请问三氟甲磺酸钠、三氟甲磺酸镨如何检测含量,急需

【作者】 管清香; 林天慕; 王恩思;【机构】 吉林大学药学院; 吉林大学生命科学学院;【摘要】 目的:建立测定甲磺酸地拉韦啶薄膜衣分散片含量的方法。方法:采用Diamonsil(TM) C18(150mm×4.6mm,5μm),乙腈-50mmol·L-1pH4.6磷酸二氢钠(55∶45)为流动相,检测波长为300nm。结果:甲磺酸地拉韦啶在12.54~62.70mg·L-1范围内呈良好线性关系(r=0.9999)。低、中、高3种浓度的平均回收率分别为99.5%,99.9%和101.5%(n=3)。结论:本方法简便、快速、专属性强,可用来测定甲磺酸地拉韦啶分散片的含量。 【谱图】

10,抽取5个版友);中奖名单:yifan1117(注册ID:yifan1117)捌道巴拉巴巴巴(注册ID:v3082413)20071940xu(注册ID:20071940xu)dyd3183621(注册ID:dyd3183621)zengzhengce163(注册ID:zengzhengce163)http://ng1.17img.cn/bbsfiles/images/2017/02/201702061500_01_1610895_3.jpghttp://ng1.17img.cn/bbsfiles/images/2017/02/201702061500_02_1610895_3.jpg【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================HPLC法测定甲磺酸多拉司琼的含量方法:HPLC基质:动物提取物应用编号:102760化合物:甲磺酸多拉司琼固定相:Diamonsil C18色谱柱/前处理小柱:Diamonsil C18, 250 x 4.6mm色谱条件:色谱柱:Diamonsil C18 250 mm× 4.6 mm, 5μm(Cat#:99903) 流动相: 乙腈- 水- 1mo l # L - 1 甲酸铵( 450:440:110), 用三乙胺调pH 至8.0 流速: 1.0 mL/min 柱温: 室温 进样量: 20μL 检测器: UV 285nm文章出处:中国药房 2007,18(22):1731-1732关键字:甲磺酸多拉司琼, Diamonsil C18, 钻石一代, 高效液相色谱法, HPLC, 含量测定谱图:摘要:目的:建立以高效液相色谱法测定甲磺酸多拉司琼含量的方法。方法:色谱柱为Diamonsil C18,流动相为乙腈-水-1mol.L-1甲酸铵(450∶440∶110),用三乙胺调pH至8.0,流速为1.0mL.min-1,检测波长为285nm,进样量为20μL。结果:甲磺酸多拉司琼检测浓度的线性范围为24~56μg.mL-1(r=0.9996);平均加样回收率为99.67%(RSD=0.74%)。结论:本方法操作简便、灵敏度高、重现性好,可用于甲磺酸多拉司琼的质量控制。http://www.dikma.com.cn/Public/Uploads/images/93-6.JPG

遗传毒性杂质,现在是药学研究的焦点之一。甲磺酸、苯甲磺酸等磺酸盐类物质与微量的低级醇在合成反应中生成烷基磺酸酯类,这些物质可与DNA发生烷基化反应,从而可能成为引发癌症的诱因。欧洲医药评价署、美国食品和药品管理局及国际药品注册协调会议等先后对基因毒性杂质做出限度规定。具体到我们的甲磺酸加贝酯产品,需要对其中的甲磺酸乙酯的限度进行控制。溶液制备:对照品溶液 取甲磺酸乙酯适量,精密称定,用乙腈制成每毫升含1.5μg/ml的溶液,精密移取2ml加入顶空瓶中,加入3ml水,6g碘化钠后,扎盖密封。供试品溶液 取甲磺酸加贝酯适量,精密称定,按供试品100mg与乙腈1ml的比例配制供试品溶液,溶液经超声、波膜过滤处理后,精密移取2ml加入顶空瓶中,加入3ml水,3g碘化钠后,扎盖密封。色谱条件:Agilent 7890色谱仪,顶空进样器,FID检测器;色谱柱,月旭WEL-PEG20M,30m*0.32mm*0.25μm(Cat. NO:01918-32001;Ser. NO:GC20131102);进样口温度为110℃,检测器温度为260℃,氢气流速为30ml/min,空气流速为350ml/min,进样量为1mL,分流比为0.1:1。升温程序,起始温度为40℃,维持10min,然后以20℃/min的升温速率,升温至160℃,维持1min。顶空瓶平衡温度为80℃,平衡时间为30min。结果:对照液色谱图: http://ng1.17img.cn/bbsfiles/images/2014/07/201407021418_503868_1609327_3.jpg其中,时间为1.982min的保留峰为甲磺酸乙酯的衍生物。供试液色谱图:http://ng1.17img.cn/bbsfiles/images/2014/07/201407021538_503896_1609327_3.jpg由色谱图中可以看出,样品中未检出甲磺酸乙酯。讨论:出峰时间非常的快,但是理论塔板数、分离度、对称因子等却非常给力!既得到了良好的分离效果,又尽可能的节约了分析时间。
请问三氟甲磺酸钠、三氟甲磺酸镨如何检测含量,急需
我的样品时硫酸镍溶液,镍含量和硫酸根的含量都很高,要检测氟离子、氯离子和硝酸根的含量,怎样进行前处理最好?
【前言】:现行硫酸根离子测定的方法有:重量法,电位滴定法,光度法,离子色谱法。重量法:样品溶液调至弱酸性,加入氯化钡溶液生成硫酸钡沉淀,沉淀经过滤、洗涤、烘干、称重,计算硫酸根含量。该方法实验过程繁琐,人工操作步骤多,而且耗时较长,一个样品需要几个小时才能完成。电位滴定法:氯化钡与样品中硫酸根生成难溶的硫酸钡沉淀,过剩的钡离子用EDTA标准溶液滴定,间接测定硫酸根。该方法虽然效率高,但对于基质复杂的样品,例如金属离子含量多,干扰因素多,造成结果不准确,电极需要定期维护。光度法:样品溶液中加入铬酸钡悬浮液生成硫酸钡沉淀,硫酸根离子置换的铬酸根离子以分光光度法测定,间接求出硫酸根离子含量。该法对于高含量的样品需要多次稀释,容易造成误差,而且试剂需要专用试剂,仪器运行成本高。离子色谱法:通过离子色谱柱分离出硫酸根离子,电导检测器检测其含量。该方法测定时间长,而且不适于高含量硫酸根的检测。【网络会议】:第五种硫酸根离子测定方法——温度滴定法测定硫酸根含量【讲座时间】:2015年07月10日 14:00【主讲人】:龚雁(国家纳米技术与工程研究院清华平台色谱组:开展硕士研究生课题的研究工作; 清华大学分析测试中心: 开展硕士研究生课题的研究工作;北京化工大学分析化学专业 硕士毕业;全国产品经理,有丰富的理论和客户实操经验。)【会议介绍】您样品中的硫酸根是如何测定的?还在用复杂而又耗时的重量分析法吗?日常分析中,您使用电位滴定时,遇到过下面的情况吗?----电位电极维护复杂,寿命短、分析人员希望不同滴定只用一个传感器??NOW,瑞士万通859 tiamo 温度滴定系统解决您上面所有问题!本次讲座主要内容:1、温度滴定简要介绍;2、硫酸根离子常见测定方法;3、温度滴定测定硫酸根离子的方法以及检测限;4、温度滴定测定硫酸根离子的方法优势;5、该应用涉及行业: 测定钒电池电解液中的硫酸根含量 检测硫酸铝铵溶液中铝离子、铵离子和硫酸根 测定电镀 Cr 溶液中SO42-含量 测定复合肥中硫酸根含量 检测火电厂脱硫浆液中的硫酸根含量 测定复合肥中硫酸根含量 测定化工产品中硫酸根含量 测定酯化反应液中的硫酸根 -------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名并参会用户有机会获得100元手机充值卡一张哦~3、报名截止时间:2015年07月10日 14:004、报名参会网址:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/15145、报名及参会咨询:QQ群—379196738http://ng1.17img.cn/bbsfiles/images/2015/04/201504071009_540886_2507958_3.jpg

[align=center][b]甲磺酸伊马替尼片的中试质量研究[/b][/align][align=center]王淑华,臧恒昌[/align](山东大学药学院)[b]摘要:[/b]甲磺酸伊马替尼片是一种小分子靶向抑制剂,用于治疗费城染色体阳性的慢性髓性白血病的慢性期、加速期或急变期和不能切除和/或发生转移的恶性胃肠道间质瘤的成人患者。甲磺酸伊马替尼由瑞士诺华公司2001年在美国首研上市,作为肿瘤的首个靶向治疗药物面世开创了肿瘤分子靶向治疗的新时代,目前已经在全球90多个国家获得批准,美国、欧盟和其它国家还批准甲磺酸伊马替尼片用于胃肠基质瘤患者的治疗。2005年进口到中国,中文商品名是格列卫。本文按照现行药品注册法规的要求对甲磺酸伊马替尼片的制备工艺进行研究,在小试工艺处方的基础上进行中试放大,对粉碎、混合、制粒、总混、压片、包衣的工艺参数进行研究确定,并确定中试设备,用中试产品与格列卫进行全面的质量对比试验,并进行影响因素试验考察10天的研究。开发出与原研药具有相同质量的甲磺酸伊马替尼片,实现甲磺酸伊马替尼片的可工业化生产。[b]关键词:[/b]甲磺酸伊马替尼片;开发;制备工艺;[b]1 实验材料和仪器[/b]1.1实验材料[align=center][img=,572,220]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251640111718_2529_3389662_3.png!w572x220.jpg[/img][/align]1.2实验仪器[align=center][img=,573,287]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251642146696_8860_3389662_3.png!w573x287.jpg[/img][/align][b]2方法与结果 [/b] 研究内容包括中试放大3批,批量为5000片,筛选各项工艺参数、进行影响因素考察、与原研药进行全面的质量对比,最终确定了中试规模的处方、工艺、工艺参数、设备及场所。[b]2.1 中试3批样品的制备[/b]为了充分验证处方及制备工艺的可行性,优化各项工艺参数,中试制备了三批甲磺酸伊马替尼片(批号20111205、20111228、20120104),每批5000片,三批产品处方见表3[align=center][img=,555,295]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251643270416_551_3389662_3.png!w555x295.jpg[/img][/align]制备工艺:取甲磺酸伊马替尼,用万能粉碎机粉碎,筛网目数为100目,粉碎后称取处方量,备用。粉碎过筛后的甲磺酸伊马替尼、微晶纤维素、羟丙甲纤维素、交联聚维酮、二氧化硅和硬脂酸镁分别称取处方量备用;将甲磺酸伊马替尼、微晶纤维素(Ⅰ)、羟丙甲纤维素同置湿法混合制粒机中,混合9min,搅拌转速20Hz,剪切转速30Hz;在HLSG-10型混合制粒机中边搅拌边加入纯化水制软材,搅拌转速15HZ,剪切转速15HZ,时间5min,取出后摇摆制粒机20目筛制粒;湿颗粒置60℃热风循环干燥箱中干燥,至水分为2.5%以下时停止;干燥完的颗粒取出,用摇摆制粒机24目筛整粒;整粒后的颗粒,加入交联聚维酮、微晶纤维素(Ⅱ)和二氧化硅,置SH-20三维混合机中混合,转速为9rpm,时间为20min,然后加入硬脂酸镁,继续混合10min,出料。取样检测中间体含量,计算理论片重;将上述总混粉用ZPW-21B型旋转压片机压片,Ф9mm圆形双凸冲模,控制平均片重为理论片重±3%,硬度50-70N;LDCS型高效包衣机,出风温度:38℃;锅体转速:5-10 rpm;喷液泵转速:5-10 rpm;雾化压力:1100mbar;直喷压力:750mbar;包衣增重2%-4%;用铝塑包装机进行泡罩包装,每板10片。泡罩板外套复合膜袋。[b]2.2 工艺参数的研究2.2.1 原料药的粉碎 [/b]甲磺酸伊马替尼为水中易溶的药物,粉碎的粒度对药物溶出的影响不大,因此,确定使用湿法制粒的常规工艺参数:即万能粉碎机粉碎,筛网为100目,备用。[b]2.2.2 混合[/b]混合采用高效湿法混合制粒机,甲磺酸伊马替尼、微晶纤维素(Ⅰ)、羟丙甲纤维素同置湿法混合制粒机中混合,搅拌转速20Hz,剪切转速30Hz,分别于3min、6min、9min和12min在不同位置取样测定甲磺酸伊马替尼的含量,计算RSD值,结果见表4,中试三批的混合参数见表5。[align=center][img=,583,304]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251644390288_1847_3389662_3.png!w583x304.jpg[/img][/align]结果显示中试样品在6min时,各个位置的含量测定结果已经没有显著差异(RSD<5%),表明这时已经混合均匀,9min和12min时,物料更加均匀(RSD<2%)。为保证工艺操作的可靠性,将中试的混合时间确定为9min。[b]2.2.3 制粒 [/b]20111205批中试样品的制粒过程:在混合制粒机中边搅拌边加入纯化水制软材,搅拌转速15Hz,根据处方筛选的结果,加入的纯化水量应为75ml,制备时先加入50ml,然后开启制粒(剪切),转速15Hz,2min后停机观察,发现软材略干,润湿不够,又加入少许,最终纯化水加入量为65 ml,制粒3min后停机观察,发现软材能够握紧成团,轻压即散,符合要求。出料后,置20目筛摇摆制粒机中制粒,湿颗粒置60℃干燥箱中干燥,至水分为2.5%以下时停止。24目筛摇摆制粒机整粒。20111228,20120104两批样品的制备均按照上述参数执行。最终确定中试的制粒参数为:搅拌转速15HZ,剪切转速15HZ,时间5min。取出后摇摆制粒机20目筛制粒。60℃干燥。水分控制小于2.5%。24目筛整粒。[b]2.2.4 总混 [/b] 由于本品制粒后需要加入较多的粉末,包括交联聚维酮、微晶纤维素、二氧化硅和硬脂酸镁,约占片芯总重的22%,所以保证粉末和颗粒的充分混合就比较关键。结合20111205批中试样品的制备,对总混时间进行了取样验证。将整粒后的颗粒与交联聚维酮、微晶纤维素(Ⅱ)和二氧化硅同置三维混合机中混合20min,转速为9rpm,然后加入硬脂酸镁,继续混合10min。分别于15min、20min、25min和30min在混合机中物料的不同部位取样6份,测定其中甲磺酸伊马替尼的含量,计算RSD值,结果见表6,三批中试批混合参数见表7。[align=center][img=,613,321]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251656004349_7538_3389662_3.png!w613x321.jpg[/img][/align]最终确定的中试混合工艺参数为:将整粒后的颗粒与交联聚维酮、微晶纤维素(Ⅱ)、二氧化硅同置三维混合机中混合20min,转速为9rpm,然后加入硬脂酸镁,继续混合10min。[b]2.2.5 中间体含量测定[/b]总混粉取样,测定其中伊马替尼的含量,按100mg/片计算理论片重,见表8。[align=center][img=,574,92]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251656591680_4007_3389662_3.png!w574x92.jpg[/img][/align][b]2.2.6 压片 [/b]参照原研药,采用Ф9mm浅圆冲压片。单独制备了一批3000片用量的总混粉,分别压制不同硬度范围的甲磺酸伊马替尼片各约800片,以确定合适的硬度,结果见表9~11及图1。[align=center][img=,555,545]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251657410320_9022_3389662_3.png!w555x545.jpg[/img][img=,512,293]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251657512292_1250_3389662_3.png!w512x293.jpg[/img][/align]试验结果显示GYYJ01批的溶出5min明显快于格列卫,10min和15min略快于格列卫,其脆碎度为0.5%,且有裂片和断片出现,脆碎度不合格;GYYJ02批溶出曲线与格列卫基本一致,脆碎度合格;GYYJ03批溶出曲线明显慢于格列卫,脆碎度合格。因此,确定压片硬度应控制在50-70N的范围之内。三批中试样品压片参数见表12。[align=center][img=,573,160]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251658297289_6842_3389662_3.png!w573x160.jpg[/img][/align][b]2.2.7 包衣[/b]取GYYJ02批的素片,进行包衣增重的研究。分别于不同时间取出部分片剂,使得它们具有不同的包衣增重。包衣条件为:取包衣粉,用纯化水配制成固含量为13%的液体,搅拌40分钟,备用;出风温度38℃,锅体转速5-10rpm,喷液泵速度5-10rpm,侧喷压力1100mbar,直喷压力750mbar。结果见表13~14及图2[align=center][img=,596,329]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251659214722_6185_3389662_3.png!w596x329.jpg[/img][img=,532,316]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251659216782_8146_3389662_3.png!w532x316.jpg[/img][/align]试验结果显示3种不同的包衣增重对溶出曲线基本无影响,因此,确定包衣增重的范围为2%~4%。中试3批包衣结果见表15[align=center][img=,593,227]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251700491390_8973_3389662_3.png!w593x227.jpg[/img][/align][b]2.2.8 包装 [/b]包衣片用铝塑包装机包装,成形温度118℃,热封温度120℃。2.2.9 中试研究工艺参数汇总[align=center][img=,533,471]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251701345335_8652_3389662_3.png!w533x471.jpg[/img][/align][b]2.3 三批中试产品数据及与原研药的对比研究[/b]结果见表17~18及图3。[align=center][img=,651,279]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251703255806_9275_3389662_3.png!w651x279.jpg[/img][img=,605,691]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251703354406_5321_3389662_3.png!w605x691.jpg[/img][/align][align=center][img=,554,639]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251705217673_2956_3389662_3.png!w554x639.jpg[/img][img=,537,641]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251705282186_7479_3389662_3.png!w537x641.jpg[/img][/align]试验结果显示20111205、 20111228两批自研产品与原研产品格列卫,在0.1M盐酸、pH6.8磷酸盐缓冲液和pH4.5醋酸盐缓冲液和水等4种溶出介质中15分钟溶出度均超过85%,判定为体外溶出行为一致。三批中试产品的各项质量指标与格列卫一致。[b]2.4 影响因素试验[/b]取20111205批中试样品,置强光照射(照度4500Lx)、高温(60℃)、高湿(RH92.5%和RH75%)条件下各放置10天,分别于0、5、10天检测吸湿增重、性状、溶出度、有关物质、含量等各项指标。同时取对照药(格列卫,100mg),置上述条件下,于0天和10天检查相应的项目,作为对比研究。影响因素试验结果表19。[align=center][img=,565,439]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251706500796_5767_3389662_3.png!w565x439.jpg[/img][/align][align=center][/align]试验结果显示自研产品和进口原研产品在高湿RH75%±5%条件下考察10天,吸湿增重均超过5%,提示产品应注意防潮。自研产品和进口原研产品在其它3个条件下各项指标均保持稳定,无显著变化。[b]2.5 中试研究试验结果[/b]2.5.1 处方(按5000片计),见表20[align=center][img=,567,322]http://ng1.17img.cn/bbsfiles/images/2018/07/201807251708221656_8624_3389662_3.png!w567x322.jpg[/img][/align][b]2.5.2 制备工艺 [/b](1)原辅料的处理取甲磺酸伊马替尼,用万能粉碎机粉碎,筛网目数为100目,粉碎后称取处方量,备用。粉碎过筛后的甲磺酸伊马替尼、微晶纤维素、羟丙甲纤维素、交联聚维酮、二氧化硅和硬脂酸镁分别称取处方量备用。(2)混合将甲磺酸伊马替尼、微晶纤维素(Ⅰ)、羟丙甲纤维素同置湿法混合制粒机中,混合9 min,搅拌转速20Hz,剪切转速30Hz。(3)制粒在HLSG-10型混合制粒机中边搅拌边加入纯化水制软材,搅拌转速15HZ,剪切转速15HZ,时间5min,取出后摇摆制粒机20目筛制粒。(4)干燥湿颗粒置60℃热风循环干燥箱中干燥,至水分为2.5%以下时停止。(5)整粒干燥完的颗粒取出,用摇摆制粒机24目筛整粒。(6)总混整粒后的颗粒,加入交联聚维酮、微晶纤维素(Ⅱ)和二氧化硅,置SH-20三维混合机中混合,转速为9rpm,时间为20min,然后加入硬脂酸镁,继续混合10min,出料。取样检测中间体含量,计算理论片重。(7)压片将上述总混粉用ZPW-21B型旋转压片机压片,Ф9mm圆形双凸冲模,控制平均片重为理论片重±3%,硬度50-70N。(8)包衣 LDCS型高效包衣机,出风温度:38℃;锅体转速:5-10 rpm;喷液泵转速:5-10 rpm;雾化压力:1100mbar;直喷压力:750 mbar;包衣增重2%-4%。(9)包装用铝塑包装机进行泡罩包装,每板10片。泡罩板外套复合膜袋。[b]2.5.3 中试研究场地[/b]固体制剂中试车间[b]2.5.4 中试设备[/b]见表2。[b]2.5.5 质量评价[/b]与原研药格列卫对比研究结果显示,中试产品的各项质量指标与格列卫相当,高温、光照、高湿三种剧烈条件下考察10天后,中试产品的各项质量指标仍与列卫相当,说明自研中试产品与原研产品质量一致。[b]3 结论[/b]因为本品原料是水溶性原料,粒度对溶出度影响不大,所以对原料前处理采用了常规机械粉碎法,过100目筛。物料混合6-12分钟都可以混匀,选择了中间点9分钟作为混合时间。根据实际情况,粘合剂水的用量由小试的2.5g/200片降到了65g/5000片。多批样品颗粒水分都小于2.5%,说明控制2.5%以下的颗粒水分适合本工艺。三批中试结果显示总混30分钟可以保证物料混合均匀。通过溶出曲线和脆碎度两个指标,考察了30-50N、50-70N、70-100N三个硬度范围,结果显示压片硬度范围在50-70N更为合理。包衣环节,考察了包衣增重2.1%、3.2%、4.2%三个梯度,对溶出曲线均无影响,最后确定包衣增重范围是2-4%。本文对甲磺酸伊马替尼片的制备工艺进行研究,用中试产品与格列卫进行全面的质量对比试验,并进行了影响因素试验考察研究,拟开发出与原研药具有相同质量的甲磺酸伊马替尼片,实现甲磺酸伊马替尼片的可工业化生产。
一下子买不到色谱纯或分析纯的甲磺酸,于是就用了化学纯的甲磺酸,只是背景稍高于以前,分离效果目前也没有什么问题,不知道以后会不会有影响。
在非水溶剂中检测甲基二磺酸亚甲酯中H离子(游离酸)的含量。(香精香料,精细化学品,定制化学品,电子化学品,表面处理剂的生产加工:)
【前言】:现行硫酸根离子测定的方法有:重量法,电位滴定法,光度法,离子色谱法。重量法:样品溶液调至弱酸性,加入氯化钡溶液生成硫酸钡沉淀,沉淀经过滤、洗涤、烘干、称重,计算硫酸根含量。该方法实验过程繁琐,人工操作步骤多,而且耗时较长,一个样品需要几个小时才能完成。电位滴定法:氯化钡与样品中硫酸根生成难溶的硫酸钡沉淀,过剩的钡离子用EDTA标准溶液滴定,间接测定硫酸根。该方法虽然效率高,但对于基质复杂的样品,例如金属离子含量多,干扰因素多,造成结果不准确,电极需要定期维护。光度法:样品溶液中加入铬酸钡悬浮液生成硫酸钡沉淀,硫酸根离子置换的铬酸根离子以分光光度法测定,间接求出硫酸根离子含量。该法对于高含量的样品需要多次稀释,容易造成误差,而且试剂需要专用试剂,仪器运行成本高。离子色谱法:通过离子色谱柱分离出硫酸根离子,电导检测器检测其含量。该方法测定时间长,而且不适于高含量硫酸根的检测。【网络会议】:第五种硫酸根离子测定方法——温度滴定法测定硫酸根含量【讲座时间】:2015年07月10日 14:00【主讲人】:龚雁(国家纳米技术与工程研究院清华平台色谱组:开展硕士研究生课题的研究工作; 清华大学分析测试中心: 开展硕士研究生课题的研究工作;北京化工大学分析化学专业 硕士毕业;全国产品经理,有丰富的理论和客户实操经验。)【会议介绍】您样品中的硫酸根是如何测定的?还在用复杂而又耗时的重量分析法吗? 日常分析中,您使用电位滴定时,遇到过下面的情况吗?----电位电极维护复杂,寿命短、分析人员希望不同滴定只用一个传感器?? NOW,瑞士万通859 tiamo 温度滴定系统解决您上面所有问题!本次讲座主要内容:1、温度滴定简要介绍;2、硫酸根离子常见测定方法;3、温度滴定测定硫酸根离子的方法以及检测限;4、温度滴定测定硫酸根离子的方法优势;5、该应用涉及行业: 测定钒电池电解液中的硫酸根含量 检测硫酸铝铵溶液中铝离子、铵离子和硫酸根 测定电镀 Cr 溶液中SO42-含量 测定复合肥中硫酸根含量 检测火电厂脱硫浆液中的硫酸根含量 测定复合肥中硫酸根含量 测定化工产品中硫酸根含量 测定酯化反应液中的硫酸根 -------------------------------------------------------------------------------1、报名条件:只要您是仪器网注册用户均可报名参加。2、报名并参会用户有机会获得100元手机充值卡一张哦~3、报名截止时间:2015年07月10日 14:004、报名参会网址:http://www.instrument.com.cn/webinar/meeting/meetingInsidePage/15145、报名及参会咨询:QQ群—379196738http://ng1.17img.cn/bbsfiles/images/2015/04/201504071009_540886_2507958_3.jpg
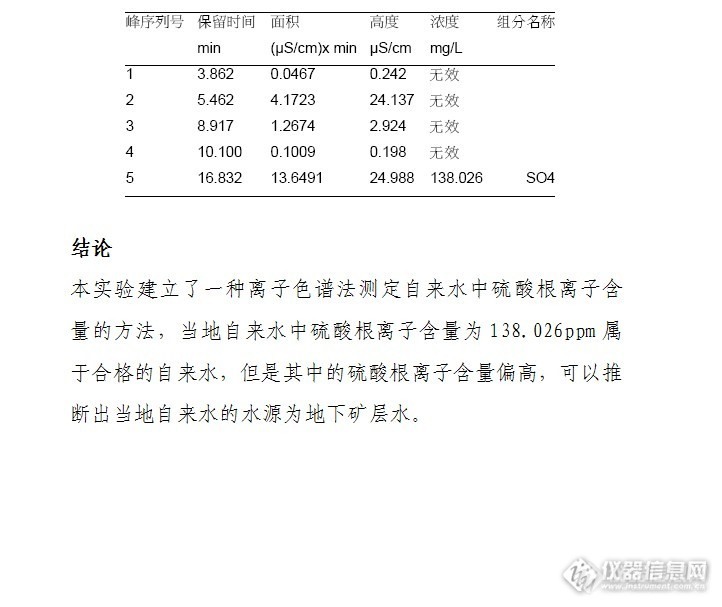
离子色谱法测定自来水中硫酸根离子的含量
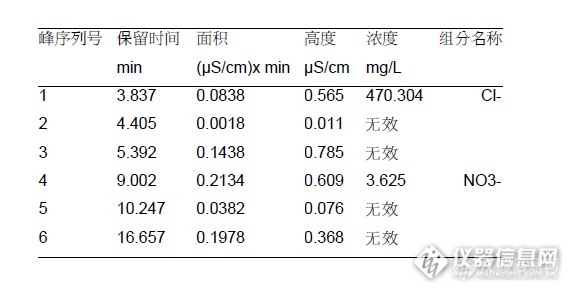
离子色谱法检测矿泉水中硝酸根的含量氮元素是构成生命体的一种重要元素,也是有机物中含量较高的一种元素,在人体内生物代谢有两种途径,其中一种是有机途径,最终产物为尿素,另外一种途径就是无机途径,其代谢的最终产物会是硝酸根。硝酸根本身没有什么危害,但是一旦进入人体,发生氧化还原反应,其危害就暴露出来了。本文中建立了一种使用离子色谱法检测矿物质水中硝酸根离子的方法。该方法能够快速准确的检测出矿物质水中硝酸根离子的含量。仪器设备本文中所使用的离子色谱仪为瑞士万通882离子色谱仪,所配的检测器为电导检测器。使用的色谱柱为Metrosep A Supp 4 - 250/4.0阴离子交换色谱柱,色谱柱的流速为1.0mL/min。淋洗液为Na2CO3和NaHCO3的混合液浓度为1.8mmol/L和1.7mmol/L。色谱的采集时间为20分钟。标准溶液配制称取相当于0.1克硝酸盐含量的硝酸铵,稀释到100毫升容量瓶中,随后移取10毫升稀释液,再次转移到100毫升容量瓶中。此时,分别吸取0.25、0.5、1.0、2.0、2.5毫升二次稀释液,定容到25毫升的比色管中。 http://ng1.17img.cn/bbsfiles/images/2013/12/201312011501_480170_2428063_3.png样品的检测谱图如下,本图中只对硝酸根离子的标准品进行了定量,所以本实验只检测硝酸根离子,检测的结果谱图如下图:http://ng1.17img.cn/bbsfiles/images/2013/12/201312011501_480171_2428063_3.png样品预处理及检测:由于矿泉水样品比较纯净,直接通过0.45um的尼龙微孔滤膜进行过滤处理即可。所使用微孔滤膜由安谱免费赞助,在此表示感谢!根据Magic软件的运算结果,该水样品中硝酸根离子含量为: http://ng1.17img.cn/bbsfiles/images/2013/12/201312011501_480172_2428063_3.png数据显示,百岁山矿泉水中硝酸根离子含量为3.625mg/L,符合国家标准中规定的含量限制,所以,该水样结果为满意结果。结论本文建立了一种离子色谱法检测矿物质水中硝酸根离子含量的方法,所选用的百岁山矿泉水中硝酸根离子含量为3.265ppm,能够达到饮用水中硝酸根的限制,属于合格产品。本实验中,标准曲线的相关系数做到了4个9和一个8是相当的不错啊!表示非常的满意!总体感觉万通的离子色谱做的真不错,赞一个!
现在我们要用气相检测甲磺酸中的甲磺酸甲酯和甲磺酸乙酯,不知道甲磺酸能不能直接进样? 对柱子和仪器有什么要求不?感谢
各位专家,有做过土壤中乙酸和甲磺酸的吗,跪求经验!
亲们,我最近在用高效液相检测透析液中醋酸根离子含量,但是标液峰面积一直在变小,样品峰面积差不多,结果与七月份检测的就相差太多,不知啥原因,求高手指导
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=24211]水质中硫酸根、硝酸根、钠离子和镁离子含量测定能力验证作业指导书等相关文件[/url]
向各位师傅请教一下,测量分析铝材阳极氧化的硫酸氧化槽中硫酸离子,铝离子,氯离子,氟离子,硝酸根离子含量的具体操作方法!谢谢!
用硝酸根离子及氯离子选择电极法分别测定各自含量时,请问分别用什么总离子强度调节剂,配比是什么?
如何用流动分析仪器准确测定烟草中硝酸根和亚硝酸根含量测定?尤其是烤烟?望大家把自己测试的心得说出来,大家交流吧,谢谢! 由于我们没有去离子水设备,所以我配给溶液时,用容器盛好蒸馏水,然后去配标准溶液,注意:所有的试剂最好用此水配置,否则会带来较大的误差,尤其是显色齐,有去离子水设备的当然就不要这么复杂了。
急需甲磺酸丁酯,哪位大虾可以指点从哪里可以购买到,急急!
本文为“离子色谱-抑制电导法快速测定葡萄酒中硫酸根离子”(http://bbs.instrument.com.cn/shtml/20130702/4827744/)的配套资料,甚少进行这方面的研究,所以问题肯定不少,请大家支持、完善。离子色谱法测定葡萄酒中可溶性硫酸根离子含量的不确定度评估摘要:应用测量不确定度评定方法,对离子色谱法测定葡萄酒中可溶性硫酸根离子含量的方法进行了不确定度评价。分析了测定过程中不确定度的来源,并对各不确定度分量及合量进行了评估,确定了对测定结果有重要影响的分量,为进一步优化测定方法、确保检测数据和结果的准确、可靠提供了依据。关键词:不确定度;离子色谱;硫酸根测量不确定度是指表征合理地赋予被测量值的分散性与测量结果相联系的参数,是表征测试结果可靠性和范围的重要参数。按照ISO/IEC17025《校准和检测实验室能力的通用要求》规定,对葡萄酒中可溶性硫酸根离子测定方法进行了不确定度分析,以确定测量中的干扰因素,了解对测量结果容易产生偏差的来源,找出不确定度中影响最大的因素,采取相应的方法减少干扰因素带来的影响。1. 实验方法1.1 主要仪器与试剂离子色谱仪:型号为ICS-3000,美国戴安公司。硫酸根(以SO42-计)标准溶液:1000 mg/L。购自国家标准物质研究中心。实验用水[size=10.
http://www.3158.com/upfiles4/2010/08/24/15/06/07/6599bb85.jpg请教各位版友,用液相法分析甲磺酸伊马替尼(见上式),其中有个中间体是没有与甲磺酸成盐的成分。在液相的溶液条件下(水相pH2.5),能把这两种物质分开吗?甲磺酸伊马替尼是可以水解的吧?这个中间体和甲磺酸依马替尼会生成同一种物质吗?要是这样的话,那这个中间体就没法分析了吧。谢谢各位版友!
请问谁有甲磺酸的检测方法,最好是简单的点的,易操作的
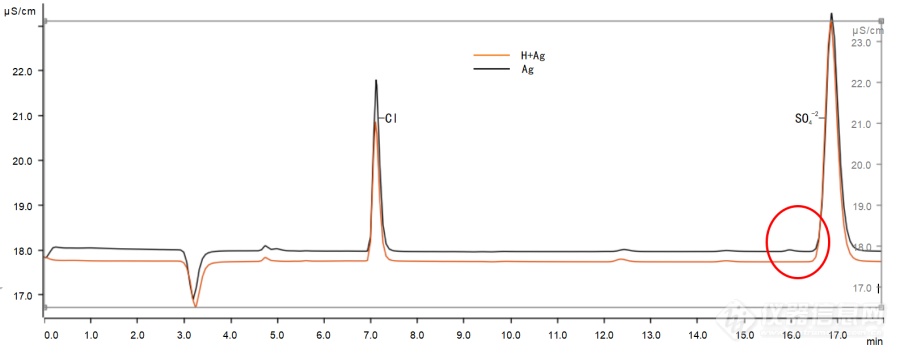
离子色谱法测定镍锰酸锂正极材料中的硫酸根含量邓 蓓*1,2,彭桂香1,2(1、宜春市锂电产业研究院,江西宜春,336000;2、江西省锂电产品质量监督检验中心,江西宜春,336000)摘 要 采用离子色谱法测定镍锰酸锂中硫酸根离子的含量,用盐酸和双氧水溶解试样,通过固相萃取柱净化后至离子色谱仪上进样测试,通过多次对比实验,优化了试验方法;在优化的试验条件下,硫酸根在0~20.0μg?L-1范围内具有良好的线性关系,其线性相关系数为0.9998。对4个不同含量的样品分别进行了11次平行测定,相对标准偏差(RSD)为1.6~5.9%。在镍锰酸锂试样中加入硫酸根标液进行加标回收实验,硫酸根离子的加标回收率在96.1%~106.3%之间。与传统的硫酸钡比浊法相比,离子色谱法操作简便快捷,方法检出限低,测量范围更宽,干扰少,测定结果可靠,再现性良好,适合大批量快速样品分析。关键词 镍锰酸锂;离子色谱法;硫酸根离子 Determination of sulfate in LiNi0.5Mn1.5O4 cathode materials by Ion chromatographyDENG Bei*1,2,PENG Guixiang1,2(1. Yichun research institute of lithium battery industry,Yichun,Jiangxi,336000,China 2.Jiangxi Province Litium Product Quality Supervision and Inspection Center, Yichun, Jiangxi, 336000, China )Abstract: The component of sulfate in LiNi0.5Mn1.5O4 were determined by Ion chromatography,The samples were dissolved in hydrochloric acid and hydrogen peroxide, purified by solid phase extraction column and injected into ion chromatography for test. The mass concentration of sulfate ion in the range of 0~20.0μg?L-1 had a good linear relationship with correlation coefficients of 0.9998.Under the optimal conditions,the relative standard deviations (RSD) of four different mass concentration samples were 1.6~5.9% for 11 parallel measurements. For recovery tests, sulfate ion were added into the LiNi0.5Mn1.5O4 samples and the recovery rates of sulfate ions were in the range of 99.4%~100.2%. Compared with the double indicator titration method, the ion chromatography method is convenient and fast. At the same time, this method has the advantages of simple operation,a lower detection limit,a wider measurement range,less interference, high precision, reliable determination results and good reproducibility. It is suitable for rapid analysis of large quantities of samples. Keywords: LiNi0.5Mn1.5O4 ion chromatography sulfate ion 摘要 随着锂离子电池在新能源汽车领域的应用扩展以及“双碳”政策的深入。人们对锂电池性能的要求越来越高。锂电池的性能很大程度上取决于正极材料,而镍锰酸锂正极材料具有资源丰富、成本低、环境友好、电压高且能量密度高等优势,在大功率领域中得到了广泛应用,从而成为了研究热点。在制备镍锰酸锂的过程中,会残留一定量的硫酸盐,因此,硫酸根含量是其成品的一项重要技术指标。现有的测定硫酸根的方法主要有:离子色谱法、分光光度法和电位滴定法。其中,分光光度法需严格控制操作条件,且测定镍锰类等有色金属化合物时,本底值有颜色需要差减法扣除,含量较低时精密度不够好;电位滴定法只适合高含量的硫酸根离子测定;而离子色谱法检出限低、操作便捷高速效率高且样品前处理简单。因此,在镍锰酸锂中测定硫酸根时选择离子色谱法。1实验部分1.1主要仪器与试剂 除非另有说明,本文中实验用水均为去离子水(18.25MΩ?cm)。 Metrohm ECO-IC离子色谱仪(瑞士万通公司);Metrosep A Supp 5-250/4.0色谱柱(瑞士万通公司);H型预处理柱:AnL Pre IC-H-1cc(宁波鸿谱仪器科技有限公司);Ag型预处理柱:AnL Pre IC-Ag-2.5cc(宁波鸿谱仪器科技有限公司);水系微孔过滤滤膜:50(mm)×0.45(μm);聚醚砜(PES)针筒式过滤器:13(mm)×0.22(μm);万分之一天平(梅特勒MS205DU);Milli-Q去离子水制备装置(德国默克密理博);真空抽滤装置(天津市津腾实验设备有限公司)无水碳酸氢钠(优级纯);无水碳酸钠(优级纯);浓硫酸(优级纯);浓盐酸(优级纯);30%过氧化氢(分析纯);7种阴离子(氟离子、氯离子、硝酸根离子、亚硝酸根离子、溴离子、硫酸根离子、磷酸根离子)混合标准溶液(质量浓度为1000mg?L-1) 国家标准物质中心。 1.2 实验方法1.2.1 标准溶液的配制 储备液的配制:用移液枪准确移取10mL混合标准溶液于100mL容量瓶中,加水稀释至终点刻度,摇匀,静置备用,放置于4℃冰箱中保存。此溶液1.00mL相当于100μg硫酸根。标准曲线溶液的配制:用移液枪分别准确移取0mL、0.50mL、1.25mL、2.50mL、5.0mL和10.0mL储备液,分别置于50mL容量瓶中,加水稀释至终点刻度,摇匀,静置备测。该系列溶液1.00mL分别相当于0.00μg、1.00μg、2.50μg、5.00μg、10.00μg、20.00μg硫酸根。在本方法确定的最佳实验条件下依次从浓度低到高的顺序进样,绘制标准曲线。 1.2.2 色谱条件 色谱柱选用 Metrosep A Supp 5-250/4.0色谱柱;流速0.7mL?min-1 ;流动相4.5mmol?L-1NaHCO3-4.5mmol?L-1Na2CO3溶液;进样体积20μL;电导检测器。1.2.3 样品测定将镍锰酸锂样品在105-110 ℃下烘干2h,然后置于干燥器内,冷却至室温,备用。准确称取 0.2000g(精确至0.0001g)镍锰酸锂试样于100 mL 烧杯中,缓慢加入3.0mL盐酸(1+1),再加入4-5滴过氧化氢,低温加热至完全溶解且蒸发至近干,冷却,加5-10mL水溶解,转移至100mL容量瓶中,用水洗涤烧杯3-5次,同时将洗涤液转移至容量瓶中,用水稀释至刻度,摇匀,待净化。 将H柱和Ag柱用10mL水活化,静置10min,将上述溶液缓慢通过活化好的H柱和Ag柱,弃去前3mL滤出液,获取后续2mL滤出液,过0.22μm聚醚砜(PES)针筒式过滤器,上机待测。 2 结果与讨论2.1 淋洗液浓度的选择 为了检验其他阴离子对硫酸根离子峰积分的干扰,选择了7种阴离子(氟离子、氯离子、硝酸根离子、亚硝酸根离子、溴离子、硫酸根离子、磷酸根离子)混合标准溶液进行分离,分别使用三种不同浓度的淋洗液对待测标准溶液进行淋洗分离。实验结果如图1所示,具体浓度等信息见表1。https://ng1.17img.cn/bbsfiles/images/2022/09/202209081603235950_1568_5824073_3.png!w690x283.jpg图1 不同浓度淋洗液的离子色谱图Fig. 1 Ion chromatograms of different concentrations of eluent表1 不同浓度淋洗液的出峰时间Table 1 Peak time of different concentrations编号No.淋洗液浓度concentrations of eluent7种阴离子分离情况Separation of 7 anionsSO42-出峰时间Peak time/minNa2CO3 /(mmol/L)NaHCO3 /(mmol/L)1#4.540.8好16.32#3.21.0好21.83#4.51.4好24.5由图1和表1可知,三种浓度的淋洗液都能很好的将7种阴离子分离,但是,从时间、资源和环境的角度,我们选择了更早出峰的1#淋洗液(4.54mmol/LNa2CO3-0.8mmol/LNaHCO3 ),这样既节省时间、节约资源,同时更少的淋洗液减少了废液的处理从而对环境更友好。 2.2 溶样方法的选择采用硝酸和盐酸加双氧水的方式溶样,称样量均为0.2g时的试验结果见表2。由表2可知,硝酸较难溶解样品,盐酸能够溶解样品但是速度缓慢,盐酸和双氧水加入样品混匀,在低温电热板上微热数分钟即可将样品完全溶解。表2 溶样方法Table 2 Methods of sample dissolving溶样介质Sample dissolving medium溶解状态Dissolved state溶样介质Sample dissolving medium溶解状态Dissolved stateHNO3难溶HCl溶解缓慢HNO3 + H2O2溶解缓慢HCl + H2O2溶解,澄清 2.3 盐酸+双氧水加入量试验 由于离子色谱仪测定时,氯离子也会出峰,浓度过大会造成拖尾并且会严重影响硫酸根离子的出峰和积分。溶解样品的盐酸中含有大量的氯离子,因此,盐酸的加入量在保证能够溶解样品的条件下应当越少越好。所以,做了盐酸和双氧水加入量的一系列试验。表3是称样量为0.2g时不同盐酸和双氧水加入量的试验结果。由表3中结果可知,当称样量为0.2g,盐酸(1+1)用量3mL、双氧水用量0.5mL时,样品在低温电热板上微热数分钟刚好能够完全溶解,满足测定分析要求。因此,当称样量为0.2g时,选择3mL盐酸(1+1)、0.5mL双氧水作为溶样体系。表3 盐酸+双氧水加入量试验Table 3 Hydrochloric acid + hydrogen peroxide addition test盐酸(1+1)加入量Addition of HCl(1+1)双氧水加入量 Addition of H2O2溶解状态Dissolved state1.00mL0.50mL部分溶解,有黑渣1.00mL2.00mL0.50mL基本溶解,有少量残余1.00mL3.00mL0.50mL完全溶解,澄清1.00mL4.00mL0.50mL完全溶解,澄清1.00mL 2.4 溶液是否蒸干再定容试验由于氯离子对硫酸根的测定有一定的干扰,而加入的盐酸可以通过加热的形式蒸发掉一部分,所以,进行了定容前是否蒸干的对比试验,其结果如图2所示。https://ng1.17img.cn/bbsfiles/images/2022/09/202209081601009376_9858_5824073_3.png!w690x324.jpg图2 样品蒸干与不蒸干的图谱Fig. 2 Ion chromatograms of dried and non dried samples由图2可知,未蒸干时的氯离子峰又宽又长,已经影响了硫酸根的出峰时间,而蒸干后的氯离子峰虽然仍旧较宽但已经明显小了不少,并且没有影响硫酸根离子的出峰时间。所以,蒸干有利于硫酸根的测定,因此,选择在定容前将溶液低温加热至近干。2.5 固相萃取柱的选择由于溶液中含有锂、钠、钾、钙、铁、铜、铬和镉等金属离子以及残留的氯离子的干扰,在溶液进样前需要过柱子净化。选取H柱去除溶液中可能存在的金属离子和Ag柱去除溶液中氯离子,其结果见图3、图4。 https://ng1.17img.cn/bbsfiles/images/2022/09/202209081600325635_5928_5824073_3.png!w690x274.jpg图3 过H柱和过H柱+Ag柱的试验结果Fig. 3 Test results of passingH column and passing H column + Ag column由图3可知,溶液只过H柱时氯离子峰特别高,严重影响了硫酸根离子的测定;而同时过H柱和Ag柱的话,氯离子峰就大大变小了,有利于硫酸根的测定。https://ng1.17img.cn/bbsfiles/images/2022/09/202209081600025629_4582_5824073_3.png!w690x271.jpg 图4 过Ag柱和过H柱+Ag柱的试验结果Fig. 4 Test results of passing Ag column and passing H column + Ag column由图4可知,只过Ag柱时硫酸根离子附近会有干扰峰,而同时过H柱和Ag柱则没有。所以综上所述,选择过H柱+Ag柱为佳。2.6 标准曲线、检出限及定量限在 1.2.2仪器工作条件下,分别测定了1.2.1中系列混合标准工作溶液。以硫酸根质量浓度(x)为横坐标,色谱峰面积(y)为纵坐标绘制校准曲线。结果表明,硫酸根离子的线性范围为0~20.0μg?L-1,线性方程为y=8.54057×10-3+7.01333×10-3x,相关系数为0.9998。依据计量规范JJG823-2014,选取相应的1.0μg?L-1硫酸根标准溶液进行11次平行测定并计算检出限为0.01μg?L-1,依据EPA SW-846方法规定采用4倍检出限浓度作为定量限为0.04μg?L-1。2.7 精密度试验选取4个不同品位的产品,按照设定的实验方法对样品中的硫酸根含量分别进行11次平行测定。得到11个测定值,计算其平均值、标准偏差及相对标准偏差,结果见表4,结果证明精密度满足分析要求。表4方法精密度实验结果Table 4 Precision tests of the method(n=11) /%样品编号Sample No.测定值(n=11)Found平均值Average标准偏差SD相对标准偏差RSD1#0.06430.05790.0567 0.0539 0.05850.3435.8650.0595 0.06130.06180.0605 0.0534 0.0554 0.0585 2#0.1120.118 0.118 0.116 0.1150.2241.9530.1140.114 0.115 0.113 0.1160.1110.115 3#0.3180.3030.308 0.317 0.3140.7772.4730.323 0.3180.313 0.316 0.3090.304 0.328 4#0.6520.632 0.655 0.650 0.6381.0581.6580.6320.6230.635 0.6430.6340.6260.638 2.8 加标回收试验由于目前还没有相应的镍锰酸锂标准样品,所以采取在样品中添加不同含量的硫酸根离子进行加标回收试验,按照试验方法进行样品处理、测定,其结果见表5。该方法加标回收率在96.1~106.3%之间,能够满足测定要求。表5 加标回收试验Table 5 Recovery tests样品编号Sample No.样品量m1Mass of Sample/g本底值SO42Mass of SO42-/μgSO42-加标量m2Spiked/μg测定值m3Measured value/μg回收率ηRecovery/%1#0.2013117.5250169.22103.40.2002117.02100213.1296.12#0.2003230.01100326.4196.40.2005231.03200426.6297.83#0.2015628.565001160.06106.30.2008629.017501367.7698.54#0.20111277.055001768.5598.30.20061276.137502041.88102.1 2.9方法比对目前,测定硫酸根的含量主要有两种方法,分别是离子色谱法和硫酸钡比浊法。对该两种方法分别进行了试验,结果如表6所示。由表6可知,含量高的时候两种方法的结果相差不大,但是含量低的时候,硫酸钡比浊法测定结果不理想。并且,硫酸钡比浊法对加入的硝酸钡状态和用量要求较高,测定前的静置时间也比较严格,在一定的时间内必须完成测定,同时对实验人员的操作手法要求严苛,因此,不适合批量测定。而离子色谱法前处理简单便捷,样品处理后稳定性良好,可用于大量样品的测定。表6 测定镍锰酸锂中的硫酸根含量Table 6 Determination of sulfate content in LiNi0.5Mn1.5O4 /% 样品编号Sample NO.离子色谱法测定值Found by ion chromatography method硫酸钡比浊法测定值Found by barium sulfate turbidimetry method1#0.0585未检出2#0.1150.0893#0.3140.3214#0.6380.650在实验过程中,从称样量、检出限和前处理等方面对比了离子色谱法和硫酸钡比浊法,具体对比结果见表7。表7 两种方法对比Table 7 Comparison of two methods序号NO.项目Content离子色谱法 Ion chromatography method硫酸钡比浊法Barium sulfate turbidimetry method1方法检出限0.01%0.1%2称样量0.2g1.0g3前处理样品稳定性常温下15天内稳定静置15-30min内完成测定4测定范围0.01%-0.7%0.1%-0.7%由表7可知,离子色谱法不仅测定结果可靠、方法检出限低,而且前处理样品稳定性好。因此,不管是从结果准确性和测定范围还是从效率上来看,离子色谱法都更胜一筹。3 结语镍锰酸锂正极材料样品用盐酸和双氧水溶解,通过固相萃取柱净化后至离子色谱仪上进样测定,较传统的硫酸钡比浊法,方法操作简单便捷,样品稳定性良好,干扰少,方法检出限低,测定范围宽,精密度较高,测定结果可靠,再现性良好,适合大批量快速样品分析。 References李 旺,周 兰,刘佳丽.镍锰酸锂正极材料及其适配性电解液研究最新进展.无机盐工业,2019,51(6):5-10.Li Wang , Zhou Lan , Liu Jiali.Latest development of LiNi0.5 Mn1.5 O4 cathode materials and adaptive electrolytes.Inorganic chemicals industry,2019,51(6):5-10 钟茂初."双碳"目标有效路径及误区的理论分析.中国地质大学学报(社会科学版).2022,22(1):10-21.ZHONG Mao-chu.A Theoretical Analysis on the Effective Path and the Misunderstanding of "Dual Carbon" Goal.Journal of China University of Geosciences (Social Sciences Edition).2022,22(1):10-21. 王静,吴比赫,林伟庆,等.锂离子电池高电压正极材料LiNi0.5Mn1.5O4-----研究进展.厦门大学学报:自然科学版,2015,54(5):630—642. Wang Jing,Wu Bihe,Lin Weiqing,eta1.Research progress in high—voltage LiNi0.5Mn1.5O4 for lithium ion batteries.Journal of Xia’men University:Natural Science,2015,54(5):630—642. 金彦章,王永和,刘强,等.高电压正极材料LiNi0.5Mn1.5O4制备及性能研究.无机盐工业, 2017, 49 ( 6 ): 45-49. Jin YanzhangWang YongheLiu Qiang,eta1.Study on synthesis and performance of high-voltage LiNi0.5Mn1.5O4 cathode material for lithium-ion battery.Inorganic Chemicals Industry.2017, 49(6):45-49 Rahaye I,Noviyanti A R,Rakmawaty D,etal.Preparation of Lithium Iron Phosphate-Carbon Composite as a Cathode for Lithium Ion Battery.Materials Science Forum,2019,966:392-397. Wen Weicheng,Yang Xiukang,Wang Xianyou,etal.Improved electrochemical performance of the spherical LiNi0.5Mn1.5O4 particles modified by nano-Y2O3 coating.Journal of Solid State Electrochemistry,2015,19(4):1235-1246. Nageswaran S,Keppeler M,Kim S J,etal.Morphology controlled Simodified LiNi0.5Mn1.5O4,microspheres as high performance high voltage cathode materials in lithium ion batteries.Journal of Power Sources,2017,346:89-96. 杨强强 , 崔雅茹,李 倩,等.共沉淀法合成镍锰酸锂正极材料前驱体.矿冶工程.2020,40(5):134-138YANG Qiang-qiang,GUI Ya-ru,LI Qian,etal.Synthesis of Precursor of LNMO Cathode Material by Coprecipitation Method.Mining and metallurgical engineering. 2020,40(5):134-138刘春峰. 离子色谱法测定镍钴锰氢氧化物中硫酸根离子含量.中国无机分析化学.2013,3(z1).LIU Chunfeng.Determination of Principal Components in Industrial and Battery Lithium Carbonate by Automatic Potentiometric Titration MethodDetermination of sulfate ion in Nickel Cobalt Manganese Hydroxide by ion chromatography.Chinese Jorunal of Inorganic Analytical Chemistry.2013,3(z1).张枫华,雷仲存,顾红琴.离子色谱法测定水中硫酸根离子浓度范围的探讨.冶金动力.2013,161(7):56-60.ZHANG Fenghua,LAI Zhongcun,GU Hongqin.Discussion on suitable concentration range of sulfate ion in water determined by ion chromatography.Metallurgical Power,2013,161(7):56-60. GB/T 11064.9-2013碳酸锂、单水氢氧化锂、氯化锂化学分析方法 第9部分 硫酸根量的测定 硫酸钡浊度法.GB/T 11064.9-GB/T 11064.9-2013 Methods for chemical analysis of lithium carbonate, lithium hydroxide monohydrate and lithium chloride - Part 9:determination of sulfate content Barium sulfate nephelometry method. 刘月菊,宋明明,邸卫利,等.电位滴定法测定钒电池电解液中硫酸根.冶金分析.2019,39(4):75-79.LIU Yue-ju,SONG Ming-ming,DI Wei-li,etal.Determination of sulfate in vanadium battery electrolyte by potentiometric titration.Metallurgical Analysis,2019, 39(4):75-79 JJG823-2014离子色谱仪检定规程.JJG823-2014Verification Regulation of Ion Chromatographs:.2013 Methods for chemical analysis of lithium carbonate, lithium hydroxide monohydrate and lithium chloride - Part 9:determination of sulfate content Barium sulfate nephelometry method. 刘月菊,宋明明,邸卫利,等.电位滴定法测定钒电池电解液中硫酸根.冶金分析.2019,39(4):75-79.LIU Yue-ju,SONG Ming-ming,DI Wei-li,etal.Determination of sulfate in vanadium battery electrolyte by potentiometric titration.Metallurgical Analysis,2019, 39(4):75-79 JJG823-2014离子色谱仪检定规程.JJG823-2014Verification Regulation of Ion Chromatographs:.
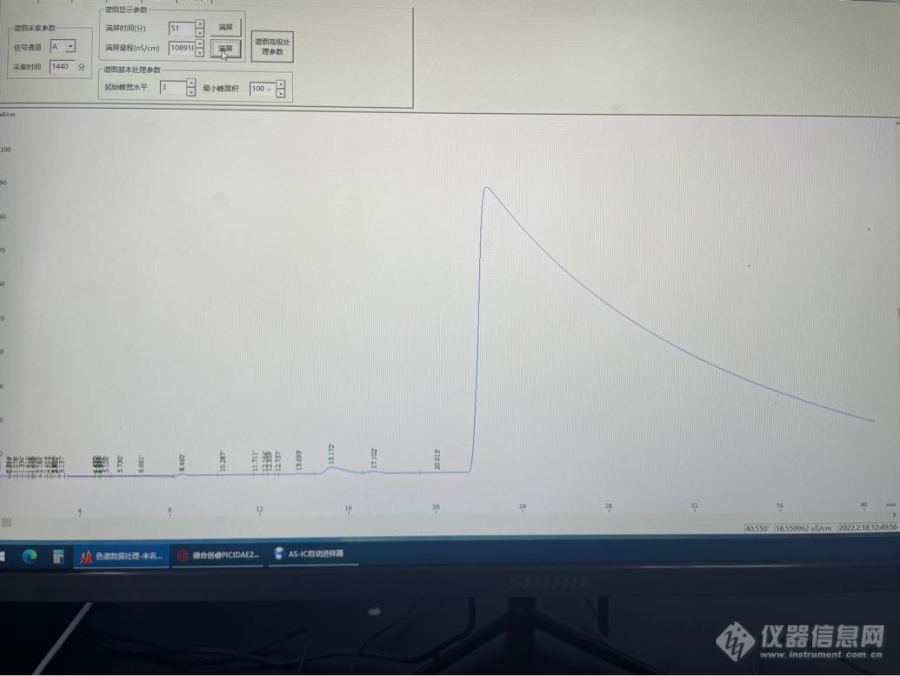
利用[url=https://insevent.instrument.com.cn/t/3p][color=#3333ff]离子色谱[/color][/url]法测定磷酸二氢锂固体中氯离子和硫酸根离子含量时,发现氯离子前面出现负峰,很容易影响氯离子的定量结果。在后面还会出现很大的一个峰,但不是在磷酸根出峰时间出的。样品的处理方式主要为称取一克固体样品溶解在100毫升水中,之后将样品溶液过氢柱后进样测定。求助一下,有做过类似的实验,或者出现过类似情况的吗?[img=,690,517]https://ng1.17img.cn/bbsfiles/images/2022/02/202202181331448878_8181_3920308_3.png[/img]
求助三氟甲磺酸酐气相测试方法我们用HP-5测试 不知道测试出来的是不是主峰,并且杂质较多,求助测试方法?是否可以做硅化测试?
请问专家:我想检测污水中的柠檬酸等有机酸根阴离子含量,但是该水中重金属含量也比较高,跟柠檬酸有络合作用,请问我应该采取什么方法比较好?请各位指点,谢谢!
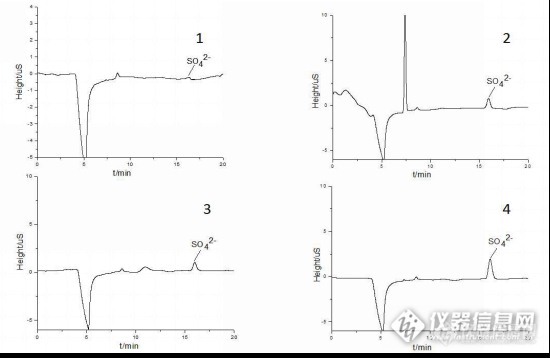
在线固相萃取-离子色谱法测定四种芳环磺酸盐中的硫酸根离子摘要:建立一种在线固相萃取离子色谱法测定四种芳环磺酸盐中硫酸根离子含量的新方法,将自装填的PGC-SPE柱应用于离子色谱系统对样品进行在线前处理,样品经过PGC-SPE柱处理后进入500μL定量环,通过阀切换-大体积进样模式使硫酸根进入阴离子检测系统。固相萃取流路以1.5mmol/L Na2CO3在0.8mL/min的流速对基体在线富集;分析柱采用SH-AC-3 (4.0×250mm) + SH-AG-3 (4.0×50mm),在6mmol/L Na2CO3 +4mmol/L NaHCO3条件下等度洗脱,柱温为35 ℃,流速为0.8mL/min,进样量20μL。结果表明:硫酸根离子在0.50-10.00 mg/L浓度范围内呈良好的线性关系,线性相关系数为0.9993,保留时间、峰高、峰面积的RSD均在0.28%~2.86%之间,方法检出限为0.0106mg/L,回收率在93.33%~105.59%之间;该方法具有良好的线性和重复性,整个在线分析过程在25min之内完成,进样量少,快速、高效。关键字:在线固相萃取,离子色谱,多孔石墨化碳,自装填技术,硫酸根离子1 前言多孔石墨化碳(PGC)作为一种新型色谱固定相,因其表面为纯碳结构、平整且无接枝的功能基团而具有耐强酸强碱、耐高温、性能稳定的特点,广泛应用于液相色谱和离子色谱中。PGC是二维结构、表面完全非极性且有可自由移动的π电子,因此与它化合物之间存在着疏水作用和电子间作用等多重作用力,从而在一定条件下对极性化合物、非极性化合物、异构体、聚合物等都具有保留性且对强极性化合物表现出强保留性。在样品前处理特别是复杂样品前处理过程中,通常利用固相萃取对样品进行净化与浓缩富集,除去干扰性的杂质,从而提高待测物分离度并保护色谱柱。Thermo公司首先推出以多孔石墨化碳填料为固定相的hypercarb 分析柱。在固相萃取技术应用中已有商品化的离线多孔石墨化碳固相萃取小柱(PGC-SPE柱),主要应用于液相色谱和离子色谱的样品的富集和基体消除。 萘磺酸盐和蒽醌磺酸盐属于多苯环磺酸盐,易溶于水的强极性离子型化合物。目前商品化的离子色谱固相萃取前处理柱,如:Onguard RP柱、H柱、Na柱以及聚合物树脂固相萃取柱等对非离子型有机化合物和常规离子基质有很好的前处理效果,但对芳环磺酸盐类的离子型强极性有机化合物保留性差,前处理效果不理想;在测定这类物质中残留的无机离子时,若不能很好地对该类物质进行基体消除,其进入分析系统后易保留在离子色谱柱上且较难洗脱,对分析柱造成损害。PGC因其特殊的表面疏水性质和带电性质使得它对该类强极性离子型化合物具有很强的保留性。本实验创新性的提出自装填可再生的多孔石墨化碳固相萃取柱并将其应用于在线离子色谱分析系统对四种芳环磺酸盐中的硫酸根离子进行测定,自装填和可再生重复使用的特点很大程度上降低了实验成本,在线基体消除较离线前处理在能保证理想的前处理效果和样品分析重复性的同时,能很大程度的减少样品污染并缩短了分析时间和样品量,更有利于复杂样品中阴离子的快速、准确的分离分析。2 实验部分1.1 仪器与试剂实验所用仪器为自组装离子色谱系统,主要部件包括:Ultimate3000 WPS-3000TSL自动进样器,ICS5000+紫外检测器,ICS5000+(SP/DP)双泵,ICS5000 TCC柱温箱(含一个六通阀和一个十通阀),ICS3000(SP)单泵,ED50A电化学检测器(DS3,带控温)Chromeleon6.8色谱工作站,(美国赛默飞世尔科技有限公司);WLK-6A阴离子抑制器(青岛仪趣仪器有限公司);AL-10电子天平(梅特勒-托利多有限公司);Milli-Q Advantage A10超纯水机(Millipore);BRANSON 2510超声清洗仪(BRANSON); 硫酸根离子标准储备液(100 mg/L,上海计量测试技术研究院);碳酸钠和碳酸氢钠(分析纯,99%,上海凌峰化学试剂有限公司);氢氧化钠 (w/w=50% ,默克公司);浓磷酸(分析纯,国药集团化学试剂有限公司);甲基磺酸(99.5%,上海阿拉丁有限公司);甲醇,乙腈(色谱纯,上海星可高纯溶剂有限公司)。 1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾(上海翰思化工有限公司提供)1.2 标准溶液的配制 分别取0.50,1.00,2.00,5.00,10.00 mL的SO42-标准储备液于100 mL容量瓶,超纯水定容后得到0.50,1.00,2.00,5.00,10.00 mg/L的SO42-标准液。 准确称取0.0242g、0.0213g、0.0248g、0.0257g的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠、蒽醌1,8二磺酸钾于50mL容量瓶,超纯水溶解定容,作为待测样品。1.3 色谱条件在线PGC-SPE柱为本实验室自装填前处理小柱(3.0×30mm),PGC填料粒径:30μm,阴离子分析柱: SH-AC-3 (4.0×250mm,9μm) + SH-AG-3 (4.0×50mm,12μm) (青岛盛翰色谱技术有限公司),柱温:35 ℃,流速:0.8mL/min;进样量20μL;定量环:500μL;淋洗液:6mmol/LNa2CO3 +4mmol/LNaHCO3; 分析时间25min;1.4 On-line SPE-IC系统的构建 On-line PGC-SPE-IC系统构建如图1 所示,实验建立了阀切换-大体积进样抑制电导离子色谱方法测定四种芳环磺酸盐中的硫酸根离子。系统中泵1用于在线固相萃取和进样,泵2用于阴离子分析,泵3用于SPE柱的在线清洗再生;利用一个十通阀和一个六通阀实现样品的在线固相萃取、大体积进样和在线前处理柱的再生,抑制电导检测器对硫酸根离子进行检测。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131609_609526_3137073_3.jpg 图1 在线SPE-IC系统2 结果与讨论2.1 PGC-SPE柱的装填和活化及柱容量 实验中所使用的SPE柱为本实验室自装填前处理小柱,采用湿法装填方法,以乙醇为分散剂,将填料以流动相的形式利用高压泵进行装填,装填过程先是填料在低流速低压条件下缓慢泵入柱体内,一段时间后再缓慢将流速调节为1mL/min装填1h以上,最后加大流速在高压条件下将填料压实;装填好的SPE柱压力约为120 psi。将装填好的PGC-SPE柱进行一下酸碱活化过程:酸冲洗(100mmol/LHCl, 1mL/min,30min)——水冲洗(1mL/min,20min)——碱冲洗(100mmol/LNaOH, 1mL/min,30min)) —— 水洗(1mL/min,20min)。酸碱冲洗的作用在于对小柱进行活化同时溶解填料中本身残留的一些酸碱可溶性杂质,避免其进入色谱系统损害色谱柱和抑制器。 利用紫外检测基线突越的方法测定PGC-SPE柱对该四种化合物的柱容量。分别配置500mL浓度为230.0、236.4、129.0、258.0mg/L的1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾溶液作为流动相,以0.1mL/min的流速流过SPE柱,记录基线开始采集和发生突越的时间间隔,根据公式计算(柱容量=物质浓度*流速*时间)得:1-萘磺酸钠、2-萘磺酸钠、蒽醌1,5-二磺酸钠和蒽醌1,8二磺酸钾的柱容量分别是0.8912mg,0.8307mg,0.2354mg和0.4373mg。2.2 色谱分析条件的选择2.2.1 硫酸根在SPE柱上洗脱条件(泵1淋洗液) 在中性条件下, 由于电荷的相互作用,PGC-SPE柱对硫酸根具一定的保留性,而酸性或者碱性条件下将其洗脱;实验中考虑到仪器系统兼容性和耐碱程度,选择低浓度碳酸钠作为洗脱液。由于使用500uL定量环大体积进样方式,流速为0.8mL/min,则SO42-的峰展宽必须控制在0.625min之内。根据实验结果可知(见图2),在0.5mM,1mM,1.5mM碳酸钠条件下,阴离子完全洗脱时间分别是1.10-2.20min,0.75-1.45min,0.65-1.20min;1.5mM浓度下的硫酸根离子在0.55min之内洗脱,因此Pump1选择1.5mM碳酸钠作为洗脱液。http://ng1.17img.cn/bbsfiles/images/2016/09/201609131601_609519_3137073_3.jpg 图
请问:如何用液相色谱(C18色谱柱、紫外检测器)测定水中的硝酸根离子含量?谢谢!







 我要推广仪器
我要推广仪器
 下载APP
下载APP