重磅!《新型冠状病毒抗原快速检测专家共识(2022)》发布!
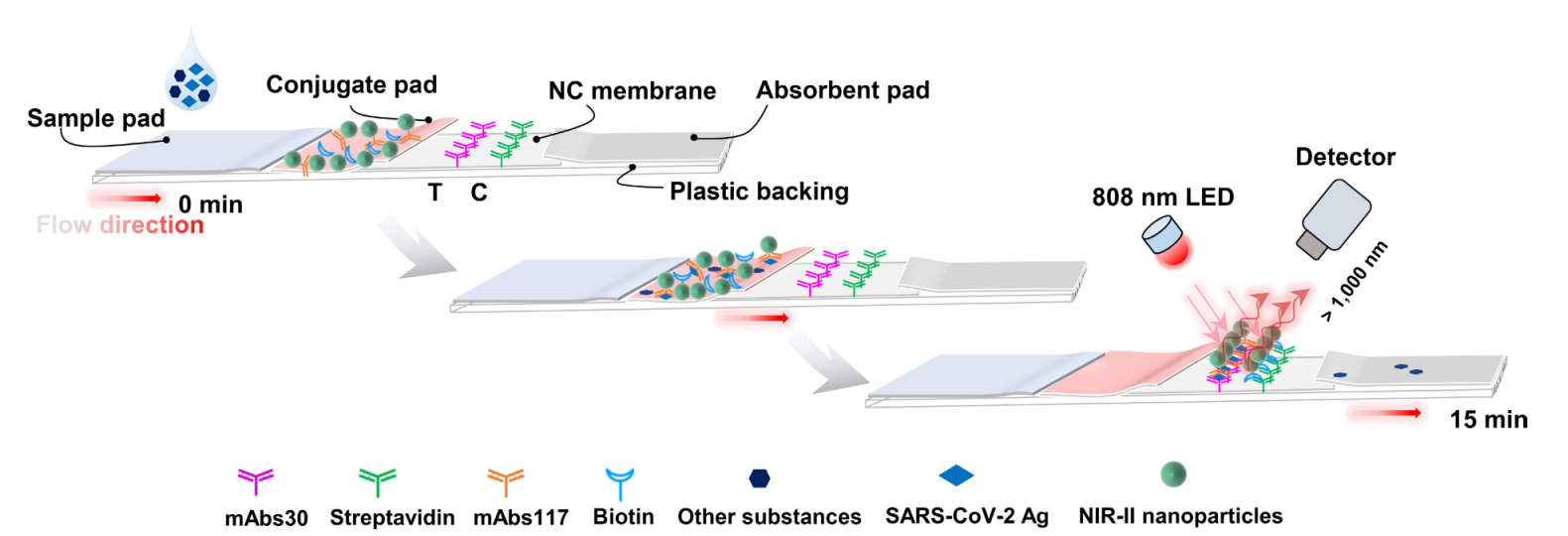
中国医院协会临床微生物实验室专业委员会通信作者:徐英春1,2,胡继红3作者单位:北京协和医院1检验科2疑难重症及罕见病国家重点实验室 3国家老年医学中心 中国医学科学院老年医学研究院 北京医院 国家卫生健康委临床检验中心项目基金:中国医学科学院医学与健康科技创新工程(2021-I2M-1-038)文章来源:协和医学杂志, 2022,13(3):402-411.独家访谈:徐英春教授新型冠状病毒肺炎(COVID-19)是由新型冠状病毒(SARS-CoV-2)感染引起的一类呼吸道传染性疾病,自2019年12月疫情暴发以来,截至目前仍在全球广泛流行,疫情防控工作任重道远。SARS-CoV-2实验室检测可为疫情防控方案的制订提供重要参考,其中核酸检测阳性是SARS-CoV-2感染者的确诊依据[1]。为进一步优化SARS-CoV-2检测策略,根据疫情防控需要,国务院应对新型冠状病毒肺炎疫情联防联控机制综合组于2022年3月10日印发了《新冠病毒抗原检测应用方案(试行)》[2],决定推进“抗原筛查、核酸诊断”的监测模式,在核酸检测基础上增加抗原检测作为补充,以提高SARS-CoV-2感染者“早发现”的能力[3]。全球范围内,SARS-CoV-2抗原快速检测已获得广泛认可,截至2022年4月7日,我国国家药品监督管理局(NMPA)已批准23款抗原快速检测试剂[4],通过世界卫生组织(WHO)紧急使用清单(EUL)[5]、美国食品药品监督管理局(FDA)紧急使用授权(EUA)[6]及欧盟(CE)认证[7]的抗原检测试剂盒分别有5款、48款和203款。基于目前实验室检测SARS-CoV-2积累的经验和常见问题,并结合国内外最新研究进展和应用实践,中国医院协会临床微生物实验室专业委员会组织相关领域专家共同制定了《新型冠状病毒抗原快速检测专家共识(2022)》。本共识明确了目前SARS-CoV-2抗原快速检测方法的性能特点,可为不同场景下的快速抗原检测应用、结果解读及处置提供技术支撑,促进疫情防控中抗原快速检测的专业化、规范化和标准化。共识核心意见(1) SARS-CoV-2抗原检测主要靶标为N蛋白,以保证较高的检测敏感性、特异性和对病毒变异株较高的包容性(共识度:100%)。(2) 抗原快速检测主要采用免疫层析技术(共识度:100%)。(3) 抗原检测特异性较高,敏感性受患者疾病严重程度、采样时机、样本类型、样本处理技术、样本病毒载量、病毒变异情况和检测试剂盒差异等因素的影响(共识度:100%)。(4) 抗原检测方法主要推荐用于发病早期、病毒载量较高的感染者(共识度:100%)。(5) 抗原检测主要适用标本类型为鼻腔拭子、鼻咽拭子和口咽拭子等(共识度:100%)。(6) 抗原、核酸和抗体检测结果解读及处置建议,根据被测者临床症状、疫苗接种史及接触史不同而采取不同措施(共识度:98.63%)。(7) 对于疑似病例,抗原检测结果无论阳性或阴性,均应进行核酸检测确认(共识度:98.63%)。(8) 抗原检测推荐在高风险、高流行区域人群中应用,一般人群不建议进行抗原检测(共识度:94.52%)。(9) 抗原检测主要适用场景:基层医疗卫生机构就诊的有症状人员、隔离观察人员、有自我检测需求的社区居民等(共识度:98.63%)。(10) 抗原检测其他适用场景:高风险工作人员监测,封闭/半封闭场所人员监测,疫情流行期间发热门诊、急诊聚集患者快速分流管理等(共识度:98.63%)。(11) 抗原检测应选择经NMPA审批的试剂盒,严格按试剂说明书操作(共识度:100%)。(12) 医疗机构开展抗原检测应进行试剂性能评价和质量控制(共识度:100%)。(13) 建议对抗原自测流程及结果进行有效管理,以保障检测的准确性和对疫情的及时监控(共识度:100%)。1 共识形成方法本共识由中国医院协会临床微生物实验室专业委员会发起,共识专家组由专委会委员及其推荐的相关领域专家共同组成,并通过共识形成会议法达成共识意见[8]。2022年2月专家组召开线上会议就SARS-CoV-2抗原快速检测主题拟订关键性问题和提纲,并进行任务分工,组建共识撰写组、编审组、外审组和秘书组,成员来自医学检验、临床医学、感染控制、公共卫生及体外诊断产品研发等领域的相关专家。共识撰写组以“新型冠状病毒抗原”“新型冠状病毒肺炎”“severe acute respiratory syndrome corona-virus 2 antigen”“SARS-CoV-2 antigen”“coronavirus disease 2019”和“COVID-19”为关键词,检索2019年12月至2022年3月PubMed、Embase、Cochrane Library、MedRxiv、中国知网、万方数据知识服务平台和维普数据库中的中、英文文献,以及国家卫生健康委员会、NMPA、WHO、FDA、欧洲疾病预防与控制中心(ECDC)及国内外学术组织现行的感染控制、生物安全相关标准、指南和共识等,经去重处理及阅读摘要和/或全文,最终纳入符合本共识主题的文献45篇。2022年3月至4月,撰写组经多次会议讨论及反复修订,统一意见后形成共识草案,然后由编审组专家对草案进行函审并提出书面意见,共收到函审意见407条(含重复意见),全部意见经撰写组逐条讨论确认,采纳其中的284条,草案修订后再次提交外审组专家审核。共形成13条共识核心意见,由撰写组、编审组和外审组专家进行“背对背”无记名投票,投票等级分为:a)完全同意;b)基本同意;c)不确定;d)不太同意;e)完全不同意。如a、b、c、d中任一项票数超过50%,或a+b、c+d票数超过70%,即视为达成共识;其余情况均视为未达成共识,相关意见进入下一轮投票。以a+b或d+e票数占回收有效票数的百分比,作为共识意见的专家共识度[9]。限于目前对新冠病毒抗原快速检测的认知程度及参与共识编写人员的专业背景,本共识的制订可能存在局限性。2 新型冠状病毒抗原检测方法及主要性能2.1 技术原理抗原检测是指应用特异性抗体直接对样本中的病原体抗原进行检测,检测结果可作为发病早期是否感染的直接证据。SARS-CoV-2的主要结构蛋白包括核衣壳蛋白(N)、刺突蛋白(S)、胞膜蛋白(E)和囊膜蛋白(M),部分研究表明SARS-CoV-2存在血凝素酯酶蛋白(HE),但目前仍存在争议[10-11]。SARS-CoV-2抗原检测的主要靶标是N蛋白,极少数针对S蛋白[12]。N蛋白是SARS-CoV-2的核心成分,其与病毒基因组RNA结合后将RNA包装为核糖核蛋白(RNP)复合体。由于N蛋白合成数量多,具有多个特异性抗原决定簇且稳定性高,因此N蛋白是理想的抗原检测靶标,具有较高的检测敏感性和特异性[13-14]。S蛋白由S1、S2两个功能亚基组成,其中S1亚基关键结构域S1-受体结合域(RBD)和/或S1-N末端结构域(NTD)可与宿主细胞的血管紧张素转换酶2(ACE2)受体结合,将病毒和宿主细胞的细胞膜融合,介导病毒进入宿主细胞[15-16]。SARS-CoV-2变异株突变位点多位于S蛋白,采用抗N蛋白联合抗S蛋白双抗体检测,虽可提高检测的敏感性,但如果病毒突变速度较快、类型较多,针对变异株S蛋白的抗原检测敏感性即会下降。因此,选择对SARS-CoV-2变异株具有更高包容性的检测靶标,对于保证检测的高敏感性尤为重要。此外,由于SARS冠状病毒(SARS-CoV)与SARS-CoV-2的N蛋白序列相似度高达91%[17],二者存在抗原与抗体交叉反应。有研究对7款SARS-CoV-2抗原检测试剂性能评价后发现,所有试剂均对SARS-CoV有交叉反应,但对其他4种人类流行冠状病毒(HcoV-229E、HCoV-NL63、HCoV-OC43和HCoV-HKU1)和中东呼吸综合征冠状病毒(MERS-CoV)基本无交叉反应[18]。由于目前并非SARS-CoV的流行期,SARS-CoV-2抗原试剂仍具有较高的特异性。2.2 技术分类与特点SARS-CoV-2抗原检测主要采用基于免疫层析技术的快速检测方法,具有检测快速、操作便捷等特点。非专业人员亦可操作,通过目视或使用便携设备即可判读结果,可快速、便捷地对有症状疑似人员、隔离观察人员和有自我检测需求的人员进行筛查,以便及时采取相应措施,助力疫情防控。根据检测试剂所使用的标记物,可分为胶体金免疫层析法、乳胶免疫层析法和荧光免疫层析法(表1)。此外,基于新型传感器和生物传感器技术的快速检测方法可实现数十秒至数分钟内完成SARS-CoV-2抗原检测,并保证检测的高敏感性[19-20],但目前尚未进入商品化与临床验证阶段。表1常用新型冠状病毒抗原快速检测方法及技术特点2.3 技术性能SARS-CoV-2抗原检测试剂具有较高的特异性,但敏感性在不同临床评价研究中存在较大差异,其影响因素包括患者疾病严重程度以及采样时机、样本类型、样本处理技术、样本病毒载量、不同时期变异株流行情况、检测试剂盒差异等[21-23]。抗原检测敏感性与感染者病毒载量呈正相关,对于传染性强的高病毒载量样本,检测的敏感性更高[23]。系统综述和Meta分析显示,抗原检测对SARS-CoV-2核酸扩增循环数(Ct)<20、<25、≥25和≥30样本的总体灵敏度分别为96.5%(95% CI:92.6%~98.4%)、95.8%(95% CI:92.3%~97.8%)、50.7%(95% CI:35.6%~65.8%)和20.9%(95% CI:12.5%~32.8%)[24]。因此,SARS-CoV-2抗原检测主要推荐用于发病早期、病毒载量高的感染者[14]。此外,可通过提高检测频次的方式提高阳性检出率,有研究显示检测频率为每3天一次或更高时,抗原检测与核酸检测对感染早期患者的总体灵敏度相当(均>98%);对处于病毒培养阳性阶段的感染者,若每天进行抗原检测,其灵敏度约为90%[25]。2.4 不同类型样本间的检测性能差异SARS-CoV-2抗原检测的样本类型主要包括鼻腔拭子、鼻咽拭子和口咽拭子等,针对下呼吸道样本、唾液、口腔液、血清等样本的抗原检测研究正在开展中[14]。不同类型样本的SARS-CoV-2抗原检出率存在差异,其中鼻腔拭子和鼻咽拭子的检测敏感性最高[24,26]。近期一项荟萃分析显示,常见样本的SARS-CoV-2抗原检测敏感性由高至低分别为鼻腔拭子(83%)、鼻咽拭子(71%)、口咽拭子(69%)和唾液(68%)[26]。血清中的SARS-CoV-2抗原一般在感染急性期(抗体产生前)出现,相较呼吸道样本,血清样本可降低样本采集因素对于检测结果的影响,具有一定诊断价值[27-28]。目前,在国内获批上市的SARS-CoV-2抗原检测试剂适用样本类型为鼻腔拭子、鼻咽拭子和口咽拭子,建议依据产品说明书推荐的标本类型进行检测。不推荐使用棉拭子,鼻腔拭子采样时应注意从双侧鼻腔采集。2.5 变异株抗原检测性能SARS-CoV-2变异株抗原检测的性能主要受病毒蛋白变异、检测试剂靶点及受检者人群中变异株流行情况等影响。SARS-CoV-2变异株突变位点多发生于S蛋白编码基因,N蛋白具有更高的稳定性,且N蛋白突变位点多集中在N-末端,而抗原检测试剂靶点多位于N蛋白C-末端,对检出率的影响较小。目前,对于阿尔法(Alpha)、贝塔(Beta)、德尔塔(Delta)等国际流行变异株,未发现抗原检测试剂对其检测性能产生显著影响[29-30]。奥密克戎(Omicron)变异株于2021年11月首次在南非发现,该变异株在S蛋白处发生了30多个突变,其中15个突变位点位于RBD,变异后的病毒传染性显著提升[31-32]。作为目前的主要流行株,尚未出现Omicron变异株 N蛋白编码基因存在明确影响抗原检测性能的缺失(如Del31-33)和突变(如P13L)。虽然少量研究提示,抗原检测试剂对Omicron变异株的检测敏感性有所下降,但多数研究显示多种抗原快速检测试剂对Omicron和Delta变异株的检测性能相当[33-35],灵敏度分别为22.2%~88.9%和52.9%~91.2%[34]。由于不同抗原检测试剂间存在性能差异,实验室应对所采用试剂进行充分评估,并密切关注SARS-CoV-2毒株变异趋势及对抗原检测可能产生的影响。2.6 假阴性与假阳性影响因素假阴性影响因素[36]:(1)样本病毒载量较低;(2)检测时间处于窗口期;(3)标本采集、运输、保存或裂解不当;(4)检测试剂敏感性低;(5)检测过程中操作不当;(6)未严格按试剂说明书规定的时间判读结果;(7)钩状效应(抗原抗体比例不合适)。假阳性影响因素[37-38]:(1)检测试剂的特异性受限,发生了交叉反应;(2)样本或试剂污染;(3)试剂阳性设定的判定值过低;(4)检测过程中操作不当;(5)在试剂说明书规定时间之后超时判读结果。2.7 抗原、核酸和抗体检测结果解读及处置建议采用抗原、核酸和抗体检测进行SARS-CoV-2感染实验室诊断时,其结果解读及后续处置建议详见表2。表2疫情流行期间新型冠状病毒抗原、核酸、抗体检测结果解读及处置建议[1-2]由于抗原检测的方法学局限性,其检测敏感性低于核酸检测,因此抗原检测不能用于疫情“清零”策略的筛查目的。核酸检测阳性是SARS-CoV-2感染的确诊依据[1],如被测者核酸阳性,无论抗原与抗体检测结果是阳性还是阴性,均需按照SARS-CoV-2感染者或COVID-19确诊患者的标准采取相应措施[2]。3 抗原检测应用策略SARS-CoV-2抗原检测(包括自我检测)能有效提高检测覆盖范围,将关口前移,是对公共卫生防控的有益补充,已成为多个国家或地区疫情防控的重要手段[1,29,37]。依据《新冠病毒抗原检测应用方案(试行)》[2]和《关于印发新型冠状病毒肺炎诊疗方案(试行第九版)》[1],SARS-CoV-2抗原检测主要适用场景、应用及处置策略见图1。图1新型冠状病毒抗原快速检测主要应用场景与处置建议[1-2]需特别注意,虽然SARS-CoV-2抗原检测具有较高的特异性,但在疫情低流行区域,广泛的人群抗原检测可能出现一定比例的假阳性,因此抗原检测推荐在高风险、高流行区域人群中应用,一般人群不建议进行抗原检测。对于已出现呼吸道、发热等临床症状的人员,仍建议至医疗机构就诊、核酸检测;明确密接、次密接等流行病学史人员,应尽快主动按当地疫情防控管理要求进行上报。3.1 基层医疗卫生机构检测受限于实验室场地、人员配备、检测设备和物流运输等条件,乡村诊所、乡镇卫生院、社区卫生服务中心等基层医疗机构常无法在短时间内进行SARS-CoV-2核酸检测,并快速获得检测结果,而抗原快速检测试剂可常温储运,且操作便捷、检测快速,为可疑人群的快速筛查提供了便利工具。虽然抗原检测的敏感性不如核酸,但可有效提高对疑似感染者的排查效率,对于疫情的有效防控具有重要意义[39-40]。抗原检测适用人群:至基层医疗卫生机构就诊,伴呼吸道、发热等症状且出现症状的时间不足5 d的人员。具体技术人员、环境与设施要求、人员配置与防护、样本采集流程与方法、样本管理规范、抗原检测流程、结果报告与处置等参见《新冠病毒抗原检测应用方案(试行)》[2]及其附件1《基层医疗卫生机构新冠病毒抗原检测基本要求及流程》[41]。目前,SARS-CoV-2抗原检测试剂在我国尚处于应急研发和特定情形人员的应用阶段,为满足疫情防控需求,NMPA紧急批准了部分SARS-CoV-2抗原检测试剂的使用。医疗机构在开展抗原检测时,需加强质量管理,应选择经NMPA审批且相关研究证实、性能符合临床检测要求的产品。在临床应用前,原则上应按定性免疫学试剂要求进行性能评价[42]。目前各级临床检验中心暂未开展室间质评活动,国内尚无SARS-CoV-2抗原商品化第三方质控品,必要时可使用中国食品药品检定研究院研发的“新型冠状病毒抗原检测试剂国家参考品”作为第三方质控品。出于生物安全考虑,不建议生物安全防护BSL-2级及以下实验室使用患者的阳性样本作为质控品。对于不具备试剂性能评价条件的基层医疗机构,可在检测中通过试剂内置质控(C线)评价试剂的有效性,质控合格方可报告,失控结果应更换试剂进行复检[14]。3.2 隔离观察人员自测抗原检测适用人群:隔离观察人员,包括居家隔离观察、密接与次密接、入境隔离观察、封控区和管控区人员。对于隔离观察期人员,在相关管理部门组织协调下,按现行疫情防控方案要求开展核酸检测,并在隔离前5 d每天进行一次抗原自测[2]。具体应用条件、注意事项、自测操作流程(包括自测前准备、样本采集、检测方法、废弃物处理)、结果处置等要求参见《新冠病毒抗原检测应用方案(试行)》[2]及其附件2《新冠病毒抗原自测基本要求及流程》[43]。3.3 社区居民自测有自我检测需求的社区居民,可自行购买经NMPA审批上市、可用于自测的抗原快速检测试剂盒进行自我抗原检测。建议居民自测时选择通风良好的房间或角落,远离人群。具体自测试剂选择原则、注意事项、自测流程与结果处置要求参见《新冠病毒抗原检测应用方案(试行)》[2]及其附件2《新冠病毒抗原自测基本要求及流程》[43]。虽然研究显示非专业人员进行抗原自测的敏感性与专业人员操作的结果相当[44],但仍需进行有效管理,以保障检测结果的准确性和公共卫生体系对于疫情的及时监控。信息化平台可能成为一种潜在的辅助手段,通过智能手机等移动设备与监管部门信息化连接,同步上传检测结果、采样/检测过程视频等,可实现自测结果的可溯源管理,并为公众提供便捷、专业的咨询渠道。3.4 其他应用场景WHO和ECDC等卫生组织和一些研究团队在旅行者、高风险工作人员、学校师生监测、隔离策略调整及卫生经济学评价等方面,也开展了SARS-CoV-2抗原检测的应用研究和实践[14,29,45]。结合我国疫情防控经验,SARS-CoV-2抗原快速检测在以下场景也具有潜在应用价值。3.4.1 高风险工作人员监测冷链物流、隔离区等高风险工作人员更易发生SARS-CoV-2感染,其中的无症状感染者由于病情隐匿、病毒传染性强,更易成为超级传播者。有研究建模显示,连续检测模式能有效弥补单次抗原检测敏感性不足的缺陷,在感染早期进行连续抗原检测,有助于早期发现无症状感染者从而阻断病毒传播链[40],推荐用于高风险工作人员的初筛。目前,抗原检测已成为中国国际航行船舶船员到岸前远端防控的检测手段之一[46]。3.4.2 封闭/半封闭场所人员监测幼儿园、学校、养老机构等封闭/半封闭场所[14]一旦发生疫情,传播速度快,对场所内人群(如儿童、青少年、老年人等)健康产生较大威胁。建议在相应场所内常备SARS-CoV-2抗原快速检测试剂,对出现发热、有呼吸道症状的人员立即进行抗原检测,一方面可及时发现潜在感染者,以迅速分流、隔离、转诊并进行核酸确认,另一方面有助于对密切接触者进行快速排查。此外,在疫情流行阶段,抗原快速检测可用于封闭/半封闭场所工作人员定期监测,但需注意抗原检测不适用于环境监测。3.4.3 发热门诊、急诊患者快速分流管理对于疫情流行地区,发热门诊和急诊常面临大量有症状疑似患者,急需进行快速鉴别诊断与分流管理。尤其对于不具备快速开展SARS-CoV-2核酸检测条件的基层医疗机构,抗原快速检测可作为核酸检测的有益补充,以便在更短的时间内快速筛查并管控阳性病例,从而减少病毒在人群密集的医疗场所进一步传播的风险。但需注意,抗原检测阴性并不能作为排除SARS-CoV-2感染的证据,对于有临床症状和流行病学史的疑似病例,仍需进行核酸检测确认。目前,该策略已在我国部分地区开展试点应用[47]。4 小结虽然COVID-19疫情在全球范围内蔓延已超过两年时间,累计感染者超过5亿,但人类对该病毒的了解仍存在较大局限性,尤其变异株层出不穷,给感染者的临床诊断、治疗、感染控制和疫情防控带来了巨大挑战,急需开展更多基础和临床研究,探索更高效、更准确、更便捷的诊断方法,以提升临床和实验室诊断能力。随着对SARS-CoV-2认识的进一步深入和疫情形势的动态变化,我国疫情防控策略和诊疗规范亦在不断调整,且愈加科学、完善。实验室检测是疫情防控的重要方面,检验人员应积极与临床沟通,结合患者流行病学史、临床表现和影像学改变,综合分析不同方法学检测结果,为疫情防控、病例诊断和治疗提供技术支撑。其中抗原检测作为核酸检测的有力补充,有助于提高对SARS-CoV-2感染者“早发现”的能力,可用于疑似病例快速筛查、隔离观察人员监测和社区居民自测,尤其适用于医疗条件有限的基层地区或医疗机构,可快速发现感染者,对降低疫情传播风险具有重要意义。但SARS-CoV-2抗原检测在我国尚处于起步阶段,应用策略仍需不断优化,其临床意义以及在现阶段和未来疫情防控中的作用,仍需进一步验证。参考文献[1]国家卫生健康委员会办公厅, 国家中医药管理局办公室. 关于印发新型冠状病毒肺炎诊疗方案(试行第九版)的通知(国卫办医函〔2022〕71号)[EB/OL]. (2022-03-14)[2022-04-10]. http://www.gov.cn/zhengce/zheng ceku/2022-03/15/content_5679257.htm.[2]国务院应对新型冠状病毒肺炎疫情联防联控机制综合组. 关于印发新冠病毒抗原检测应用方案(试行)的通知(联防联控机制综发〔2022〕21号)[EB/OL]. (2022-03-11) [2022-04-10]. http://www.nhc.gov.cn/yzygj/s7659/202203/d4d7fb72088447f7a4f9cd10966a67eb.shtml.[3]医政医管局. 新冠病毒抗原检测应用方案(试行)政策解读[EB/OL]. (2022-03-11) [2022-04-10].http://www.nhc.gov.cn/yzygj/s7659/202203/4573dfb9cca244509 c29b964ba287889.shtml.[4]国家药品监督管理局. 医疗器械数据查询[EB/OL]. (2022-04-07) [2022-04-10]. https://www.nmpa.gov.cn/datasearch/ search-result.html.[5]World Health Organization. WHO Emergency Use Listing for in vitro diagnostics (IVDs) detecting SARS-CoV-2[EB/OL]. (2022-02-23) [2022-04-10]. https://extranet.who.int/pqweb/sites/default/files/documents/220223_EUL_SARS-CoV-2_product_list.pdf.[6]U.S. Food & Drug Administration. In Vitro Diagnostics EUAs-Antigen Diagnostic Tests for SARS-CoV-2[EB/OL]. (2022-04-07) [2022-04-10]. https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas-anti-gen-diagnostic-tests-sars-cov-2.[7]European Commission Directorate-General for Health and Food Safety. EU health preparedness: A common list of COVID-19 rapid antigen tests A common standardised set of data to be included in COVID-19 test result certificates and A common list of COVID-19 laboratory based antigenic assays[EB/OL]. (2022-03-30) [2022-04-10]. https://ec.europa.eu/health/system/files/2022-03/covid-19_rat_common-list_en_1.pdf.[8]范曼如, 申泉, 王丹琦, 等. 临床实践指南制订方法:形成推荐意见的共识方法学[J]. 中国循证心血管医学杂志, 2019,11:647-653.[9]北京协和医院罕见病多学科协作组, 中国罕见病联盟. 多准则决策分析应用于罕见病药品临床综合评价的专家共识(2022)[J]. 协和医学杂志, 2022,13:126-145.[10]Kandimalla R, John A, Abburi C, et al. Current Status of Multiple Drug Molecules, and Vaccines: An Update in SARS-CoV-2 Therapeutics[J]. Mol Neurobiol, 2020,57:4106-4116.[11]Zandi M, Soltani S. Hemagglutinin-esterase cannot be considered as a candidate for designing drug against COVID-19[J]. Mol Divers, 2021,25:1999-2000.[12]Scheiblauer H, Filomena A, Nitsche A, et al. Comparative sensitivity evaluation for 122 CE-marked rapid diagnostic tests for SARS-CoV-2 antigen, Germany, September 2020 to April 2021[J]. Euro Surveill, 2021, 26: 2100441.[13]Peng Y, Du N, Lei Y, et al. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design[J]. EMBO J, 2020, 39: e105938.[14]世界卫生组织. 抗原检测用于诊断SARS-CoV-2感染:临时指导文件[EB/OL]. (2021-10-06) [2022-04-10].https://apps.who.int/iris/bitstream/handle/10665/345948/ WHO-2019-nCoV-Antigen-Detection-2021.1-chi.pdf.[15]刘巨钊, 杨玉萍, 徐建波, 等. 新冠病毒S蛋白RBD突变侵染性增强潜在分子作用机制[J/OL]. 生物学杂志, 2022:1-8. [2022-04-09]. http://kns.cnki.net/kcms/detail/34.1081.q.20220321.1514.002.html.[16]徐本锦, 范蕾, 杜淼, 等. 新冠病毒核衣壳蛋白结构与功能的生物信息学分析及原核表达[J/OL]. 中国免疫学杂志, 2021:1-25.http://kns.cnki.net/kcms/detail/22.1126.R.20210924.0101.002.html.[17]Bates TA, Weinstein JB, Farley S, et al. Cross-reactivity of SARS-CoV structural protein antibodies against SARS-CoV-2[J]. Cell Rep, 2021, 34:108737.[18]Corman VM, Haage VC, Bleicker T, et al. Comparison of seven commercial SARS-CoV-2 rapid point-of-care antigen tests: a single-centre laboratory evaluation study[J]. Lancet Microbe, 2021,2:e311-e319.[19]Seo G, Lee G, Kim MJ, et al. Rapid Detection of COVID-19 Causative Virus (SARS-CoV-2) in Human Nasopharyn-geal Swab Specimens Using Field-Effect Transistor-Based Biosensor[J]. ACS Nano, 2020,14:5135-5142.[20]Mahari S, Roberts A, Shahdeo D, et al. eCovSens-Ultrasensitive Novel In-House Built Printed Circuit Board Based Electrochemical Device for Rapid Detection of nCovid-19 antigen, a spike protein domain 1 of SARS-CoV-2[J]. bioRxiv,2020.doi:10.1101/2020.04.24.059204.[21]Dinnes J, Deeks JJ, Berhane S, et al. Rapid, point-of-care antigen and molecular-based tests for diagnosis of SARS-CoV-2 infection[J]. Cochrane Database Syst Rev, 2021,3: CD013705.[22]Bruzzone B, De Pace V, Caligiuri P, et al. Comparative diagnostic performance of rapid antigen detection tests for COVID-19 in a hospital setting[J]. Int J Infect Dis, 2021,107:215-218.[23]Chaimayo C, Kaewnaphan B, Tanlieng N, et al. Rapid SARS-CoV-2 antigen detection assay in comparison with real-time RT-PCR assay for laboratory diagnosis of COVID-19 in Thailand[J]. Virol J, 2020,17:177.[24]Brümmer LE, Katzenschlager S, Gaeddert M, et al. Accur-acy of novel antigen rapid diagnostics for SARS-CoV-2: A living systematic review and meta-analysis[J]. PLoS Med, 2021,18:e1003735.[25]Smith RL, Gibson LL, Martinez PP, et al. Longitudinal Assessment of Diagnostic Test Performance Over the Course of Acute SARS-CoV-2 Infection[J]. J Infect Dis, 2021,224:976-982.[26]Khalid MF, Selvam K, Jeffry A, et al. Performance of Rapid Antigen Tests for COVID-19 Diagnosis: A Systematic Review and Meta-Analysis[J]. Diagnostics (Basel), 2022,12:110.[27]Deng Q, Ye G, Pan Y, et al. High Performance of SARS-Cov-2N Protein Antigen Chemiluminescence Immunoassay as Frontline Testing for Acute Phase COVID-19 Diagnosis: A Retrospective Cohort Study[J]. Front Med (Lausanne), 2021,8:676560.[28]Yokoyama R, Kurano M, Nakano Y, et al. Association of the Serum Levels of the Nucleocapsid Antigen of SARS-CoV-2 With the Diagnosis, Disease Severity, and Antibody Titers in Patients With COVID-19: A Retrospective Cross-Sectional Study[J]. Front Microbiol, 2021,12:791489.[29]European Centre for Disease Prevention and Control. Options for the use of rapid antigen tests for COVID-19 in the EU/EEA-first update[EB/OL]. (2021-10-26)[2022-04-09].https://www.ecdc.europa.eu/en/publications-data/options-use-rapid-antigen-tests-covid-19-eueea-first-update.[30]Jungnick S, Hobmaier B, Mautner L, et al. Detection of the new SARS-CoV-2 variants of concern B.1.1.7 and B.1.351 in five SARS-CoV-2 rapid antigen tests (RATs), Germany, March 2021[J]. Euro Surveill, 2021,26:2100413.[31]Pulliam J, van Schalkwyk C, Govender N, et al. Increased risk of SARS-CoV-2 reinfection associated with emergence of Omicron in South Africa[J]. Science, 2022,376: eabn4947.[32]Saxena SK, Kumar S, Ansari S, et al. Characterization of the novel SARS-CoV-2 Omicron (B.1.1.529) variant of concern and its global perspective[J]. J Med Virol, 2022,94:1738-1744.[33]Soni A, Herbert C, Filippaios A, et al. Comparison of Rapid Antigen Tests' Performance between Delta (B.1.61.7 AY.X) and Omicron (B.1.1.529 BA1) Variants of SARS-CoV-2: Secondary Analysis from a Serial Home Self-Testing Study[J]. medRxiv, 2022. doi:10.1101/2022.02.27.22271090.[34]Bekliz M, Perez-Rodriguez F, Puhach O, et al. Sensitivity of SARS-CoV-2 antigen-detecting rapid tests for Omicron variant[J]. medRxiv, 2021. doi:https://www.medrxiv. org/content/10.1101/2021.12.18.21268018.[35]Deerain J, Druce J, Tran T, et al. Assessment of the Analytical Sensitivity of 10 Lateral Flow Devices against the SARS-CoV-2 Omicron Variant[J]. J Clin Microbiol, 2022, 60: e247921.[36]Bullard J, Dust K, Funk D, et al. Predicting Infectious Severe Acute Respiratory Syndrome Coronavirus 2 From Diagnostic Samples[J]. Clin Infect Dis, 2020,71:2663-2666.[37]U.S. FOOD & DRUG ADMINISTRATION. Potential for False Positive Results with Antigen Tests for Rapid Detection of SARS-CoV-2-Letter to Clinical Laboratory Staff and Health Care Providers[EB/OL]. (2020-11-03)[2022-04-10].https://www.fda.gov/medical-devices/letters-health-care-providers/potential-false-positive-results-antigen-tests-rapid-detection-sars-cov-2-letter-clinical-laboratory.[38]Mouliou DS, Gourgoulianis KI. False-positive and false-negative COVID-19 cases: respiratory prevention and management strategies, vaccination, and further perspectives[J]. Expert Rev Respir Med, 2021,15(8):993-1002.[39]Larremore DB, Wilder B, Lester E, et al. Test sensitivity is secondary to frequency and turnaround time for COVID-19 screening[J]. Sci Adv, 2021,7: eabd5393.[40]Mina MJ, Parker R, Larremore DB. Rethinking Covid-19 Test Sensitivity-A Strategy for Containment[J]. N Engl J Med, 2020, 38: e120.[41]国务院应对新型冠状病毒肺炎疫情联防联控机制综合组. 基层医疗卫生机构新冠病毒抗原检测基本要求及流程[EB/OL]. (2022-03-10)[2022-04-10]. http://www. nhc.gov.cn/yzygj/s7659/202203/d4d7fb72088447f7a4f9cd 10966a67eb/files/b62d9eeeca8242b3a22e78a25ef 9fd92.pdf.[42]中国合格评定国家认可委员会. 2019临床免疫学定性检验程序性能验证指南:CNAS-GL038[S]. 2019.[43]国务院应对新型冠状病毒肺炎疫情联防联控机制综合组. 新冠病毒抗原自测基本要求及流程[EB/OL]. (2022-03-10) [2022-04-10]. http://www.nhc.gov.cn/yzygj/s7659/202203/d4d7fb72088447f7a4f9cd10966a67eb/files/8db5dd22ccb84b6296bc4257ba7db9c0.pdf.[44]Stohr J, Zwart VF, Goderski G, et al. Self-testing for the detection of SARS-CoV-2 infection with rapid antigen tests for people with suspected COVID-19 in the community[J]. Clin Microbiol Infect, 2021,S1198-743X(21)00434-1. doi: 10.1016/j.cmi.2021.07.039.[45]Maya S, Kahn JG. Cost-effectiveness of antigen testing for ending COVID-19 isolation[J]. medRxiv, 2022,2022.03.21.22272687. doi: 10.1101/2022.03.21.22272687.[46]交通运输部, 外交部, 海关总署. 关于做好国际航行船舶船员新冠肺炎疫情远端防控的公告(交通运输部公告2022年第14号)[EB/OL]. (2022-01-28) [2022-04-10]. http://www.gov.cn/zhengce/zhengceku/2022-02/13/content_5673345.htm.[47]天津市卫生健康委员会. 市卫生健康委转发市防控指挥部关于印发天津市新冠病毒抗原检测应用阶段性 实施方案(试行)的通知(津卫医政〔2022〕151号)[EB/OL]. (2022-03-29) [2022-04-10].http://wsjk.tj.gov.cn/ZWGK3158/ZCFG6243_1/GZWJ625/202203/t20220329_5842885.html.作者贡献徐英春牵头制订共识框架,组建共识制订工作组,组织工作组讨论、修订并审阅定稿;胡继红组织工作组复习文献,起草、修订共识并审阅定稿;所有成员参与讨论、修订并形成共识意见;王瑶、何书宇、邹嘉琪对共识进行撰写、修订和审校。执笔人邹嘉琪(广州万孚生物技术股份有限公司),王瑶(中国医学科学院北京协和医院),康可人(广州万孚生物技术股份有限公司),胡继红(国家老年医学中心 中国医学科学院老年医学研究院 北京医院),徐英春(中国医学科学院北京协和医院)所有参与本共识制订的人员编审组(按姓氏首字母排序):阿祥仁(青海省人民医院),褚云卓(中国医科大学附属第一医院),多丽波(哈尔滨医科大学附属第二医院),高春海(临沂市人民医院),顾兵(广东省人民医院),郭大文(哈尔滨医科大学附属第一医院),郭鹰(重庆医科大学附属巴南医院),韩崇旭(江苏省苏北人民医院),侯锐钢(山西医科大学第二医院),胡辛兰(福建省立医院),胡云建(北京医院),贾伟(宁夏医科大学总医院),康梅(四川大学华西医院),柯江维(江西省儿童医院),李彬(福建医科大学附属协和医院),李刚(宁夏医科大学总医院),李俊明(南昌大学第一附属医院),廖康(中山大学附属第一医院),林宁(江苏省淮安市第一人民医院),林勇平(广州医科大学附属第一医院),刘文恩(中南大学湘雅医院),刘小平 (北京大学深圳医院),刘勇(中国医科大学附属盛京医院),卢志明(山东第一医科大学附属省立医院 山东省立医院),罗春华(湖北省宜昌市中心人民医院),罗春玉(赤峰学院附属医院),罗燕萍(中国药师学会),马小军(中国医学科学院北京协和医院),马筱玲(中国科技大学附属第一医院),毛小琴(云南省第一人民医院),木克代斯米尔地洋(新疆维吾尔自治区人民医院),穆红(天津市第一中心医院),潘艳(江苏省涟水县人民医院),单斌(昆明医科大学第一附属医院),沈瀚(南京大学附属鼓楼医院),沈继录[安徽省公共卫生临床中心 安徽医科大学第一附属医院(北区)],王晶(大连医科大学附属第一医院),魏莲花(甘肃省人民医院检验中心),吴洁(中国医学科学院北京协和医院),谢丽(广西医科大学第二附属医院),徐雪松(吉林大学中日联谊医院),杨滨 (福建医科大学附属第一医院),杨青(浙江大学医学院附属第一医院),喻华(四川省医学科学院 四川省人民医院),张利侠 (陕西省人民医院),张义(山东大学齐鲁医院),张樱(解放军总医院第一医学中心),赵建宏(河北医科大学第二医院 河北省临床检验中心),周泽奇[丹娜(天津)生物科技股份有限公司],朱镭(山西省儿童医院)外审组(按姓氏首字母排序):曹东林(广东省第二人民医院),陈浪(北京金沃夫生物工程科技有限公司),戴二黑(石家庄市第五医院),戴俊(广州海关技术中心),段朝晖(中山大学孙逸仙纪念医院),关伟杰(广州医科大学附属第一医院 广州呼吸健康研究院),郝晓珂(西安区域医学检验中心),胡凤玉(广州市第八人民医院),李六亿(北京大学第一医院),李一荣(武汉大学中南医院),梁皓钧(香港大学公共卫生学院),马学军(中国疾病预防控制中心病毒病预防控制所),秦晓松(中国医科大学附属盛京医院),苏建荣(首都医科大学附属北京友谊医院),汤一苇(丹纳赫诊断平台/赛沛中国),陶志华(浙江大学医学院附属第二医院),王华梁(上海市实验医学研究院),王云峰(美国亚特兰大格雷迪纪念医院) ,张国军(首都医科大学附属北京天坛医院),赵锐(北京电力医院),郑磊(南方医科大学南方医院),周海卫(中国食品药品检定研究院),卓超(广州医科大学附属第一医院)秘书组(按姓氏首字母排序)::何书宇(广州万孚生物技术股份有限公司),卢国萍[梅里埃诊断产品(上海)有限公司],汪小芳(广州万孚生物技术股份有限公司),杨文航(中国医学科学院北京协和医院),杨洋(中国医学科学院北京协和医院),张戈(中国医学科学院北京协和医院),邹嘉琪(广州万孚生物技术股份有限公司)通信作者北京协和医院 检验科徐英春研究员检验科主任,主任医师,教授,博士生/博士后导师,北京协和医学院临床检验诊断学系主任。北京市重点实验室主任,北京市国际科技合作基地负责人,国家卫生健康委员会抗菌药物临床应用与耐药评价专家委员会委员兼办公室主任,国家卫生健康委员会全国真菌病监测网国家中心主任,全国细菌耐药监测网质量管理中心负责人,全球华人临床微生物感染病学会理事长,欧洲EUCAST华人药敏试验委员会主任委员。国家卫生健康委临床检验中心胡继红教授国家卫生健康委临床检验中心微生物室主任,主任技师。负责全国临床及疾控中心微生物室间质评、临床微生物检验标准化、实验室生物安全等培训工作。主持完成3项临床检验行业标准、国家“863”“十二五”重大传染病防治专项分课题负责人。中国医院协会临床微生物实验室专委会副主委,中国医疗保健国际交流促进会临床微生物与感染分会副主委,国家病原微生物实验室生物安全专委会委员,中华医学会检验分会临床微生物学组副组长,《中国抗生素杂志》编委等。研究方向:临床微生物检验质量控制及病原诊断和药敏方法学、病原微生物基因诊断标准化、细菌感染所致RNA氧化及作用机制研究。


























 我要推广仪器
我要推广仪器
 下载APP
下载APP