破千人!第三届抗体药物研发与质量分析网络会议成功举办
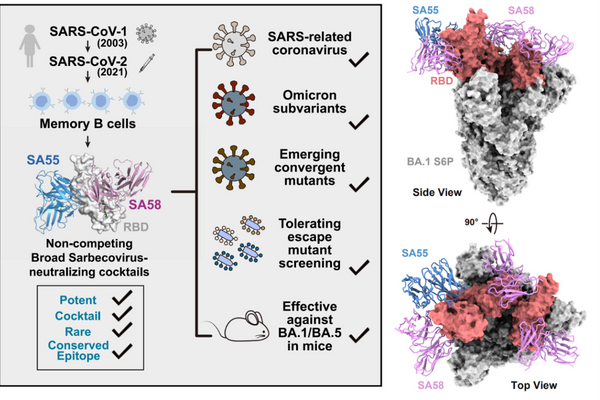
p style="text-indent: 2em "strong仪器信息网讯 /strong2020年9月10日-9月11日,由仪器信息网举办的第三届“抗体药物研发与质量分析”主题网络研讨会成功举办,会议为期2天,旨在为从事抗体药物研发生产的从业人员分享技术干货。本次会议共吸引近1300位来自全国各地制药企业、高校及科研院所及政府单位的用户报名参加。相较于前两届,本次抗体药物从专家阵容到会议规模均取得了极大进步!/pp style="text-indent: 2em "为方便未能参加会议直播的用户学习,仪器信息网特将本次会议内容视频剪辑整理,点击报告图片即可进入相应主题报告视频观看页面。/pp style="text-align: center"a href="https://www.instrument.com.cn/webinar/video_113521.html" target="_blank"img style="width: 550px height: 413px " src="https://img1.17img.cn/17img/images/202009/uepic/66bbe262-6dea-4927-b546-2ccab8049f79.jpg" title="李玉玲.jpg" width="550" height="413" border="0" vspace="0" alt="李玉玲.jpg"//a/pp style="text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""抗体药物研发的科学探索与质量控制/span》/pp style="text-align: center "strong报告嘉宾/strong:李玉玲 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""浙江健新原力制药有限公司 CEO/span/pp style="text-align: center "a href="https://www.instrument.com.cn/webinar/video_113522.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/43b95a1a-3a99-4ef6-98f7-2f5ef4b6dfaf.jpg" title="杨晓明.jpg" width="550" height="413" border="0" vspace="0" alt="杨晓明.jpg" style="text-align: center white-space: normal width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""创新生物药候选分子的评价和筛选不可缺失的重要环节和指标 - 商业化产品的适应性 Developability/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:杨晓明 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""杭州奕安济世生物药业有限公司 CDMO业务总经理/span/pp style="text-align: center "a href="https://www.instrument.com.cn/webinar/video_113519.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/c8f6372f-d9eb-42b9-afa9-2d74c8ae5c8d.jpg" title="张娟.jpg" width="550" height="413" border="0" vspace="0" alt="张娟.jpg" style="text-align: center white-space: normal width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""CD-24,不仅仅是“别吃我”/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:张娟 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""中国药科大学 教授/span/pp style="text-align: center "a href="https://www.instrument.com.cn/webinar/video_113524.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/74b2fdbb-6afd-46fb-9aed-af17a799143a.jpg" title="孙乐.jpg" width="550" height="413" border="0" vspace="0" alt="孙乐.jpg" style="text-align: center white-space: normal width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""新冠病毒ADE与抗体药去ADCC/ADCP技术平台/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:孙乐 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""京天成生物 总经理/span/pp style="text-align: center"a href="https://www.instrument.com.cn/webinar/video_113533.html" target="_blank"img style="max-width: 100% max-height: 100% width: 550px height: 413px " src="https://img1.17img.cn/17img/images/202009/uepic/31b9c45e-5393-46cd-9319-58c7c37f1e57.jpg" title="易继祖.jpg" alt="易继祖.jpg" width="550" height="413" border="0" vspace="0"//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""蛋白药物的糖基化修饰及表征研究/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:易继祖 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""友芝友生物 副总裁/span/span/pp style="text-align: center"a href="https://www.instrument.com.cn/webinar/video_113526.html" target="_blank"img style="width: 550px height: 413px " src="https://img1.17img.cn/17img/images/202009/uepic/ef4b7991-9d8b-45d4-b66b-60f66c723566.jpg" title="李国荣.jpg" width="550" height="413" border="0" vspace="0" alt="李国荣.jpg"//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""高效液相色谱法评估单抗药物的关键质量属性(CQA)/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:李国荣 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""苏州美极医疗科技有限公司 总裁/span/pp style="text-align: center"a href="https://www.instrument.com.cn/webinar/video_113525.html" target="_blank"img style="width: 550px height: 413px " src="https://img1.17img.cn/17img/images/202009/uepic/f07c5d7f-2f24-4fb0-a735-9d450680e437.jpg" title="施立明.jpg" width="550" height="413" border="0" vspace="0" alt="施立明.jpg"//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""QbD approach to analytical method development and optimization/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:施立明 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""杭州奕安济世生物药业有限公司 分析科学、质量控制、工艺与产品开发运营副总裁/span/pp style="text-align: center "a href="https://www.instrument.com.cn/webinar/video_113517.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/631a258a-db66-4543-8fe4-a13938f0132d.jpg" title="屈锋.jpg" width="550" height="413" border="0" vspace="0" alt="屈锋.jpg" style="text-align: center white-space: normal width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""毛细管电泳与抗体药物分析/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:屈锋 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""北京理工大学 教授/span/pp style="text-align: center "a href="https://www.instrument.com.cn/webinar/video_113518.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/dff88282-eb15-4bea-aada-52ea3f55e066.jpg" title="赛默飞刘晓达.jpg" width="550" height="413" border="0" vspace="0" alt="赛默飞刘晓达.jpg" style="text-align: center white-space: normal width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""色谱分析新技术加速推进单克隆抗体药物研发》/span/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:刘晓达 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""赛默飞世尔科技(中国)有限公司 应用顾问/span/pp style="white-space: normal text-align: center "a href="https://www.instrument.com.cn/webinar/video_113531.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/b76e07ee-e7dd-48b6-8b10-dcb9231ad679.jpg" title="张睿.jpg" width="550" height="413" border="0" vspace="0" alt="张睿.jpg" style="width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strongspan style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:="":《分子互作技术在抗体药研发与质控中的应用》/span/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:张span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""睿 Cytiva(思拓凡) 分子互作产品专家/span/pp style="white-space: normal text-align: center "a href="https://www.instrument.com.cn/webinar/video_113520.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/c9834186-c746-4ae4-8d66-de3e4a0ccb0e.jpg" title="luninex常青.jpg" width="550" height="413" border="0" vspace="0" alt="luninex常青.jpg" style="width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""不同类型流式细胞仪在抗体药物研发与质控中的创新应用不同类型流式细胞仪在抗体药物研发与质控中的创新应用》/span/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:常青 span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:="" Luminex公司 中国区流式业务市场部经理/span/pp style="white-space: normal text-align: center "a href="https://www.instrument.com.cn/webinar/video_113528.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/f02060a1-b393-4e89-afee-e2255980f1f7.jpg" title="岛津.jpg" width="550" height="395" border="0" vspace="0" alt="岛津.jpg" style="width: 550px height: 395px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""岛津质谱在抗体药物质量控制中的应用》/span/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""邵锴 岛津企业管理(中国)有限公司 应用工程师/span/pp style="text-align: center"a href="https://www.instrument.com.cn/webinar/video_113530.html" target="_blank"img style="width: 550px height: 413px " src="https://img1.17img.cn/17img/images/202009/uepic/0eeb515f-6c69-4522-b6d4-b49a4d3dfd5e.jpg" title="吴姗.jpg" width="550" height="413" border="0" vspace="0" alt="吴姗.jpg"//a/pp style="white-space: normal text-align: center "strong报告主题/strong:span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""《Qsep毛细管电泳仪和INB-D200生物标志物分析仪在抗体药物研发质控中的应用》/span/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""杭州厚泽生物/span/pp style="text-align: center"a href="https://www.instrument.com.cn/webinar/video_113527.html" target="_blank"img style="width: 550px height: 413px " src="https://img1.17img.cn/17img/images/202009/uepic/82ca5526-f2a9-47e3-802e-c66e20b94baf.jpg" title="姚金龙.jpg" width="550" height="413" border="0" vspace="0" alt="姚金龙.jpg"//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""span style="color: rgb(51, 51, 51) font-family: " hiragino="" sans="" microsoft="" helvetica="" background-color:=""抗体药物生产中细胞生长状态和抗体制剂的质控方法/span/span》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:姚金龙 贝克曼库尔特 生命科学细胞活力分析应用专家/pp style="text-align: center"a href="https://www.instrument.com.cn/webinar/video_113532.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/d7cc3b58-95f0-4951-ab73-91b5ea571fa7.jpg" title="锐欧森.jpg" width="550" height="413" border="0" vspace="0" alt="锐欧森.jpg" style="text-align: center white-space: normal width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《微量真实粘度技术在生物医药中的应用》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:刘兵 锐欧森 锐欧森中国业务总经理/pp style="white-space: normal text-align: center "a href="https://www.instrument.com.cn/webinar/video_113529.html" target="_blank"img src="https://img1.17img.cn/17img/images/202009/uepic/eb0d85c7-b794-4861-8235-df6c4b48715f.jpg" title="默克.jpg" width="550" height="413" border="0" vspace="0" alt="默克.jpg" style="width: 550px height: 413px "//a/pp style="white-space: normal text-align: center "strong报告主题/strong:《默克Supelco液相色谱柱技术在单克隆抗体药物分析方面的应用》/pp style="white-space: normal text-align: center "strong报告嘉宾/strong:田海玉 默克生命科学 产品经理/pp style="text-indent: 2em "因部分报告嘉宾的内容涉及本单位研发数据,不便回放,敬请理解。同时,报告老师也表示欢迎各位网友就相关问题通过其他方式联系沟通。如有问题可反馈至仪器信息网抗体药物技术交流群,或邮箱lizk@instrument.com.cn。/pp style="text-indent: 2em "strong回放视频集锦:/stronga href="https://www.instrument.com.cn/webinar/video/collection/10645" target="_blank"stronghttps://www.instrument.com.cn/webinar/video/collection/10645/strong/a/pp style="text-indent: 2em "span style="color: rgb(192, 0, 0) "strong精彩会议预告/strong/spanbr/ 错过抗体药物会议的朋友们,9月24日-25日,仪器信息网将举办“2020药典-药物分析检测技术”主题网络 研讨会,会议邀请了二十余位来自政府药品检验单位、高校及科研院所和制药企业的科研专家及技术专家将为大家详细介绍药物研发及生产过程中分析检测技术,精彩内容,不要错过!/pp style="text-align: center "a href="https://www.instrument.com.cn/webinar/meetings/pharmaanalysis" target="_blank"img style="max-width: 100% max-height: 100% width: 382px height: 182px " src="https://img1.17img.cn/17img/images/202009/uepic/b8647dad-1757-4e59-ad68-39fc1f410b2c.jpg" title="64030020200826_wps图片.jpg" alt="64030020200826_wps图片.jpg" width="382" height="182" border="0" vspace="0"//a/p























 我要推广仪器
我要推广仪器
 下载APP
下载APP