各位好!我实验室有个项目难题要攻克,现在没做出来都懵了,希望大家能帮忙解决问题给些意见 我们在做西洋参提取物物的总皂甙含量测定,之前我们是用过柱子紫外测定的方法,但方法的平行性和耐用性都不好。 现在用饱和正丁醇萃取总皂甙紫外测定的方法,结果平行性很差。不知道各位大侠有没有这方面的经验,能知道传授给小弟的!
求助,关于银杏叶提取物的总银杏酸含量测定。请问各位有没有做过的,我怎么做了几次都做不出来呢。条件都是按照药典要求做的,对照品都不出峰。我都要疯了。有谁做过帮忙分析一下或者上传一张图谱,谢谢。
植物提取物中多菌灵含量的检测,用什么基质的固相萃取小柱?
请问是否能检测银杏叶提取物中银杏酸的含量?
请问在哪里能检测人参茎叶提取物中人参皂甙单体含量?最好是官方的.谢谢!
硝酸铝显色法测银杏提取物总黄酮含量,以芦丁为对照,紫外波长扫描结果显示芦丁对照品在500nm左右有最大吸收峰,而供试品在400-600nm范围内均无最大吸收峰,而是呈上升趋势。特此求助,谢谢!

葛花及葛花提取物中总酚酸含量的测定 葛花(Flos Puerariae)为豆科植物野葛的干燥花蕾。广泛分布于湖南、河南、广东等省。具有解酒醒脾保肝的功效,主治伤酒发热烦渴、不思饮食、呕逆吐酸、吐血、肠风下血等症。葛花是中国的传统中药,用于解酒缓解酒后呕吐等症状,临床上具有显著疗效。总酚酸具有抗血栓形成、溶纤和抗脂质过氧化的作用,是治疗心肌缺血、心绞痛、心肌梗死等心血管疾病的有效成分。葛花中含有多种有效成分,但国内鲜有关于葛花中总酚酸方面的报道。因此测定葛花及葛花提取物中总酚酸的含量有其研究的价值。目前在我国含葛花的解酒方剂主要有葛花解酒汤、酒速愈用于临床,以及葛花茶、葛花露等保健品上市。葛花这一传统中药还有待进一步研究,以期开发出新的药物剂型和保健品,使其在医疗、保健等方面开辟更广阔的用途。本文利用分光光度法测定葛花及葛花提取物中总酚酸的含量,方法简单快捷,重现性好,可作为总酚酸检测的质量标准。实验部分1. 药品与仪器药品葛花:购自药店。没食子酸对照品:中国药品生物制品检定所。浓盐酸,95%乙醇,十二烷基硫酸钠,FeCl3,K3,均为分析纯试剂。仪器HW2-II电热恒温水温箱 (北京市医疗设备厂)SHZ-D3循环水式真空泵(上海双迅仪器有限公司)YHG-9023A台式电热恒温鼓风(上海姚氏仪器设备厂)分析天平(万分之一)(梅特勒)721分光光度计(上海索域设备有限公司)2 实验方法对照品溶液的配制精密称取干燥至恒重的没食子酸对照品23.2mg,加入100mL无水乙醇稀释至0.232mg·mL-1,移取10mL,加入90mL乙醇稀释十倍,制成23.2μg·mL-1溶液,用乙醇1:1倍稀释后作为对照品溶液备用。供试品溶液的制备生药溶液的制备取药材约0.1g精密称定置于25mL比色管中,用蒸馏水浸泡1.5h,沸水浴提取40min,冷却后倒出上清液。残渣再重复上述操作二次。合并三次提取液,和药渣一起转移到100mL容量瓶中,用水定容到刻度,摇匀,过滤,弃去初滤液,取续滤液10.00mL,用无水乙醇定容到20mL备用。葛花提取物的制备称取葛花[size=14p
我公司生产一保健品含有银杏叶提取物、沙棘提取物等多种含黄酮类物质,请问如何测定此保健品的总黄酮含量?? 用液相以槲皮素做对照品还是用紫外以卢丁做对照品测定呢?? 或有更好的办法? [em40] [em43] [em46]
1.多糖:加糊精2.多酚:加鞣花酸或儿茶素等3.总黄酮(紫外测含量的):加芦丁4.总黄酮(葛根、大豆、红车轴等液相色谱测含量的):加大豆甙元或染料木素等廉价单体5.当归、南瓜籽、锯叶棕:糊精分别添加少量当归油、南瓜籽油、脂肪酸6.刺蒺藜:加水杨甙提取物7.淫羊藿、水飞蓟、人参等:并非传统意义的假货,主要是混淆概念,如淫羊藿双甙、单甙;水飞蓟素、水飞蓟宾。不清楚多种成分之间的区别,很容易上套。8.比例提取物:糊精肯定要添加,主要是看加多少;还有就是使用伪品原料,或是已经没有含量的料渣j进行提取,应了那句话:只要缸里有一只鳖,你就不能说我造的不是鳖精。9.化学添加剂:为了防腐、防止快速吸潮等,有时添加苯甲酸钠、硬脂酸镁等化学品,背离了天然提取物的初衷10.廉价替代品:杜仲提取绿原酸冒充金银花绿原酸,莨菪浸膏冒充颠茄浸膏,南五味子冒充北五味子,人参茎叶提取物冒充人参根提取物等,很多总之,紫外法测定含量的,80%都可能造假,实际流通的紫外产品中造假的也达到了50%左右。带个多,或者总的产品都要提防了,这意味着比较精确的液相法可能测不了。有些产品连检测方法都没有,那更要当心了。再次对造假提出自己的观点不是为了打击谁,也不想改变什么,更不是说谁对谁错。有人说:你不上车,但你不要档着车走。就是这个道理,更何况我在车上。其实搞生产的90%以上都知道这些猫腻,搞技术的基本也都清楚,业务员和客户可能就欠缺这方面的鉴别能力,也算是提个醒吧,大冬天的,活跃一下气氛也不错有感于近年来植提产品造假越来越严重,市场形象很差的现状,本人根据自己的一些经验,谈一些常见的造假手段,希望能帮助大家一起提高。
请问有没有权威部门检测银杏叶提取物中银杏酸的含量?在哪里检测?谢谢!
作者:金智利; 袁文婧;哈药集团三精制药股份有限公司;摘要:目的:建立高效液相色谱法测定金银花连翘提取物中绿原酸、咖啡酸的的含量。方法:采用迪马(钻石)C18(250mm×4.6mm,5μm)色谱柱,流动相为甲醇-水-冰乙酸(20:80:1),流速1.0ml/min,检测波长:324nm。结果:金银花连翘提取物中绿原酸、咖啡酸能达到有效分离,绿原酸在9.44μg/ml~141.60μg/ml范围内,浓度同峰面积呈良好的线形关系,仪器精密度RSD为0.03%,方法的重复性RSD=0.24%。咖啡酸在5.36μg/ml~80.40μg/ml范围内,咖啡酸峰面积值与浓度有良好的线性关系,仪器精密度RSD为0.03%,方法的重复性RSD=0.33%。结论:该方法简便、准确、可行。可用于金银花连翘提取物的质量控制。
作者:仉晶, 金智利(哈药集团三精制药股份有限公司)摘要:目的:建立高效液相色谱法测定金银花连翘提取物(注射用)中连翘苷的含量。方法:采用迪马(钻石)C18(250mm×4.6mm,5μm)色谱柱,流动相为甲醇-水-冰乙酸(20:80:1),流速1.0ml/min,检测波长:324nm。结果:金银花连翘提取物中连翘苷能达到有效分离,在10.04μg/ml g~150.60μg/ml范围内,浓度与峰面积呈良好的线性关系。仪器精密度RSD为0.03%,方法的重复性RSD=0.96%。结论:该方法简便、准确、可行。可用于金银花连翘提取物的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/07/201207171451_378237_1606903_3.jpg
为了方便,买了该种中药10:1和20:1的提取物,但是该提取物成份有哪些,还有含量啥的都不清楚,求各位老师指点思路,应该怎么入手?小生初次接触这些,各位老师,指导一下,不甚感激啊
怎样用气相准确测出混合物中甲醇基甲苯的准确含量?混合物为甲醇甲苯的混合物,只含这两种物质,我按甲醇3份甲苯一份的比例混合后,用气象色谱面积归依法测定的含量为,甲醇56%甲苯44%,不对劲啊。
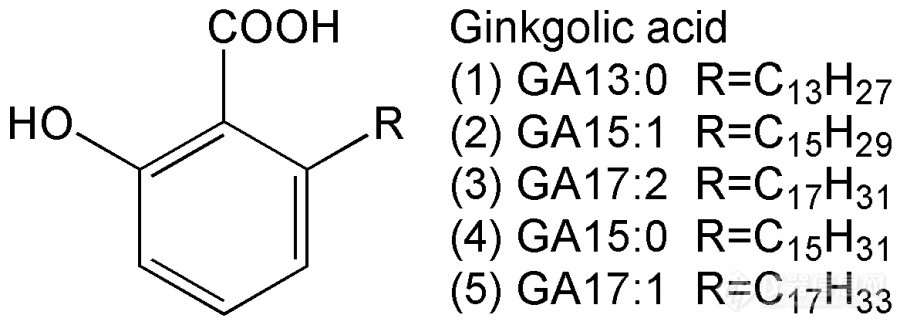
[b]1 引言[/b] 银杏叶提取物是目前临床广泛应用的天然产物药。西方学术界对该药物的成分和疗效也广泛接受。目前银杏叶提取物执行的标准基本上都源自德国EGB-761,其中除规定有效成分含量外,还对银杏酸含量进行了限定。按欧盟药典【European Pharmacopoeia 8.0.Monograph: Ginkgo dry extract. Refined and quantified. [b]2008[/b]】、美国药典【United States Pharmacopeia.Thirty-seventh Revision: Powdered Ginkgo Extract. [b]2015[/b].】要求,总银杏酸含量不得超过5 mg/Kg。我国执行的标准略为宽松,2015版药典【中华人民共和国药典[b]2015[/b], 一部:416-417页】规定总银杏酸含量不得超过10 mg/Kg。.[img=,690,248]http://ng1.17img.cn/bbsfiles/images/2018/07/201807292217347240_855_2204387_3.jpg!w690x248.jpg[/img][b]图1银杏酸的结构式.[/b] 银杏酸的分子结构如图1其中GA13:0、GA15:1、GA17:2是药典规定需要检测的项目。银杏酸的色谱分离并无难点,但是要测定银杏叶提取物中的银杏酸却存在两方面的问题:一是银杏酸限量很低,几乎接近仪器检出限;二是体系中共存的黄酮、内酯、糖类、蛋白质等成分复杂,基体干扰严重。目前通行的检测方法有欧盟药典法、中国药典旧版(2010版)方法、中国药典2015版方法,对比见表1。[img=,690,306]http://ng1.17img.cn/bbsfiles/images/2018/07/201807292218248285_199_2204387_3.png!w690x306.jpg[/img] 色谱分离方面,各种方法均采用反相色谱,条件略有差异,但无本质不同。提取方面,欧盟药典采用甲醇溶剂,提取较为完全,但共存的大量极性组分也一起提取进入溶液中,形成严重的基体干扰。而我国2010版药典方法采用石油醚提取,极性组分溶解少、干扰有所降低,但石油醚对极性试样的浸润性和渗透性不好,提取不完全,因此出现结果偏低的现象。相关研究人员【姚建标, 等.药物分析杂志, [b]2015[/b], 35 (11): 2041-2044.】借鉴了欧盟药典的提取方法提出了2015版药典方法,但是将试样量从0.5g增加到2g。增加样品量是为了弥补检测灵敏度的不足,因为2015版药典方法采用的紫外检测波长为310nm,这一波长下银杏酸的吸收强约为210nm下的五分之一。当然,在310nm下共存杂质的吸收更小、色谱基线漂移的问题也有所缓解,但是取样量增大使得更多的基体杂质进入色谱柱,基体干扰问题有增无减。. 众所周知,通过液液萃取、固相萃取等前处理手段可以起到减少基体干扰、富集目标物的作用,相关研究也有一定报道【Stefan U, Iuliana D S, Victor D,Andrei M. [i]J. Liq. Chromatogr. R. T.[/i], [b]2010[/b], 33 (1): 133-149. Ji W, Ma X, XieH, Chen L, Wang X, Zhao H, Huang L. [i]J.Chromatogr. A[/i], [b]2014[/b], 1368 (1):44-51.】。但是这些前处理过程较为繁琐,不利于推广。也有通过超高效液相色谱-串联质谱方法进行银杏叶提取物中银杏酸的检测【孙健, 等. 色谱, [b]2016[/b], 34 (2): 184-188.】,灵敏度和基体干扰问题都能较好的解决,但仪器昂贵,也难以推广。.[b] 本课题组在进行银杏活性成分提取研究的过程中也遇到了上述测试问题。为了改进现有方法,我们将简便有效的“分散液液微萃取”方法应用到样品的前处理过程中,有效解决了基体干扰问题,灵敏度也显著提高,同时该前处理方法十分简便,样品和试剂的用量也很少。相关研究前沿和创新内容已经撰文投稿,现将较为基础的实验方法介绍如下。[/b].[b]2 实验方法2.1 仪器与试剂[/b] 大连依利特230型高效液相色谱仪,安捷伦HC-C18(4.6*150 mm,5 μm)色谱柱。 色谱纯甲醇、乙腈为上海国药产,纯水为石英亚沸蒸馏。萃取剂为ACS级三氯乙烯,其余试剂为分析纯。 银杏叶提取物试样购于湖北某公司,相关制剂为市售药品(天保宁、金纳多)。 混合银杏酸标准品购于武汉某公司,总含量不低于99%,其中GA13:1占10.1%,GA15:1占50.5%,GA17:1占34.7%,GA17:2占1.7%,GA15:0占3.0%。用甲醇溶解配置成总银杏酸浓度为1.000 g/L的标准储备液,4℃保存。使用时用甲醇稀释成所需浓度的工作标液。.[b]2.2 实验方法 a)提取 准确称取0.5 g试样,加10 mL甲醇,常温超声提取10 min,冷却后定容、摇匀。 上述溶液用微孔滤膜过滤,收集滤液。 b)萃取 取上述滤液3.00 mL移入尖底离心管中,加入萃取剂三氯乙烯0.150 mL混匀,此时不分层。 加入水溶液(含10%无水硫酸钠 + 0.2%三氟乙酸)7.00 mL,加塞、摇匀,此时应得到浑浊液。 4000转离心3min,三氯乙烯萃取剂沉到底层。 c)取样 用注射器吸取三氯乙烯层0.100 mL移入样品瓶,氮气吹干,加0.100mL甲醇溶解。 d)色谱测定 条件见2.3。.2.3 色谱条件[/b] 采用Agilent HC-C18(4.6*150mm,5μm)色谱柱,甲醇-水-三氟乙酸体系(体积比92:8:0.1)为流动相,流速1.00mL/min,检测波长242nm,进样量50.0μL。.[b]2.4 定量分析[/b] 银杏酸工作标液直接进样测定,建立工作曲线,外标法定量。计算待测样时需乘以萃取的富集因子。富集因子为20。.[b]3 实验结果3.1 色谱图[/b] 银杏酸标样、银杏叶提取物试样、试样加标的色谱图见图2,分离效果较好,基线平直、基体干扰小。[img=,690,642]http://ng1.17img.cn/bbsfiles/images/2018/07/201807292222523305_3418_2204387_3.jpg!w690x642.jpg[/img].[b]3.2 方法验证[/b] 将银杏酸的甲醇标液按试样同样方法萃取和检测,考察了方法的重现性、线性范围、定量限(按S/N=10计算),测定了萃取过程的萃取率和富集因子,结果见表2。方法重现性2.9% ~4.8%。方法定量限0.007 ~ 0.020 mg/L,测定上限为5.00 mg/L。按取样0.5g定容至10mL计算,对试样的定量限为0.14 ~0.40 mg/Kg,对试样的测定上限为100 mg/Kg。 萃取率达到95.4% ~98.3%,富集因子达到19.1 ~19.7。[img=,690,231]http://ng1.17img.cn/bbsfiles/images/2018/07/201807292224269505_3788_2204387_3.png!w690x231.jpg[/img][b]3.3 样品测定[/b] 银杏叶提取物试样的测定结果见表3,进行了加标回收实验(总银杏酸加标量为5.00mg/Kg),加标回收率94.0% ~ 100.8%。以欧盟药典方法进行对照试验,结果与本方法一致。药品制剂试样的测定结果见表4,同样进行了加标回收实验和对照实验,结果也较好。[img=,690,306]http://ng1.17img.cn/bbsfiles/images/2018/07/201807292225141669_6540_2204387_3.png!w690x306.jpg[/img][img=,690,312]http://ng1.17img.cn/bbsfiles/images/2018/07/201807292225188364_2915_2204387_3.png!w690x312.jpg[/img].[b]4 相关讨论[/b] (1)试样体积与萃取剂体积之比影响萃取率和富集因子。此比值大时富集因子高但萃取率明显低于100%。反之则导致萃取率接近100%但富集因子很低。本方法选择3.00 mL试样加0.150 mL萃取剂,是兼顾两个方面考虑的结果。此时萃取率达到95.4% ~98.3%,接近100%。富集因子达到19.1 ~19.7,接近体积比计算结果,因此在定量计算时近似认为富集因子为20。 (2)萃取剂要选择低沸点、高密度、弱极性、难溶于水的有机溶剂,符合条件的主要是卤代烃。对比表明三氯乙烯效果最好。 (3)试样溶剂为甲醇,与萃取剂能够互溶,不能分成两相。但加入适量水之后可以实现分层。极性强的基质进入水相,从而使干扰减小。极性弱的银杏酸进入三氯乙烯层实现富集。水中加入10%硫酸钠起盐析作用,加0.2%三氟乙酸可以抑制银杏酸电离,都有利于提高萃取率。不加时萃取率显著降低。 (4)萃取过程形成浑浊的乳状液,两相接触面积大,因此很快达到平衡。加水溶液后震荡30秒到1min即可。萃取时间对结果几乎不影响。 (5)萃取在室温下进行,温度变化对结果影响不大。 (6)银杏酸的紫外吸收有3个特征峰,分别为210 nm、242 nm、310 nm。使用210 nm虽然灵敏度高,但干扰大。本方法使用242 nm检测,实际信噪比高于210 nm。使用310 nm检测的信噪比略有降低。 (7)由于萃取减少了基体干扰、实现了富集,本方法使用等度洗脱进行色谱分离也能获得很好的效果,并且没有基线漂移。本方法灵敏度也显著由于药典方法,定量限达到0.14 ~0.40 mg/Kg,而药典方法的检出限为0.5 mg/Kg。.[b]5 结论[/b] 采用“分散液液微萃取”方法对银杏叶提取物样品进行前处理,具有简单快捷的优点,同时减少基体干扰、实现富集的效果较好,能有效提高信噪比,降低检出限和定量限。方法准确可靠,易于推广。
甲醇甲苯混合物怎样测定?我想测定混合物中甲醇、甲苯的准确含量,有什么办法吗?

黄芩提取物-黄芩苷的测定http://ng1.17img.cn/bbsfiles/images/2012/12/201212091347_411149_2369266_3.jpg http://ng1.17img.cn/bbsfiles/images/2012/12/201212091349_411153_2369266_3.jpg 黄芩茎叶 http://ng1.17img.cn/bbsfiles/images/2012/12/201212091348_411151_2369266_3.jpg http://ng1.17img.cn/bbsfiles/images/2012/12/201212091350_411154_2369266_3.jpg黄芩根部http://ng1.17img.cn/bbsfiles/images/2012/12/201212091350_411155_2369266_3.jpg黄芩提取物-黄芩苷黄芩提取物-黄芩苷C21H18O11446.37黄芩苷药理作用:⑴抗炎抗变态反应,黄芩甙、黄芩甙对豚鼠离体气管过敏性收缩及整体动物过敏性气喘,均有缓解作用;⑵抗病毒、抗微生物(菌)作用;⑶镇静、解热、解痉作用;⑷抗癌、降压、利尿作用;⑸对血脂及血糖上升的作用;⑹利胆、保肝作用;⑺可降低乙醇所致的甘油三酸酯水平等。实验室测定方法名称:黄芩提取物-黄芩苷的测定-高效液相色谱法应用范围:本方法采用高效液相色谱法测定黄芩提取物中黄芩苷的含量。本方法适用于黄芩经加工制成的提取物。方法原理:本品加甲醇溶解、稀释,摇匀,滤过,续滤液进入高效液相色谱仪进行色谱分离,用紫外吸收检测器,于波长280nm处检测黄芩苷的吸收值,计算出其含量。试剂:1.甲醇2.磷酸仪器设备:高效液相色谱仪(配带紫外检测器)色谱条件:1.色谱柱:Xtimate C18柱2.流动相:甲醇 水 磷酸 =47 53 0.23.流速:1.0ml/min4.检测波长:280nm5.柱温:室温样品的制备:1.对照品溶液的制备精密称取黄芩苷对照品适量
我是一名检验员,在检验功劳木含量时,用的是功劳木提取物作为对照品,有四个成分,计算时我认为应该四个成分分别计算出含量再相加,而我同事认为可以以四个成分峰的总和计算,我们不能说服对方,求高人指点,最简单明了的解释方式
因为我们有GC和TD-GC/MS,因为这种方法没有做过,所以想请教一下,测试液体试剂中的甲苯、二甲苯含量,用GC 做还是GC/MS做?那个更好?怎么做,因为没有标准的。谢谢!
如何用内标法测定二甲苯中的苯含量?内标物的选择?谢谢!本人新手!
在使用uv的前提下,测定一个未知溶剂中甲苯的含量。
近日,我国输日植物提取物产品中因含有甜蜜素而遭到了日本海关的通报。为最大限度削弱该事件对我植物提取物出口贸易的冲击,检验检疫部门提醒辖区内相关出口企业应了解自身产品的特性,在确保产品不含违禁添加剂的前提下,跨越出口门槛。 甜蜜素,其化学名称为环己基氨基磺酸钠,是食品生产中常用的添加剂。有实验表明,消费者食用超过安全摄入量的甜蜜素,咽喉会有刺激反应,甚至会出现咽喉水肿或肿痛,继而引起疾病,损害人体健康。因此,目前世界上有包括美国、英国、日本等国在内的40多个国家禁止使用甜蜜素作为食品甜味剂。而我国、欧盟、澳大利亚、新西兰在内的80多个国家则允许在食品中添加甜蜜素。 植物提取物是以植物为原料,定向获取和浓集植物中的某一种或多种有效成分,用于药品、保健食品、烟草、化妆品的原料或辅料等。由于国内外对植物提取物的标准并不一致,导致了我上述产品出口频遭通报、召回、退运等,如何实现我国植物提取物产品在国际市场的快速准入问题,已成亟待破解的一大难题。 为此,检验检疫部门建议植物提取物出口企业积极采取应对措施:一方面需建立一套科学的符合产品提取规格、标准和质量的生产体系,严格控制产品的生产过程,避免在生产过程中造成产品添加剂、重金属等污染;另一方面,提高植物提取物检测技术,实现精细指标监控,把好原料安全关;最后,应及时掌握信息,对相关动向保持高度敏感,一旦国外对植物提取物法规有新进展,便应加强研究,以最快速度寻找应对之策。'而我国、欧盟、澳大利亚、新西兰在内的80多个国家则允许在食品中添加甜蜜素。'所以还难控制啊!
本人正在做重金属方面的综述,急需欧盟、美国、日本等关于药用植物提取物方面关于重金属含量限度的规定。拜托各位能不能给个答复。不胜感谢!
请问大家谁知道木材中苯、甲苯、二甲苯含量测定的方法是什么?或者相关的标准是什么?紧急求助,不胜感激!
我们买的工业甲苯溶剂,甲苯标称纯度约为90-95%,里面含有其他苯系物(约0.02-5%)。现在想检测甲苯和其他苯系物的准确浓度,请问可以用GC直接检测吗?使用毛细管柱可以吗?使用MS检测器可不可以?还是需要配FID之类的检测器?要不要配顶空?还有我们有送过一次工业环己酮溶剂(标称纯度95%)到外面的测试机构测试,他们用静态顶空测试(大概是取10ml样品,顶空加热80度,保持30min,取顶空气体分析),测得顶空气体约含有55%空气,45%的样品蒸汽,其中样品蒸汽中含有环己酮55%,还有10%的甲醇、5%的丙酮...(加起来100%),请问这种测试结果可不可以代表样品中的成分?还有如果测试涂料、油漆(已经涂在塑料表面固化的涂料或油漆)中的VOC,是不是必须用顶空?
我们买的工业甲苯溶剂,甲苯标称纯度约为90-95%,里面含有其他苯系物(约0.02-5%)。现在想检测甲苯和其他苯系物的准确浓度,请问可以用GC直接检测吗?使用毛细管柱可以吗?使用MS检测器可不可以?还是需要配FID之类的检测器?要不要配顶空?还有我们有送过一次工业环己酮溶剂(标称纯度95%)到外面的测试机构测试,他们用静态顶空测试(大概是取10ml样品,顶空加热80度,保持30min,取顶空气体分析),测得顶空气体约含有55%空气,45%的样品蒸汽,其中样品蒸汽中含有环己酮55%,还有10%的甲醇、5%的丙酮...(加起来100%),请问这种测试结果可不可以代表样品中的成分?发错位置了!版主能不能帮我移到“[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]”版?
大家测甲苯时一般苯的含量是多少?
广东省质检院顺德基地组织的涂料中苯,甲苯,二甲苯,乙苯含量的测定,QQ交流群191160216
我现在测叔甲苯的含量,原料有甲苯、异丁烯,柱子是KB-1,非极性柱子,规格是30m*0.25mm,我设的柱温120,气化室220,检测器220,进样量0.2微升,氢气流量40ml/min,空气流量是400ml/min,连续做了好几次,出来的峰型都不好,最大的峰就是叔甲苯的,我实在不知道是哪的原因,希望各位老师解答一下。还有一些小峰,自动积分积不出来,用手动积分的话,误差会不会很大?各位老师,做完谱图,让它自动积分,然后结果就准确吗?还用不用手动积分?[img=,690,407]https://ng1.17img.cn/bbsfiles/images/2019/10/201910081139092668_5569_1658948_3.jpg!w690x407.jpg[/img]
科罗索酸产品名称: 科罗索酸 产品类别: 主打产品 产品用途: 用于减肥原料及治疗糖尿病 点击次数: 1249 发布时间: 2010-03-02 产品介绍 产品名称:科罗索酸别 名:2α-羟基熊果酸(2-alpha- hydroxyursolic acid)商品名称:植物胰岛素.提取物名:巴拉巴提取物、大花紫薇提取物 规格: 科罗索酸1% 科罗索酸2% 科罗索酸10% 科罗索酸20%-98% 科罗索酸标准品CAS 号:4547-24-4分子式及分子量:C30H48O4 结 构式: 来 源:大花紫薇(俗称巴拿巴,盛产于菲律宾群岛)的提取物。 科罗索酸(corosolic acid),又名2α-羟基熊果酸,是存在于大叶紫薇中的一种三萜化合物。近年来研究发现其具有降血糖、减肥、抗肿瘤、抗炎、抗病毒和抗心血管疾病作用,尤其是其较好的降血糖活性,具有类似胰岛素的生理作用,被称为“植物胰岛素”,受到人们广泛关注,目前在国内外已被开发出多种保健品。为更好地开发利用科罗索酸,下面就科罗索酸降血糖活性进行相关介绍。 1 科罗索酸的理化性质科罗索酸为白色无定型粉末(甲醇),可溶于石油醚、苯、氯仿、吡啶等有机溶剂,不溶于水,可溶于热乙醇、甲醇。在无水条件下,与强酸(硫酸、磷酸、高氯酸)、中强酸(三氯乙酸)或Lewis氏酸(氯化锌、三氯化铝、三氯化锑)作用,会出现颜色反应或萤光。科罗索酸属α-香树脂醇型或乌斯烷型五环三萜类化合物,其基本骨架为多氢蒎的五环母核。科罗索酸在植物体内可游离存在,也可以皂甙的形式存在。在植物体内,其常常与其同分异构体山楂酸(2α-羟基齐墩果酸)共同存在,结构和化学性质相似,分离较难。 2 降血糖作用的研究科罗索酸降血糖作用的研究源于对菲律宾、马来西亚等东南亚国家传统降血糖保健饮料原料大叶紫薇叶的研究。1940年,Garcia采用以1~2g大叶紫薇干叶/kg体重的剂量喂养普通家兔,发现每24h家兔的血糖水平平均下降16~49mg/L。1956年,Garcia又采用大叶紫薇叶提取物进行临床实验,发现其具有降低糖尿病病人血糖的效果。1993年,Murakami等从大叶紫薇的叶中分离出2种三萜酸科罗索酸和山楂酸,发现科罗索酸具有降血糖的效果。目前,对科罗索酸降血糖的作用,已从体外细胞实验、体内动物实验和临床实验等方面进行了系统的研究。 在体外实验中,1993年,Murakami等以肿瘤细胞进行体外培养,发现大叶紫薇提取物可刺激细胞膜的葡萄糖运输通道,增强细胞对葡萄糖的利用,进而降低血糖含量,此作用与胰岛素降糖作用机理相似。体内动物实验中,1999年,Hamamoto等制备了一种含1%(w/w)科罗索酸的大叶紫薇提取物胶囊,将其喂养小鼠,喂养后90min,小鼠的血糖水平比对照组明显下降。此外,在兔子体内也进行了科罗索酸降糖作用实验。对禁食24h正常的兔子喂食科罗索酸,考察喂食一段时间后血糖含量,结果表明,大剂量服用科罗索酸引起的血糖降低与服用两个单位的胰岛素相似。大剂量服用科罗索酸可以带来57mg/ml的血糖降低。正常兔子口服科罗索酸会带来16~49mg/ml的血糖降低。2个多小时后再次给药会使血糖保持在低水平上,甚至略微有所下降,此过程会保持5个多小时。大剂量服用克罗索酸会带来40~58mg/ml的血糖降低。血糖降低的峰值期出现在服药后2~4h,6~10h后血糖恢复到正常水平。血糖下降明显和科罗索酸摄入量有关。临床实验中,1999年Ikeda等采用含大叶紫薇提取物的胶囊对糖尿病病人进行临床实验,发现其具有治疗糖尿病的作用。William等将含1%(w/w)科罗索酸的大叶紫薇甲醇提取物制备为软胶囊和硬胶囊,对56名II型糖尿病人进行了临床观察,结果表明,每人每天供给剂量32mg和48mg的两组,两周后,他们的血糖水平有明显下降,服用软胶囊组的病人,其血糖水平平均下降30%,服用硬胶囊组的病人,其血糖水平平均下降20%,表明两种胶囊都具有降血糖的效果,且软胶囊的降糖效果优于硬胶囊。 科罗索酸还有减肥作用,临床研究发现服用该药后能够调节体内胰岛素和血糖含量,具有明显的的减肥趋势(月平均减重0.908-1.816Kg),该过程比较缓且无需节食。 目前该天然产物已在美国作为营养补充剂上市,同时进行治疗糖尿病的三期临床试验,近期将通过FDA认证。其用于治疗糖尿病的作用效果与注射胰岛素相比较,具有口服效果显著、毒副作用小、使用方便等优点,作用效果与注射胰岛素相当。 应用展望: 科罗索酸作为防治肥胖症和II型糖尿病植物新药和功能性天然保健食品医药原料,目前已经成为市场的热门产品,产品供不应求。但目前在提取、制备上还存在一定的问题,随着研究的不断深入,该产品将具有非常广阔的市场前景。







 我要推广仪器
我要推广仪器
 下载APP
下载APP