用50%乙醇洗脱树脂柱得到130ml的洗脱液,取出一定体积计算其所含有的目标物质量,约为95%,把这瓶洗脱液拿去冷冻干燥后,再定量检测,目标物质含量怎么只有75%了?是什么原因啊?
用50%乙醇洗脱树脂柱得到130ml的洗脱液,取出一定体积计算其所含有的目标物质量,约为95%,把这瓶洗脱液拿去冷冻干燥后,再定量检测,目标物质含量怎么只有75%了?是什么原因啊?
在测量土壤项目时,需要土壤的干物质含量,有的项目标准中明确规定用新鲜土壤干物质含量,有的项目没有说明用哪一种土壤的干物质含量,有什么依据去判断是用新鲜土壤的干物质含量,还是用风干土壤的干物质含量,求各位大神帮忙解惑,万分感谢!!!
专家您好!“某物质的含量是100%”我所配制的染料:要测的那种物质含量肯定在2%-4%之间,所以我还是想请教专家!谢谢
要测釜残某物质的含量,它的含量很低,含着较大的水分,用气相怎么做呢?想知道它的含量
如何通过气相面积归一法测得物质的面积和含量来求物质转化率和选择性。例如 A生成了B+C+D+F 目标产物为B 反应后生成的含量A有20% B20% C20% D20% F20% ,需要校正因子什么的么。我是初学者
请教各位专家,用液相色谱方法检测一物质的含量,面积归一化法,用该方法测定物质的含量约在98%,请问应该怎样做此方法验证的准确度呢?很困惑……在此先感谢!
我的情况是这样的:我在植物性饮料中添加了其中的一种添加剂(阿拉伯胶、卡拉胶、环状糊精、琼脂),都是外源性添加剂,然后我需要知道其在饮料中的含量,我该如何测定,用什么方法?或者我只要求定性里面是否含有这些物质,然后才是一个测定这种物质的含量问题?请同行的朋友指点下!先谢谢了!
我的情况是这样的:我在植物性饮料中添加了其中的一种添加剂(阿拉伯胶、卡拉胶、环状糊精、琼脂),都是外源性添加剂,然后我需要知道其在饮料中的含量,我该如何测定,用什么方法?或者我只要求定性里面是否含有这些物质,然后才是一个测定这种物质的含量问题?请同行的朋友指点下!先谢谢了!
我的情况是这样的:我在植物性饮料中添加了其中的一种添加剂(阿拉伯胶、卡拉胶、环状糊精、琼脂),都是外源性添加剂,然后我需要知道其在饮料中的含量,我该如何测定,用什么方法?或者我只要求定性里面是否含有这些物质,然后才是一个测定这种物质的含量问题?请同行的朋友指点下!先谢谢了!
请问 如何检测盐酸纳洛酮注射液中的EDTA二钠含量最近在盐酸纳洛酮注射液中加入EDTA二钠 需检测一下该物质的含量请问大家有什麽简便的检测方法吗 多谢
针对样品体系中某物质的含量未知,或者随着取样时间的不同,该物质的含量是变化的,在做方法验证时,加标回收实验应该如何设计加标水平呢?如何设计加标水平较为合理呢?
大家好,请问一下哪些物质不适合用高效液相来进行含量测定?
含量的高低会不会影响有关物质的高低,含量高,有关物质就会低,含量与有关物质之间的关系,有没有大佬解释一下,萌新化验员
现在有一个问题想请教大家,原先做的有关物质和含量液相条件都一样,所以做含量液相条件方法学时就不存在色谱条件确定这项了,但现在有关物质和含量液相色谱条件不一样?含量的色谱条件也点进行确定,我现在想问,含量的色谱条件也点做耐用性、专属性、检测限、溶液稳定性试验吗?

[img=,690,251]https://ng1.17img.cn/bbsfiles/images/2020/10/202010100838096525_5513_1699457_3.jpg!w690x251.jpg[/img]我用KBr压片法制作了三个切片,就一种目标物,含量也一样,为什么做出来的吸光度会有差别?求大神指点。谢谢!
致癌物质丙烯酰胺含量最近英国食物标准局对248份食品样品进行检测,发现13种食品中含有的致癌物质丙烯酰胺含量有上升趋势。其中亨氏、雀巢等许多知名食品公司都遭到英国食物标准局的警告,产品涉及薯条、速溶咖啡和谷类食物等。 世卫组织的专家指出,丙烯酰胺已被证实与多种癌症有关联。2002年,科学家便开始催促食品行业降低含量。但是英国食物标准局检测表明,包括薯片、即饮咖啡、面包、饼干、油炸土豆片、早餐麦片、幼儿食品在内的多款食品中的丙烯酰胺含量并未降低。其中被点名的有亨氏的香蕉儿童手指饼干、雀巢的金牌速溶咖啡等。
[color=#444444]想用动态顶空的结合GC-MS的方法来检测植物芳香挥发物,然后测定其中某种具体物质的含量,比如说用10克材料进行动态顶空吸附12h后,之后解吸附,上[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用仪[/color][/url],该如何来进行准确定量呢?加内标,之后用测定物质的标样作标准曲线来定量么?然后可以得到物质的含量为 X ug/10g材料/12h,对这一块的实验不懂,望指教,谢谢![/color]
有关物质分析方法验证的可接受标准简介摘要:本文介绍了在对有关物质检查所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。 关键词:有关物质检查 分析方法验证 可接收标准 药品中的有关物质泛指在药品的生产与储存过程中产生的工艺杂质或降解产物。由于这些有关物质的存在会影响到药品的纯度,进而可能会产生毒副作用,所以有关物质的控制是药品研发的一个重要方面,也是我们在药品审评中一直重点关注的要点之一。而要对有关物质进行严格的控制,就离不开专属性强、灵敏度高的分析方法,这就涉及到分析方法的筛选与验证。从现有的申报资料看,药品研发单位已基本上意识到分析方法验证的重要性,但是对验证时各具体指标是否可行尚没有一个明确的可接受标准,从而难以对验证结果进行评判。为解决这一问题,本文结合国外一些大型药品研发企业在此方面的要求,提出了在对有关物质检查方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。 1.准确度 该指标主要是通过回收率来反映。验证时一般要求根据有关物质的定量限与质量标准中该杂质的限度分别配制三个浓度的供试品溶液各三份(例如某杂质的限度为0.2%,则可分别配制该杂质浓度为0.1%、0.2%和0.3%的杂质溶液),分别测定其含量,将实测值与理论值比较,计算回收率,并计算9个回收率数据的相对标准差(RSD)。该项目的可接受的标准为:各浓度下的平均回收率均应在80%-120%之间,如杂质的浓度为定量限,则该浓度下的平均回收率可放宽至70%-130%,相对标准差应不大于10%。 2.线性 线性一般通过线性回归方程的形式来表示。具体的验证方法为:在定量限至一定的浓度范围内配制6份浓度不同的供试液,分别测定该杂质峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.990,Y轴截距应在100%响应值的25%以内,响应因子的相对标准差应不大于10%。 3.精密度 1)重复性 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于15%。 2)中间精密度 配制6份杂质浓度(一般为0.1%)相同的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于20%。 4.专属性 可接受的标准为:空白对照应无干扰,该杂质峰与其它峰应能完全分离,分离度不得小于2.0。 5.检测限 杂质峰与噪音峰信号的强度比应不得小于3。 6.定量限 杂质峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液杂质峰保留时间的相对标准差应不大于2.0%,峰面积的相对标准差应不大于5.0%。 7.耐用性 分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、检测波长变化±5nm、流速相对值变化±20%以及采用三根不同批号的色谱柱进行测定时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:各杂质峰的拖尾因子不得大于2.0,杂质峰与其他成分峰必须达到基线分离;各条件下的杂质含量数据(n=6)的相对标准差应不大于2.0%,杂质含量的绝对值在±0.1%以内。 8、系统适应性 配制6份相同浓度的杂质溶液进行分析,该杂质峰峰面积的相对标准差应不大于2.0%,保留时间的相对标准差应不大于1.0%。另外,杂质峰的拖尾因子不得大于2.0,理论塔板数应符合质量标准的规定。 9.溶液稳定性 按照分析方法分别配置对照品溶液与供试品溶液,平行测定两次主成分与杂质的含量,然后将上述溶液分别贮存在室温与冰箱冷藏室(4℃)中,在1、2、3、5和7天时分别平行测定两次主成分与杂质的含量。 可接受的标准为:主成分的含量变化的绝对值应不大于2.0%,杂质含量的绝对值在±0.1%以内,并不得出现新的大于报告限度的杂质。含量测定分析方法验证的可接受标准简介摘要:本文介绍了在对含量测定所用的分析方法进行方法学验证时,各项指标的可接受标准,以利于判断该分析方法的可行性。关键词:含量测定 分析方法验证 可接收标准在进行质量研究的过程中,一项重要的工作就是要对质量标准中所涉及到的分析方法进行方法学验证,以保证所用的分析方法确实能够用于在研药品的质量控制。为规范对各种分析方法的验证要求,我国已于2005年颁布了分析方法验证的指导原则。该指导原则对需要验证的分析方法及验证的具体指标做了比较详细的阐述。但是文中未涉及各具体指标在验证时的可接受标准,国际上已颁布的指导原则中也未发现相关的要求。另一方面,大多数药品研发单位在进行质量研究时,已逐步认识到分析方法验证的必要性与重要性,大都也在按照指导原则的要求进行分析方法验证,但验证完后却因没有一个明确的可接受标准,而难以判断该分析方法是否符合要求。本文结合国外一些大型药品研发企业在此方面的要求,提出了在对含量测定方法进行验证时的可接受标准,供国内的药品研发单位在进行研究时参考。1.准确度该指标主要是通过回收率来反映。验证时一般要求分别配制浓度为80%、100%和120%的供试品溶液各三份,分别测定其含量,将实测值与理论值比较,计算回收率。可接受的标准为:各浓度下的平均回收率均应在98.0%-102.0%之间,9个回收率数据的相对标准差(RSD)应不大于2.0%。2.线性线性一般通过线性回归方程的形式来表示。具体的验证方法为:在80%至120%的浓度范围内配制6份浓度不同的供试液,分别测定其主峰的面积,计算相应的含量。以含量为横坐标(X),峰面积为纵坐标(Y),进行线性回归分析。可接受的标准为:回归线的相关系数(R)不得小于0.998,Y轴截距应在100%响应值的2%以内,响应因子的相对标准差应不大于2.0%。3.精密度1)重复性配制6份相同浓度的供试品溶液,由一个分析人员在尽可能相同的条件下进行测试,所得6份供试液含量的相对标准差应不大于2.0%。2)中间精密度配制6份相同浓度的供试品溶液,分别由两个分析人员使用不同的仪器与试剂进行测试,所得12个含量数据的相对标准差应不大于2.0%。4.专属性可接受的标准为:空白对照应无干扰,主成分与各有关物质应能完全分离,分离度不得小于2.0。以二极管阵列检测器进行纯度分析时,主峰的纯度因子应大于980。5.检测限主峰与噪音峰信号的强度比应不得小于3。6.定量限主峰与噪音峰信号的强度比应不得小于10。另外,配制6份最低定量限浓度的溶液,所测6份溶液主峰的保留时间的相对标准差应不大于2.0%。7.耐用性分别考察流动相比例变化±5%、流动相pH值变化±0.2、柱温变化±5℃、流速相对值变化±20%时,仪器色谱行为的变化,每个条件下各测试两次。可接受的标准为:主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离;各条件下的含量数据(n=6)的相对标准差应不大于2.0%。8、系统适应性配制6份相同浓度的供试品溶液进行分析,主峰峰面积的相对标准差应不大于2.0%,主峰保留时间的相对标准差应不大于1.0%。另外,主峰的拖尾因子不得大于2.0,主峰与杂质峰必须达到基线分离,主峰的理论塔板数应符合质量标准的规定。本文为转帖!
小弟最近在用NIR做一个低含量物质,物质质量百分含量在0.01-0.50%,在建模时总是发现线性不好(不是一条45度的直线,而几乎是一条0度的水平线),我知道对于NIR来说,低含量物质不太好检测,那么,1 对于特定的组分,我怎么确定它的检测下限呢?可以通过实验还是计算来获得么?2 我是觉得这是到了物质的检测下限,所以模型做不好,不知道各位专家有没有别的意见?3 另外,由于含量比较低,仪器的系统偏差是否也会影响到模型,那么,实验的时候仪器的偏差有多大,这个有办法测量或计算么?

高效液相色谱法测定盐酸甲氯芬酯分散片含量及有关物质周岐勋 李健和 徐幸民 彭六保 曹俊华 罗霞(1.湖南省湘西自治州人民医院药剂科,湖南吉首416000;2.中南大学湘雅二医院药剂科,湖南长沙410011)【摘要】目的:建立盐酸甲氯芬酯分散片含量与有关物质测定的高效液相色谱分析方法。方法:采用Diamonsil C18柱(250 mmx4.6 mm,5um)。以乙腈_o.02 mol/ml磷酸二氢钾溶液(磷酸调pH值至4.0)(33:67)为流动相,检测波长为225 nm,流速为1.0 ml/min,柱温为25℃,进样量为20ul。结果:有关物质测定中降解产物与样品主峰能有效分离,分离度符合要求。盐酸甲氯芬酯最低检出量为2.0 ng。3批样品中有关物质的平均含量为1.56%。样品浓度在5.2—20.8斗∥ml的范围内与峰面积呈良好的线性关系(r=1.000),平均回收率为99.26%(RSD=I.12%)。结论:本检测方法专属性强.结果可靠。重复性良好,可用于盐酸甲氯芬酯分散片的质量控制。【关键词1盐酸甲氯芬酯分散片;有关物质;含量测定;高效液相色谱法http://ng1.17img.cn/bbsfiles/images/2012/07/201207232327_379311_2355529_3.jpg
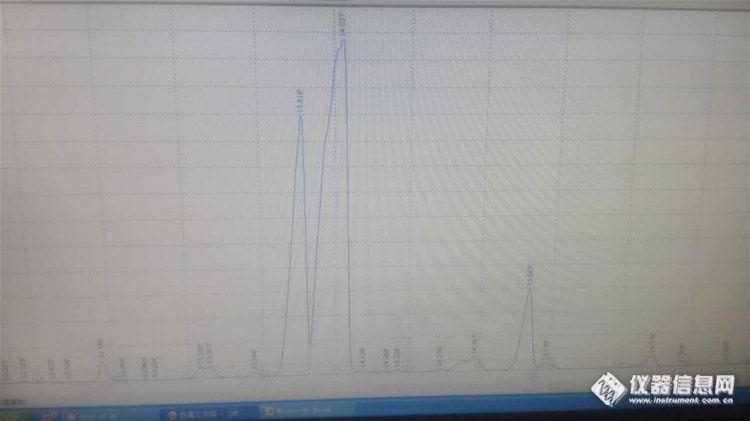
测物质含量,试样的保留时间和标样的保留时间相差0.25秒,这个试样的物质就有问题了?前面的第一个峰和标样的保留时间更接近,但是得出含量差距太大,而第二个大峰虽然计算含量符合,但是保留时间不符,请教大家,是不是第二个大峰就不是要测的那个物质了好友回复:只能依靠保留时间定性,其他都是猜的,理论站不住,多进几针看看怎么回事大家看看怎么回事呀http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_668253_3111590_3.jpg
请问专家循环水中如何测定苯酚和硝基酚类物质的含量?
求助:如何在线检测粉浆干物质含量测量介质是酒精发酵来料:小麦粉和玉米、木薯粉混合粉浆,要求控制来料干物质含量。目前采用的是化验室取样,物理干燥法,烘干水分测得干物质含量(质量百分比)。粉浆是物料加水常温混合,实际应是悬浊液,取样放置一会儿即固液分离沉淀了。我想请教对这种介质有没有在线检测或现场快速检测干物质含量的仪器或方法呢,谢谢!

样品制备制备方法:【有关物质】取本品适量,加流动相A溶解并稀释至每1ml中含1.0mg的溶液,滤过,取续滤液作为供试品溶液;精密量取1ml,置100ml量瓶中,用流动相A稀释至刻度,摇匀,作为对照溶液。取7-氨基去乙酰氧基头孢烷酸对照品和α-苯苷氨酸对照品各约10mg,精密称定,置同一100ml 量瓶中,加pH7.0磷酸盐缓冲液约20mL超声使溶解,再用流动相A稀释至刻度,摇匀。精密量取2 ml,置20 ml量瓶中,用流动相A稀释至刻度,摇匀,作为杂质对照品溶液。【含量测定】系统适应性试验: 取供试品溶液适量,在80℃水浴中加热60min,取20μl,测定,头孢氨苄峰与相邻杂质峰的分离度应符合要求。含量测定法:取装量差异项下的内容物,混合均匀,精密称取适量(相当于头孢氨苄0.1g),置100 ml量瓶中,加流动相适量,充分振摇,使头孢氨苄溶解,再用流动稀释至刻度,摇匀,滤过,精密量取续滤液10mL,置50mL量瓶中,用流动相稀释至刻度,摇匀,取20μl,注入液相色谱仪。分析条件【有关物质】色谱柱:Spursil C18,150×4.6 mm,5um,Cat#:(82001)流动相:流动相A为0.2mol/L磷酸二氢钠溶液(用氢氧化钠调pH至5),流动相B为甲醇洗脱方式线性梯度流速:1mL/min柱温:30 ℃检测器:UV 220nm进样量:20 μL【含量测定】色谱柱:Spursil C18,150×4.6 mm,5um[/fon
各位兄弟姐妹: 大家平时做有关物质、含量样品检测时时怎么操作的?我们 含量测定样品称取各两份,一份进两针,一份进三针,然后五针计算RSD 有关物质只称取一份,进五针。 觉得挺麻烦的。大家都是怎么弄的?
用标准加入法测定待测物质含量需要跟外标法一样得到一个方程吗?如果也是得到一个方程,这个方程就可以用于后面精密度、加样回收率的验证了?
[color=#444444]我想用液相色谱方法测定一个混合物中主物质和杂质对氯苯胺的含量,目前只能买到对氯苯胺这个标样,主物质是没法买到标样的,试问除了归一化法,还有什么方法可以测定主物质的含量呢?可否有从对氯苯胺入手的方法?谢谢各种前辈的建议![/color]
我想检测水中有哪些物质和相关的含量,请问有没有什么方法?同时,有人知道北京哪里能测水中有机物分子量分布吗 多谢了
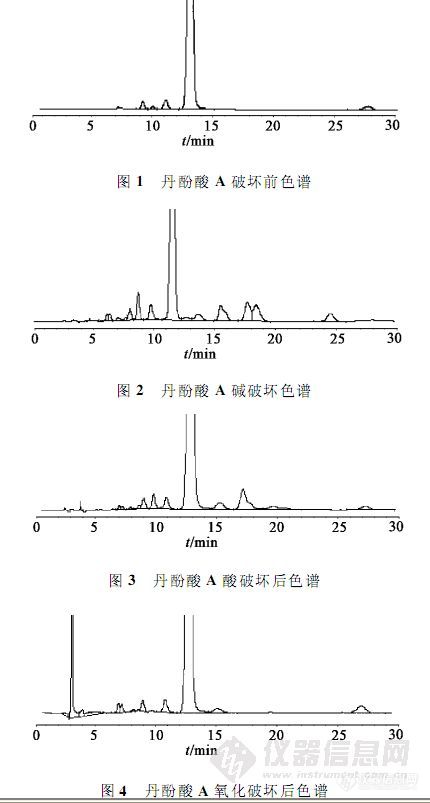
【作者】 于翠翠; 刘军锋; 车鑫; 刘珂;【机构】 烟台大学药学院; 山东靶点药物研究有限公司;【摘要】 目的:建立一种测定丹酚酸A中有关物质含量的方法。方法:用外标法测定丹酚酸C的含量,采用Diamonsil C18柱(4.6 mm×250 mm,5μm),乙腈-0.2%磷酸水溶液(30∶70)为流动相,流速1 mL.min-1,检测波长285 nm,柱温30℃,以主成分自身对照法控制其他杂质的总含量。结果:丹酚酸C的含量均1.0%,其他杂质峰面积和3.0%。结论:该方法专属性强,灵敏度高,简单准确,能有效控制丹酚酸A中有关物质的含量。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207231559_379228_2379123_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP