标准溶液。精确吸取标准溶液0.0、0.2、0.4、0.6、0.8、1.0、1.2mL,分别置于10mL容量瓶中,加入 5%NaNO20.4mL,摇匀,放置6min;加入10%A1(NO3)3 0.4mL,摇匀,放置6min;加入5%NaOH4.0mL,再加水至刻度,摇匀,放置15min。以试剂空白作为参比溶液。用1cm比色皿,在500nm波长处测定吸光度,绘制标准曲线。二、总黄酮的提取把新鲜的银杏叶低温烘干,使水分小于8%,制成干粉。精确称取干粉1.0g,置于 100mL容量瓶中,加入70%乙醇30mL,浸泡24h。超声波提取30min,过滤,滤液用70%乙醇定容于100mL容量瓶中,得到黄酮提取液,待用。三、测定吸取黄酮提取液1.00mL, 置于10mL容量瓶中,加入5%NaNO30.4mL,摇匀,放置6min;加入10%A1(NO3)3 0.4mL,摇匀,放置6min;加入5%NaOH4.0mL,再加水至刻度,摇匀,放置15min。以试剂空白作为参比溶液。用1cm比色皿,在500nm波长处测定吸光度,由标准曲线法计算总黄酮含量。注意事项对于某些热敏成分的提取,采用超声波破碎法效果较为理想。由于此过程是一个物理过程,浸提过程中无化学反应,被浸提的生物活性物质在一定时间内保持不变。







 我要推广仪器
我要推广仪器
 下载APP
下载APP