我们实验室是公司里为公司的客户医院提供质控品的实验室,医院做完质控后,我们将数据收回后,需要对数据进行质控分析。那么,问题就来了,对于不同医院收回的数据,我们该如何选择分析方法呢?很令人头疼,愿有高手能一解我的困惑。
求助!求助!求助!——来自刚入门的实验室小白 关于水质实验分析,标线已经绘制好,但是不知道是否准确,需要做质控样验证分析。那么请问:1.质控样的浓度如何选取2.导入比色管中是否需要稀释,稀释多少倍3.例如氨氮,总磷,总氮,六价铬质控样的做法都是大致相似吗,能否以氨氮质控样的做法为例详细讲讲谢谢大佬了,新手小白,实验室老员工也不教,只让做,质控也没有像国标一样的指导书,快被这些搞疯了,来个善良帅气的大神或者美丽可爱的小仙女为我指点迷津吧,谢谢啦
1。不知你们有否分析过稀土元素,我分析了好几次了,每次带我们买来的质控样茶叶进行分析,总是发现Sc钪,Tb铽,Lu镥比质控样的数值偏低好多。而其它元素倒是与质控样的数值相吻合的。造成这种现象的原因是因为茶叶样品前处理的问题呢还是因为ICP-MS仪器的调试问题啊2。为什么稀土元素中的Eu,我选择151和153这两个同位素来分析,151的同位素较接近质控样的数值,而选择153Eu来分析的话,则偏高好多,为什么,按道理来说151和153分析出来的数值要差不多才对啊,难道是有153Eu对于茶叶的样本来说有基体干扰,因为如果是标准溶液的话,151和153这两个同位素的CPS值就差不多了
国家标准物质,元素成分分析质控样中有些元素的定值没有给出不确定度,只给了参考值(就是有括号的),这种情况能当做质控样吗?毕竟是很难做到与它一样的数值的,如GSB-27大葱、GSB-28大虾中锑Sb。另外,大家可以提供下关于锑的质控样?
分析标准质控样品,分析结果当如何评价?用样品的参考值±不确定度进行评价合理吗?
内部质控分析报告如何编制?
老师同学们有无这样的经历?对已知化合物定量分析,标准品和质控样分别出自不同的厂商,皆在有效期内,定量过程线性良好,RSD合规,但是多种化合物合成的质控样一直有一个化合物无法进入合理区间范围?多次尝试无果?这个时候考虑标准品有问题还是指控样有问题?还是分析方法有问题?从哪里入手?类似问题咋解决???
我在分析质控样时,结果总是偏高,在质控最大限值左右,是说明曲线不好吗?大家一般在做质控样时,结果怎样?
现求空气分析项目中氨的质控样值:氨质控样编号206905,其测定范围值是多少?
请教各位大佬,水质分析一般采用的质控手段有哪些?
好多分析实验最后都败在质控样品上
空气样品分析,其中例如铅尘的消解是可以加入标准物质一起消解,后来计算消解前后的结果来看消解过程中的回收率,从而达到质控要求。那么请问,其他用吸收液分析的样品,或者是重量法分析的样品,是如何做好质控的呢?请大家畅所欲言。
有哪位大侠知道 生物成分分析标准物质是 质控品吗?谢谢
在日常分析中,质控手段是咱分析员的“亲兄弟”,要让咱们分析出来的数据有说服力,就得靠咱的“亲兄弟”说话了,但是,如果我们没有选择恰当的质控手段,“亲兄弟”可能就说“假话“了,会让我们产生误判。[b]平行[/b]平行样是最基本的质控手段了,虽然基础,但作用依然不容小觑。分享一个我个人的经历:[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测地表水铁,水样清澈,过滤后就上机测试了,过滤了两个平行,测试的时候发现本该平行的却不平行,A浓度约为0.210mg/L,A平行浓度约为0.090mg/L,由于没有复杂的前处理步骤,于是直接拿样品瓶测试,结果令人大吃一惊,未检出!反复思量,认为是过滤时,未用纯水充分冲洗滤纸,且滤纸受到了污染(印象中那一叠滤纸没有在塑封膜里),自此以后,我个人养成了一个习惯,凡是过滤,先以纯水冲洗滤纸三遍。当时我若选择的是质控样品,而不做平行,质控样断然不会像样品一样过滤,质控样若在范围,就不会怀疑这个数据了,也就可能造成误判。[b]标准曲线校正点[/b]样品量较大的时候,对于仪器分析,有必要每10个样进行一次曲线校正。仪器进行大量样品分析时,容易发生漂移现象,也由于样品的复杂,可能对仪器进样系统产生一定的影响,如[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原吸[/color][/url]易堵塞毛细管,造成雾化效率降低,响应值略微偏低,不易察觉。[b]加标回收率[/b]在第六届原创作品中,《我对加标回收实验的一点理解》一文透彻讲解了加标回收率,感谢这位前辈的文章,帮助了我这样的后生理解加标回收率。加标回收实验是经典的质控手段,针对污染物繁多,基体复杂的水样,相比于标准样品,加标回收实验更有实际意义。水质标准样品,通常基体简单,在玻璃器皿洁净度、药品试剂纯度、稀释手法没有太大问题的情况下,准确测定标准样品并不是太难。将一定浓度的标液加入到样品中,同步前处理,如此才能反应前处理过程中的损失、污染、干扰等情况。诚如该前辈文中所说,“加标回收率100%不能说明测定结果没有问题”,但回收率的高低,可以给我们解决实验事故提供参考。[b]标准样品[/b]标准样品是重要的质量控制手段,尤其是对于土壤、食品、药品等领域的分析,而加标回收率对于这些领域,就显得乏力了。标液与土壤、食品、药品等难以充分混匀,待测组分部分存在于特殊晶体结构当中,标准溶液中的待测组分存在于晶体结构之外。加标就好比往一堆石头块(石头里藏有钻石)里放几块钻石,混在一起进行表面破碎,这几块钻石都已经存在了,无法确定石头里的钻石都破碎出来了;标准样品就好比一块已知里面有几颗钻石的石头,与未知的同时进行表面破碎,最后对比已知的那块石头,就可以知道是否完全了。[b]总结[/b]四种质控手段,平行应是必选的,标准通常也会要求10%平行样品。对于仪器分析,曲线校正点极有必要,一来可以帮助我们及时发现仪器的漂移情况,及时解决,二来原始记录中也可以体现在仪器没有发生严重漂移,提高样品分析数据的可信度;对于分光光度实验,曲线校正点则不是必要。加标回收实验和标准样品的选择,在样品与标准样品是同步进行前处理时,建议优先选择标准样品;当样品需要复杂前处理,而标准样品无需前处理时,建议优先选择加标回收实验。文中如有不恰当之处,敬请各位老师斧正。
[align=center]乳成分分析仪质控样的制备和使用[/align][align=center]作者:小王[/align][b]1 概述[/b] 乳成分分析仪一般采用红外的原理进行检测。有采用傅里叶变换的中红外技术的,比如福斯FOSS的FT系列。早期也采用滤光片技术。也有采用近红外技术的,比如布鲁克BRUKER的MPAD系列。无论哪种技术,都是为了更好的为乳品厂等客户服务,能更准确,更快速的检测样品中乳成分的含量。在检测过程中,一般会采用质控样(control sample,也有翻译成控制样)来对检测过程进行质量控制。质控样,说白了,就是有标准值的样品,我们每天在正式测定样品之前,先进行质控样的测定,质控样在容许的误差范围内,说明仪器稳定,就可以进行样品的测定。否则,要先对进行调整,调整完毕后再进行质控样的测定,符合要求后开始样品的测定。 那么问题就来了,如何对质控样进行定值?[b]2 质控样的定值[/b] 质控样的定值,严重依赖湿化学方法(俗称手工)的准确性。一般乳成分分析仪,对四个组分进行定值,脂肪(fat,F),蛋白质(protein,P),乳糖(lactose,L),全乳固体(Total Milk Solids,TS)(俗称干物质Dry matter,DM)。还有一个很重要的概念,非脂乳固体(Solids not fat,SNF)顾名思义,就是全乳固体减去脂肪。 所以,一般有如下三个公式: [b]牛奶=水+全乳固体 全乳固体=脂肪+蛋白质+乳糖+灰分 非脂乳固体=全乳固体-脂肪[/b] 这里我解释一下,灰分一般代表矿物质的含量,在牛奶中这个值比较固定,一般都在0.68-0.70%之前,一般取0.7%。[b]2.1脂肪的定值[/b] 脂肪一般采用碱水解法(原来叫罗兹-戈特里法),还有盖勃法,在这里我就不墨迹了。[b]2.2 蛋白质的定值[/b] 蛋白质一般采用凯氏定氮法。这里要注意凯氏定氮的质控,一般采用硫酸铵,不需要前处理,直接测定回收率。尿素,前处理后,测定回收率。[b]2.3 乳糖[/b] 一般采用滴定法,当然也有用液相色谱-蒸发光散射检测器或示差折光检测器检测的。[b]2.4 干物质[/b] 一般采用减量法进行检测。[b]3 质控样的使用[/b] 定值完毕以后,就可以使用了。有的客户容许误差比较低,一般为±0.03,有的要求±0.05。如果测定值在容许误差范围了,就可以开始样品的测定了,如果不在范围内,就对仪器进行调节。如果测定值偏离很大,那很遗憾,可能你的仪器故障了,这也是质控样的另一个作用,用于检查仪器的故障。 有的客户要求样品测定开始前,样品测定后都进行质控样的测定,质控样都合格了,说明两次质控样之间样品的测定都是准确的。 有的客户每个小时都进行质控样的测定,这要根据样品量的多少和实际情况来决定。
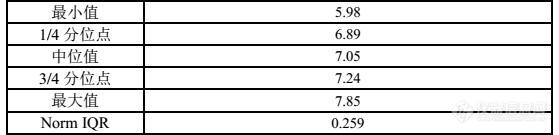
为了保证检测结果的准确性,每个实验室每年不但要做外部质量控制而且还要做内部质量控制,对于外部质量控制的样品自不必说,组织单位会做好一切准备工作,实验室只管收样测试即可。但是对于内部质量控制,实验室少不了要自己选择样品并对样品进行前处理,为了保证质量控制的有效进行,在这里有结果质控样选择的小窍门供大家参考:1.质控样选择方法A —“外源节流”,这种方法一般可以通过购买或其他有效方式直接使用有已知结果并且均匀的样品作为质控样,当然购买的话有很多组织能力验证的机构会有提供这种样品;但很多实验室为了控制成本可能不会花这些“冤枉钱”。那么我们实验室可以利用参加能力验证或实验室间比对的机会在参加能力验证够的情况下尽量对来样进行留样,将这种样品进行“节流”,自然分析报告中的结果我们也可以用到我们内部质量控制中的报告中。[img=,355,455]https://ng1.17img.cn/bbsfiles/images/2018/08/201808311428330809_3682_1954597_3.jpg!w355x455.jpg[/img]图1 外部质量控制中的留样可以作为质控样来使用[img=,556,142]https://ng1.17img.cn/bbsfiles/images/2018/08/201808311429502548_8658_1954597_3.jpg!w556x142.jpg[/img]图2 外部质量控制(能力验证或实验室间比对)中给定的中位值、Norm IQR值就是我们做质量控制时的质控数据依据。2. 质控样选择方法B — “一物多用”,这种方法是充分利用检测中的标准物质,用标准物质来作为质控样来检测,看检测结果是否在证书的范围内。这种方法也是实验室常用的一种方法。[img=,233,429]https://ng1.17img.cn/bbsfiles/images/2018/08/201808311457594747_6791_1954597_3.jpg!w233x429.jpg[/img]图3—标准物质可以作为质控样来使用[img=,276,41]https://ng1.17img.cn/bbsfiles/images/2018/08/201808311458231720_2977_1954597_3.jpg!w276x41.jpg[/img]图4—证书上的结果也是可以作为质控数据来使用的。3. 质控样选择方法C — “自给自足”,这种方法一般是针对没有上述选择的情况下,也是实验室普遍使用的方法,对于这种情况我们一般是在设备刚刚校准合格的两天内完成制备,因为这种情况下设备的准确性是最好的,质控样的选择应是实验室常规测试的同一批样品,其均匀性可以通过标准偏差来判定,这种情况下的质控样相对稳定和准确。[img=,433,549]https://ng1.17img.cn/bbsfiles/images/2018/08/201808311520359868_192_1954597_3.jpg!w433x549.jpg[/img]图5—设备校准的两天内是做质控样的最好时间。总结总结以上三种方法各有优劣:方法A: 这种“拿来主义”的方法好是好,但是一般达不到样品的数量,或者质控的重复次数有限。这种情况我们可以在样品接收时作为样品数量不够给组织单位单位多要点,但是这种情况很少,要多了人家组织单位就不给了。方法B:这种方法虽然好,一般来说实验室的样品也足够,并且使用的一般在有效期内,但是因为是标准物质价格要昂贵一些,并且对控制的项目相对有局限性。方法C:这种方法适用于任何项目的质控选择,但是相对于以上A、B两种方法准确率上会差一点。所以,不论是那种方法只有适合的才是最好的。对于质控样我们不但要选择好还要保存好。因为样品的保存对结果的影响也很大。
实验室内部质控的结果分析能引用《能力验证规则》中的方法吗?
“内部质量控制活动中不能提供质控项目的原始记录”不符合的原因分析?
购买中检科院奶粉质控样品QC-IP-701。检测结果一直都是钠低,钙高。每次检测称取0.8g,电热板处理,马弗炉500℃,5小时,冷却后取出,加1+1硝酸200ul,蒸干,马弗炉500℃,1小时,冷却取出,加1+1硝酸2.5ml,溶解,去离子水定容25ml,钠稀释50倍加氯化铯,钙稀释100倍加氧化镧,上机检测。钠曲线:0 0.5 1.0 1.5 2.0 2.5 ug/ml 钙曲线:0 1.0 2.0 3.0 4.0 5.0质控样参考范围 钠:158.0mg/100g 扩张不确定度U (k=2)17.8 钙:607.5mg/100g 扩展不确定度U (k=2)26.0每次检测钠的检测值基本上都低,130-140左右,偶尔高于140。钙则是高,每次检测值都能达到640-650.。百思不得其解,请大师们帮分析下,该从那些点着手,能够改变这一状况!!!
【摘要】本文综述了β-内酰胺类抗生素高分子聚合物测定的国内外研究进展,介绍了聚合物测定的原理和具体方法,分析了目前在聚合物测定中存在的问题,并针对问题提出了处理建议。 【关键词】高分子聚合物、质控现状、测定方法 1、概述 抗生素是临床用量最大和较易发生不良反应的药物。其中最常见的一类不良反应就是过敏反应。多年来的研究已证明,在β-内酰胺类抗生素所致的速发型过敏反应中,药物分子本身只是半抗原,药物中存在的高分子聚合物才是引发速发型过敏反应的真正过敏原,因此严格控制抗生素中高分子聚合物的含量有着重要的意义。 本文综述了高分子聚合物测定方法在国内外的研究进展,对实际测定过程中遇到的问题作简单的分析和探讨。 2、高分子聚合物的分类 抗生素中的高分子杂质系指药品中分子量大于药物本身的杂质总称,其分子量一般在1000~5000Da,个别可达10000Da左右。β-内酰胺类抗生素中的高分子杂质按其来源通常被分为两类:外源性杂质和内源性杂质。 外源性杂质包括蛋白、多肽、多糖等类杂质或抗生素和蛋白、多肽、多糖等的结合物,来源于发酵工艺,如青霉素中的青霉噻唑蛋白、青霉噻唑多肽等。 内源性杂质是指抗菌药物的自身聚合产物,聚合物既可来自生产过程,又可在储存中形成,甚至在用药时也可产生。对于现在的生产工艺,外源性的高分子杂质已经较少产生,对内源性聚合物的控制是当前抗生素高分子杂质质量控制的重点。 金少鸿等人的研究表明,高分子聚合物不仅能引发IgE介导的过敏反应,而且青霉素类的抗生素还能经胃肠道吸收而引发过敏反应。故对β-内酰胺类抗生素中的高分子聚合物进行严格的控制,可以保证临床用药的安全有效
大家好,请问大家在原子吸收中,质控方法有哪几种,每种质控方法是怎么进行的?根据我的经验和查找的资料,整理一下,我认为一般的质控方法有:(1) 空白实验进行空白实验有助于找出产生系统误差的潜在原因,比如试剂、实验室用水、仪器稳定性等条件。操作方法:取坩埚或者高型烧杯,除了不加入样品外,与样品处理步骤一样,加入相同量的酸和其他试剂,随样品一同分析,严格的来说一般每次要做两个空白实验值,而且两个空白值的相对偏差小于50%,否则要查找原因。(2) 平行样分析在样品处理时,称取两个(或两个以上)平行样进行分析,主要考察检测结果的平行性(精密度)(3) 标准曲线在原子吸收分析中,每条标准曲线不得少于5个点(除0点外),一般要求标准曲线相关系数达到0.9990以上。对于比较容易做好标线的,要求标准曲线相关系数达到0.9995以上(4) 加标回收试验加标回收试验是用加入标准溶液到试样中的方法,来检查或验证分析方法的准确度或可靠性的方法,可以用来表征方法或仪器分析测试结果的准确度操作方法:在待测样品中,一个加入已知浓度的标准溶液(加入的浓度一般为样品含量的0.5~2倍,而且加标后浓度不超出标线最高点)后进行测定,另一个(或两个)直接进行测定,然后用加入标液的样品的测得值减去样品的测得值再除以加入标准溶液的浓度,就得到加标回收率了。(5) 有证标准样品测试购买国家标准物质作为质控样品进行测试,结果的评价以方法的允许偏差或标准物质的标准值的不确定度(允许差)来判断,在方法的允许偏差或标准物质的标准值的不确定度(允许差)范围内,表示满意,否则表示不满意。(6) 留样再测试留样再测试适用于样品性质稳定,稳定性和均匀性好,耐保存,而且在保存期内待测成分不发生明显变化的样品。定期抽取一定数量的已检样品进行再检测,与原来的结果相比较,以允许差或结果复现性判定。(7) 实验室人员间比对实验室中熟悉同一种仪器和方法的不同人员,对同一个样品进行测试,以允许差或结果复现性判定。(8) 实验室间比对同一个样品分发到不同的实验室(或第三方检测机构)进行测试,结果以方法重复性或允许差进行判定。(9) 测量审核和能力验证参加国家认可的有资质,有能力的检测机构的测量审核和能力验证,由发放样品的检测机构或中国实验室认可委能力验证部判定。以上的几种方法有哪些理解不到位的,或者需要补充的,请大家批评指正和扩充,做好质控,让我们的检测结果更有说服力。
如题,像原子荧光光谱法的分析方法验证,其中的准确度,是用回收率做还是 质控样品来做呢?但可能找不到具有相同基质的质控样品,一般用回收率试验吧......
随着公众对药物安全性的日益关注,控制药物中杂质已成为控制药品质量的关键因素之一,也是困扰着广大药物分析工作者的难题之一。由于药物杂质的来源广泛,已知的杂质可以通过现有的分析手段进行定性定量,未知的杂质则成为分析的难题,因此对于药品的杂质控制首要解决的问题就是将所有杂质进行完全分离。为了让广大药物分析工作者能实现有效地药品杂质控制,全国医药技术市场协会于2012年4月10日-13日在上海市举办“2012药物研究分析中新技术、新方法应用及杂质控制研讨会”。 制药企业和新药研究机构的研发人员,各级药品检验所(院)和口岸药品检验所人员,药品生产企业研发技术与质量管理负责人,新药研发CRO实验室人员及高管,各高等院校、科研院所等相关专业人员100多人参加了此次会议。 在此次会议上,多位行业知名专家钟大放(中科院药物研究所),王洪允(协和医院临床药理中心),胡昌勤(中国食品药品检定研究院),周立春(北京市药检所),王玉(江苏省检品检验所),张尊建(中国药科大学分析测试中心)分别讲解了当前药物分析领域中各种新技术、新方法,探讨分析新技术在药品研发及药品质量控制中的应用,特别是用于生物标志物、活性成份、药物代谢等高通量、定性、定量的各种分析技术,以及新版药典对药物分析方法新要求与国外药典比较等内容。 作为全球色谱消耗品领先的制造商,迪马科技一直致力于为食品、药品检测行业提供完善的技术服务,除与参会专家进行技术交流外,迪马科技技术应用工程师还与广大与会者共同分享了《Dikma 高效液相色谱柱技术应用于药品杂质控制分析》技术报告。 对于药品中杂质控制分析,首先要借助色谱柱进行良好的分离,迪马科技在此次技术报告中重点讲解了在杂质控制中色谱柱的分离性能所起关键作用及迪马科技多款液相色谱柱:Ø Diamonsil(钻石)—通用型反相色谱柱,超高的分离性能特别适合分析复杂的样品及杂质;Ø Spursil(思博尔)—通用型极性改性反相色谱柱,耐受100%水相-100%有机相,特别适用于强碱性化合物和极性化合物的分析;Ø Endeavorsil(奋进)—1.8 μm UHPLC专用色谱柱,超高的柱效满足您UHPLC分离杂质的需求;Ø Leapsil(飞跃)—2.7μm兼容UHPLC/HPLC色谱柱,低柱压设计,高选择性可在HPLC上拥有UHPLC色谱柱的分离能力;[font=Wing
群友提问:跟大家交流下,平常大家分析样品的时候,质控措施都怎么做,我们现在主要是做空白,空白加标,标准样品还有样品平行。标准样品买不到的项目,大家质控都怎么做? 需要做两家不同厂家的标样么?因为有专家提出,做曲线的标样跟稀释带标的样是同一家不合理。有人回复(1):没有质控样品,可以把标样稀释后做;你还看可以做质量控制图有人回复(2):自己标定大家还有什么办法么?

[font=Arial][size=15px][color=#00d100]《医疗机构临床实验室管理办法》明确指出:[/color][/size][/font][font=Arial][size=15px][color=#00d100]医疗机构临床实验室应当对开展的临床检验项目进行室内质量控制,绘制质量控制图。[/color][/size][/font][font=Arial][size=15px][color=#00d100]出现质量失控现象时,应当及时查找原因,采取纠正措施,并详细记录。[/color][/size][/font][img=,690,582]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012110014638_7347_2646158_3.jpg!w690x582.jpg[/img][size=15px]这段话其实说明了两层意思:[/size][color=#ff6827][b]第一,临床实验室只要开展的检验项目就必须做室内质控。[/b][/color][color=#ff6827][b]第二,出现质量失控时,应当做三件事情:查找原因—采取纠正措施—详细记录。[/b][/color][size=15px]对于实验室工作人员来说,有一部分经验比较丰富,能够有清晰的思路去查找失控相应的原因。但是还是有一部分实验室工作人员对于发生失控时的原因查找感觉特别困难,因为引起失控的原因五花八门,不知道从哪里开始找起。[/size][size=15px]本文对失控现象可能引起的常见原因做一个简单的汇总,希望可以给实验室的工作人员一些参考。[/size][size=15px]其实失控的原因只包含2个方面:[/size][color=#ff6827][b][size=15px]检测系统和质控品[/size][/b][/color][size=15px]。检测系统又分为[/size][size=15px][color=#ff6827][b]人、机(仪器)、料(试剂、校准品)、法(SOP)、环境。[/b][/color][/size][size=15px]本文会按照顺序进行一一介绍。[b][color=#3a3a3a]人~~[/color][/b][img=,690,458]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012111535127_2008_2646158_3.jpg!w690x458.jpg[/img][size=15px]跟人员有关引起失控的常见原因有:[/size][b][size=14px]? 检验人员的变更[/size][size=14px][/size][size=14px]? 检验人员没有按照仪器说明书定期对仪器进行维护保养[/size][size=14px][/size][size=14px]? 检验人员没有按照试剂说明书混匀试剂[/size][size=14px][/size][size=14px]? 检验人员没有仔细阅读新试剂说明书导致操作错误[/size][size=14px][/size][size=14px]? 检验人员没有按照操作规程配制缓冲液、洗液等[/size][size=14px][/size][size=14px]? 检验人员把试剂放错位置[/size][size=14px][/size][size=14px]? 检验人员设定了错误的校准值[/size][size=14px][/size][size=14px]? 检验人员没有按照校准品说明书的校准时限进行校准[/size][size=14px][/size][size=14px]? 检验人员把校准品的校准时限设置过长[/size][size=14px][/size][size=14px]? 检验人员没有严格按照质控品操作SOP进行操作[/size][size=14px][/size][size=14px]? 检验人员没有掌握质控品操作SOP中关键的操作步骤[/size][size=14px][/size][size=14px]? 检验人员把质控品编号弄错[/size][size=14px][/size][size=14px]? 检验人员复融(溶)质控品没有放置到室温足够时间;[/size][size=14px]使用水浴箱以及体 温加速质控品的溶解[/size][size=14px]? 检验人员混匀质控品时使用高频振荡器,检测微量元素时颠倒混匀[/size][size=14px][/size][size=14px]? 检验人员加样量不准确(加样枪或者[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url]没有定期校准,没有垂直加样)[/size][size=14px][/size][size=14px]? 检验人员取质控品时不是吸取而是从瓶子里倒出来[/size][size=14px][/size][size=14px]? 检验人员把质控品取出后剩余部分倒回瓶里[/size][size=14px][/size][size=14px]? 检验人员对于不稳定的质控品取样方法不正确(没有采用注射器抽取引起挥发)[/size][size=14px][/size][size=14px]? 检验人员没有保持质控品瓶口的清洁[/size][size=14px][/size][size=14px]? 检验人员使用了错误的单位[/size][/b][size=15px][/size][b][size=14px][/size]仪器~~[/b][img=,690,379]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012114186666_2133_2646158_3.png!w690x379.jpg[/img][size=15px]跟仪器有关引起失控的常见原因有:[/size][b]? 近期仪器使用情况不正常,有小的故障发生? 近期没有进行过仪器维护保养? 仪器没有按照厂商规定的维护周期进行维护? 厂商规定的维护周期不适用于本实验室? 电压不稳定,没有使用UPS? 孵育箱或反应加热块的温度不恒定? 光电比色光源老化造成光强不足? 电极损坏? 离子检测稀释杯未清洗? 滤网需要清洁? 样品或试剂加样系统安装不完整? 马达安装不正确? 注射器漏液? 仪器管路漏气? 电源有断电现象? 仪器某部件的缺损,比如注射器活塞脱落? 加样针携带引起的交叉污染? 加样针部分堵塞? 仪器取质控品时加样量不够(质控品和样本管容器不一致)? LIS传输错误? 新仪器与旧仪器未做平行检测,直接使用旧仪器的质控控制限引起失控[/b][size=15px][/size][size=15px]总结:以上是对检测系统中的人和机(仪器)引起失控的常见原因的简单汇总。[/size][size=15px]下面继续汇总检测系统中料[/size][size=15px][color=#ff6827][b](试剂、校准品)、法(SOP)、环境和质控品[/b][/color][/size][size=15px]有关的引起失控的常见原因。[/size][b]试剂~~[/b][img=,690,455]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012115315968_5604_2646158_3.png!w690x455.jpg[/img][size=15px]跟试剂有关引起失控的常见原因有:[/size][b]? 试剂未混匀? 试剂超过有效期? 配套或辅助试剂超过有效期? 试剂开瓶时间长变质? 试剂开瓶时间长导致挥发,试剂浓缩? 两瓶同批号剩余试剂混合使用? 不同批号试剂混合使用? 自行配制的试剂配制错误? 试剂瓶间差大? 新旧批号试剂批间差大? 试剂储存温度不符合试剂说明书的要求? 试剂上机未用完,测试完成后未放回正确的储存温度保存? 试剂储存温度变化引起试剂变质? 试剂剩余量低于样本测试需要总量? 试剂瓶或试剂管道中有气泡? 试剂间的交叉污染? 未做试剂空白[/b][size=15px][/size][b][b]校准品[/b]~~[/b][img=,690,547]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012116198052_2948_2646158_3.png!w690x547.jpg[/img][size=15px]跟校准品有关引起失控的常见原因有:[/size][b]? 校准品未混匀? 校准品超过有效期? 校准品开瓶时间长变质? 校准品反复使用引起的污染? 校准品复溶未按说明书进行操作? 校准品复溶不完全? 校准品复溶后未及时使用(比如CO2)? 校准品复溶后反复冻融使用? 校准品与试剂不配套使用? 校准品位置放错? 校准品更换批号后校准值未重新设定? 校准品标示值与实际不符? 校准品储存温度不符合校准品说明书的要求? 校准品储存温度变化引起校准品变质(比如冰箱反复开关门或者冰箱自动化霜)? 某些项目未按要求避光保存(比如胆红素)操作方法(SOP)~~[/b][img=,690,524]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012117117262_5788_2646158_3.png!w690x524.jpg[/img][size=15px]跟SOP有关引起失控的常见原因有:[/size][b][size=14px]? SOP未规定操作内容(比如仪器维护保养周期、校准时限等)[/size][size=14px][/size][size=14px]? SOP记录内容有误(比如和仪器、试剂、校准品、质控品说明书不一致)[/size][size=14px][/size][size=14px]? SOP虽然描述了相关操作内容,但是不够具体,不能保证操作一致(比如质控品的混匀没有规定混匀时间或者混匀次数)[/size]环境~~[/b][img=,690,386]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012117598218_174_2646158_3.png!w690x386.jpg[/img][size=15px]跟环境有关引起失控的常见原因有:[/size][size=14px][b]? 实验室温度不符合要求[/b][/size][size=14px][b]? 实验室湿度不符合要求[/b][/size][size=14px][b]? 实验室用水不符合要求[/b][/size][size=14px][b]? 取水的容器不洁净或者加样的滴管或者加样枪头不洁净[/b][/size][size=14px][b]? 实验室的电压不稳定[/b][/size][size=14px][b]? 实验室间的仪器干扰[/b][/size][size=14px][b]? 实验室空气洁净度不够[/b][/size][size=14px][b]? 实验室操作台不洁净带来的交叉污染[/b][/size][size=14px][b]? 实验室被阳光照射引起的仪器异常[/b][/size][size=14px][b]? 实验室封闭性不好引起的污染(比如风的影响引起的洁净度差)[/b][/size][size=15px][/size][size=14px][b][/b][/size][b]质控品~~[/b][img=,690,872]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012118571826_6747_2646158_3.png!w690x872.jpg[/img][size=15px]跟质控品有关引起失控的常见原因有:[/size][b]? 质控品批号超过有效期? 质控品开瓶时间超过说明书规定的开瓶有效期? 质控品复溶后每次取用后未及时返回规定储存条件? 质控品储存温度不符合说明书的要求? 质控品储存温度变化引起质控品变质(比如冰箱反复开关门或者冰箱自动化霜)? 某些项目未按要求避光保存(比如胆红素)? 质控品未彻底溶解? 复溶后未及时使用? 复溶后反复冻融使用? 质控品稳定性差? 质控品瓶间差大[/b][size=15px][/size][img=,690,270]https://ng1.17img.cn/bbsfiles/images/2023/12/202312012119509400_5241_2646158_3.png!w690x270.jpg[/img][font=system-ui, -apple-system, BlinkMacSystemFont, &][size=12px]来源:[/size][/font][font=system-ui, -apple-system, BlinkMacSystemFont, &][size=12px]本文来源:bio-rad、质控学苑[/size][/font][b][color=#3a3a3a][/color][/b][/size]
接http://bbs.instrument.com.cn/shtml/20150703/5860997/后续实验室留样再测是在一段时间后,取形状较稳定的、上次结果的测定值在满意范围内的样品,由相同人员测试水平,在相同环境、设备、方法等条件下重复试验,是判断和监控实验室能力的检测情况的有效手段之一。通过内部质控措施的实施,来评价自身实验室相应的检测能力,帮助实验室发现存在问题,并及时采取措施,保证检测结果准确、可靠。一、 目的 根据ISO XXXX的要求及所规定的检测标准、方法和检测项目,通过比对及时发现问题和开展纠正措施,监控实验室内检验人员的检测能力和差异,积累和总结人员工作的经验,以做好实验室内部质控工作,不断提高和促进实验室的检测能力,进一步提高质量管理水平。二、 方法 1. 人员:参加本次质控试验有1名检测员。2. 样品:检测样品为上次测试留样ABS注塑样条,样品类型为1A型拉伸样条,样品5根,每样品给予一个编号,检测项目为拉伸强度。3. 检测方法:按照XXXXXXX执行。4. 质量监督:由质量监督员进行监督,做好监督记录。三、 判定标准http://ng1.17img.cn/bbsfiles/images/2015/07/201507061646_553722_2368716_3.bmp四.结果 序号拉伸强度(MPa)第一次测试拉伸强度(MPa)留样再测断裂强度(MPa)第一次测试断裂强度(MPa)留样再测146.647.333.632.1247.947.835.634.1347.946.935.933.2447.747.432.034.6546.947.431.733.9结果值47.447.433.833.6标准偏差0.6080.5361.960.968备注:根据测试方法,5次检测标准差<5%则可只需5次测试,反之,则需要再取5根样条进行测试。因以上四组数据的标准差(0.608、0.536、1.96、0.968)均<5%,则无需继续进行测试,取5次测试结果的平均值作为最终结果。对数据采用不同的方法进行分析如下:http://ng1.17img.cn/bbsfiles/images/2015/07/201507061652_553723_2368716_3.bmp http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif通过以上三种方式判定,本次留样再测试验数据是满意的。五.总结1、通过这次实验室内部质控工作可以看出,检测结果还是令人满意的,说明实验室人员能力能达到测试要求。2、在检测实验室中影响测试结果的因素很多,结果出现离群值的概率很大,所以在数据分析前,需要根据测试方法提供的方式或按照格拉布斯等规则剔除离群值。方法一、方法二均需要通过经验来获得一个结果参考值(如本例中5%)。方法三,此公式http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gif,适用于两次测试n值相同的情况下,适用于在不知道测量不确定度的情况下进行数据分析。--------------------文章里的公式发不上来,我干脆把文档给作为附件上传吧-----------------
实验室内部质控措施 人员比对 设备比对 留样再测 数据结果怎么分析
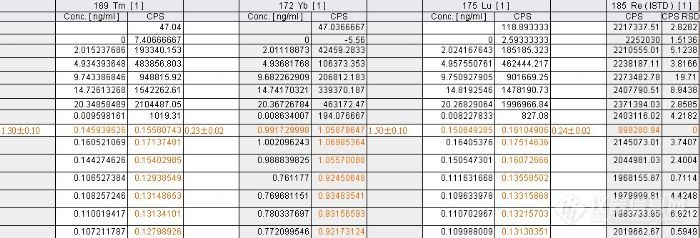
称取25mg的土壤质控样GSS07746,测三份,每份连续进两针,加2mL硝酸与2mL氢氟酸,压力溶样于190度的烘箱中2天,赶出HF,赶了二趟,再加2mL硝酸于130度下3h,取出,定容25mL。在no gas 模式下分析钡和稀土元素,内标液选用103Rh、185Re。结果有的元素还是会偏低些,为什么啊?http://ng1.17img.cn/bbsfiles/images/2012/11/201211121334_403400_2166779_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/11/201211121334_403399_2166779_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/11/201211121335_403402_2166779_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/11/201211121335_403404_2166779_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/11/201211121335_403405_2166779_3.jpg
在《职业卫生技术服务机构检测工作规范》中看到有个规定:(七)在样品测定前,应进行质控样品测定,测定结果满足质控要求后,方可进行样品测定。样品测定过程中,应根据仪器设备的稳定性,同一检测项目每分析10~30个样品应进行质控样品分析,检查分析条件的变动。质控样品测定结果应在质控标准值范围内,或在质控图控制线范围内。质控样品可直接外购或单独配制。如无质控样品,可采用加标回收率进行质量控制,加标回收率应保证在75%~105%。想请教一下大家在分析样品的时候是如何操作的?样品检测前,用质控样还是用样品加标回收做质控?或者是用中间校核点?谢谢!
实验室在实施内部质量控制计划时,用标准物质做质控样品检测时发现某项目质控结果不满意,结果偏差超出了再现性。对其原因进行分析,排查了人员、标准物质、设备后发现检测设备校准曲线过期。个人感觉将“未定期标定校准曲线”作为不符合工作去纠正并更简单方便。但是在这个不符合工作关闭后,需要另外对“质控结果不满意”去组织验证。而如果将“质控结果不满意”作为不符合的话,不符合的条款不知道写哪条合适;而且纠正和纠正措施会更复杂些,排查原因的过程算纠正措施的话形成记录比较麻烦。请教各位大佬,把质控不满意的原因作为不符合去纠正,可行不可行?







 我要推广仪器
我要推广仪器
 下载APP
下载APP