金属所张哲峰团队:金属材料拉伸与疲劳性能预测研究取得新进展
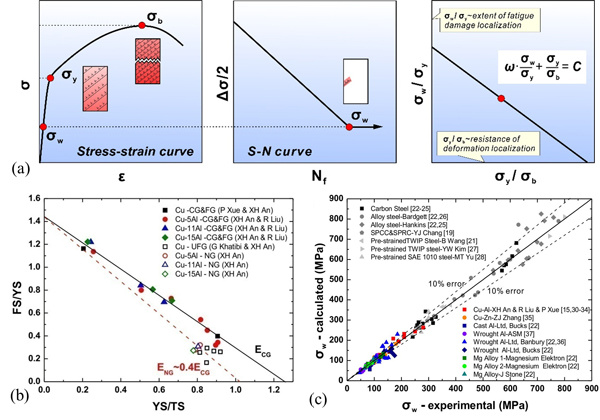
拉伸性能与疲劳性能是金属材料工程应用的关键指标,建立二者之间定量关系,实现金属材料不同力学性能之间关系的定量预测是金属结构材料领域重要研究目标之一。由于目前相关理论不够完善,基于微观变形与损伤机制的拉伸性能与疲劳性能定量预测模型并未建立起来。因此,虽有大量实验数据表明金属材料拉伸强度与塑性之间存在明确的倒置关系,拉伸强度与疲劳强度之间存在特定的关系,但至今仍缺乏定量模型来描述上述定量关系。因此,建立金属材料拉伸性能与疲劳性能定量预测具有重要科学意义。金属研究所张哲峰团队长期坚持材料疲劳与断裂基础理论研究,团队成员张振军项目研究员前期在缺陷与金属材料加工硬化关系方面进行了系统性研究,包括四类典型缺陷:1)零维缺陷:发现过饱和空位可提升合金的加工硬化能力;2)一维缺陷:在位错主导塑性形变的合金中实现了加工硬化能力回升;3)二维缺陷:在FeMnCAl系TWIP钢中实现随孪晶密度增加应变速率敏感性由负到正的转变;4)三维缺陷:在TWIP钢等强加工硬化材料中建立了微孔致颈缩判据。近来,在加工硬化微观机制研究基础上,张振军项目研究员提出了新的位错湮灭模型,并通过考虑初始组织状态与合金成分对加工硬化的影响,建立了单相金属材料普适性硬化模型-指数硬化(ESH:Exponential Strain-Hardening)模型,并据此首次推导出单相金属材料拉伸应力(σ)-应变(ε)定量关系:其中硬化指数n为位错湮灭距离(ye)的表达式反映合金成分的影响。η为初始缺陷对屈服强度(σy)非位错性贡献的比例,反映微观组织的影响;ΘⅡ为第二阶段硬化率,对同一金属合金体系为常数。该ESH模型得到了6种合金成分、100余种不同微观组织状态单相铜铝合金的实验验证,如图1所示。该ESH模型阐明了单相金属材料形变过程中一些重要规律:1)用一个参数(n)统一了五阶段加工硬化规律;2)揭示了极限强度、临界强度、真抗拉强度与成分及变形机制之间关系;3)首次推导出"屈服强度-抗拉强度-均匀延伸率"之间定量关系(公式(2-4),图2a-2c);4)定量揭示了拉伸强度-塑性同步提升的两个基本原则,即成分优化(提升位错滑移平面性)与组织优化(降低初始高能缺陷),在铜合金、镍基合金、TWIP钢、高氮钢、316L不锈钢等单相合金中均得到了系统性实验验证;5)实现了单相铜铝合金拉伸强度、塑性及拉伸应力-应变曲线的定量预测,如图2d-2f所示: 上述研究成果最近以2篇论文连载方式发表在Acta Mater 231 (2022) 117866和231 (2022) 117877上。基于该ESH模型,博士生曲展在张振军项目研究员的指导下,进一步揭示了三类变形铝合金(2xxx、6xxx、7xxx)拉伸强度和塑性随时效时间变化的共性转变规律与机制,建立了三类铝合金加工硬化指数与时效过程中析出相性质及几何特征之间的定量关系,提出了变形铝合金时效过程对加工硬化能力提升的析出相控制原理(J Mater Sci Technol 122 (2022) 54-67)。为了建立金属结构材料拉伸性能与疲劳性能之间定量关系,该团队成员刘睿博士在对铜铝单相合金拉伸性能与高周疲劳强度系统性研究的基础上,从疲劳损伤过程弹性变形与应变局部化两方面入手,通过引入合金成分、微观组织与宏观缺陷参数,建立了金属结构材料高周疲劳强度预测模型:其中参数C代表合金成分(或弹性模量)对疲劳强度的影响,强度σy和σb为微观组织对疲劳强度的影响,参数ω反映了宏观缺陷对疲劳强度的影响,如图3(a)所示;该高周疲劳强度预测模型得到了钢铁材料、铝合金、铜合金、钛合金、镁合金等20余种典型工程结构材料系统性疲劳实验验证,如图3(b)所示。该研究成果也以2篇论文连载方式发表在J Mater Sci Technol 70 (2021) 233-249和70 (2021) 250-267上。在疲劳裂纹扩展预测模型方面,最近李鹤飞博士在团队成员张鹏研究员的指导下,针对高强钢强度-韧性匹配关系,通过断裂力学理论分析,建立了以静态力学性能预测其疲劳裂纹扩展速率模型:其中σb为拉伸强度,KIC为断裂韧性,E为弹性模量,R为应力比,α为扩展速率常数。同时,为了指导关键构件材料强度-韧性优化提高疲劳裂纹扩展阻力,建立了高强度金属材料等效疲劳裂纹扩展速率模型(如图4(a)所示)。通过选择高强度金属材料强度-韧性之间匹配关系,可快速预测和降低其疲劳裂纹扩展寿命(如图4(b)所示),进而可以指导关键构件材料抗疲劳损伤容限设计。上述关于疲劳裂纹扩展速率预测模型在多种高强铝合金、钛合金及高强钢材料中得到了验证。该研究成果发表在J Mater Sci Technol 100 (2022) 46-50上。将上述金属材料拉伸性能和疲劳性能定量预测模型联合起来,可以实现通过测试金属结构材料少数组织状态的拉伸性能快速预测和优化其疲劳性能的功能,为金属结构材料疲劳性能预测与优化软件研发奠定理论基础,也为金属结构材料及工程构件抗疲劳设计与制造提供理论支撑。上述研究工作得到了国家自然科学基金重大项目(51790482)、重点项目(51331007、52130002)、面上项目(51771208、51871223)项目、中国科学院王宽诚率先人才计划"卢嘉锡国际合作团队"(GJTD-2020-09)、"青年促进会"项目(2018182、2021192)及辽宁省"兴辽计划"创新团队项目(XLYC1808027)的资助。相关成果列表及链接:1. Zhang ZJ*, Qu Z, Xu L, Liu R, Zhang P, Zhang ZF*, Langdon TG. A general physics-based hardening law for single phase metals. Acta Mater 231 (2022) 117877https://www.sciencedirect.com/science/article/pii/S1359645422002531#sec00202. Zhang ZJ*, Qu Z, Xu L, Liu R, Zhang P, Zhang ZF*, Langdon TG. Relationship between strength and uniform elongation of metals based on an exponential hardening law. Acta Mater 231 (2022) 117866.https://www.sciencedirect.com/science/article/pii/S135964542200252X3. Qu Z, Zhang ZJ*, Yan JX, Gong BS, Lu SL, Zhang ZF*, Langdon TG. Examining the effect of the aging state on strength and plasticity of wrought aluminum alloys. J Mater Sci Technol 122 (2022) 54-67.https://www.sciencedirect.com/science/article/pii/S1005030222001967?via%3Dihub4. Liu R, Zhang P*, Zhang ZJ, Wang B, Zhang ZF*. A practical model for efficient anti-fatigue design and selection of metallic materials: I. Model building and fatigue strength prediction. J Mater Sci Technol 70 (2021) 233-249.https://www.sciencedirect.com/science/article/pii/S1005030220307441?via%3Dihub5. Liu R, Zhang P*, Zhang ZJ, Wang B, Zhang ZF*. A practical model for efficient anti-fatigue design and selection of metallic materials: II. Parameter analysis and fatigue strength improvement. J Mater Sci Technol 70 (2021) 250-267.https://www.sciencedirect.com/science/article/pii/S100503022030743X?via%3Dihub6. Li HF, Zhang P*, Wang B, Zhang ZF*. Predictive fatigue crack growth law of high-strength steels. J Mater Sci Technol 100 (2022) 46-50.https://www.sciencedirect.com/science/article/abs/pii/S1005030221005053?via%3Dihub7. 张振军、张哲峰、张鹏、王强;一种金属材料拉伸性能的预测方法, 2021-7-6, ZL201711234799.0,发明。已授权8. 张哲峰、刘睿、张鹏、张振军、田艳中、王斌、庞建超;一种金属材料疲劳强度的预测方法,2021-8-10,ZL201711235841.0,发明。已授权9. 张鹏、李鹤飞、段启强、张哲峰;一种预测高强钢疲劳裂纹扩展性能的方法,2021-3-26,ZL201910030260.6,发明。已授权图1 ESH模型的建立与实验验证:(a-b) 模型推导过程;(c-d) 强度与塑性验证图2 ESH模型的应用:(a)建立"屈服强度-抗拉强度-均匀延伸率"之间定量关系;(b)实现拉伸性能及拉伸应力-应变曲线定量预测图3 高周疲劳强度预测模型的建立与验证:(a) 模型建立过程;(b,c) 系统性实验验证图4 (a)等疲劳裂纹扩展速率模型图 (b)工程材料强度-韧性与疲劳裂纹扩展速率关系

























 我要推广仪器
我要推广仪器
 下载APP
下载APP