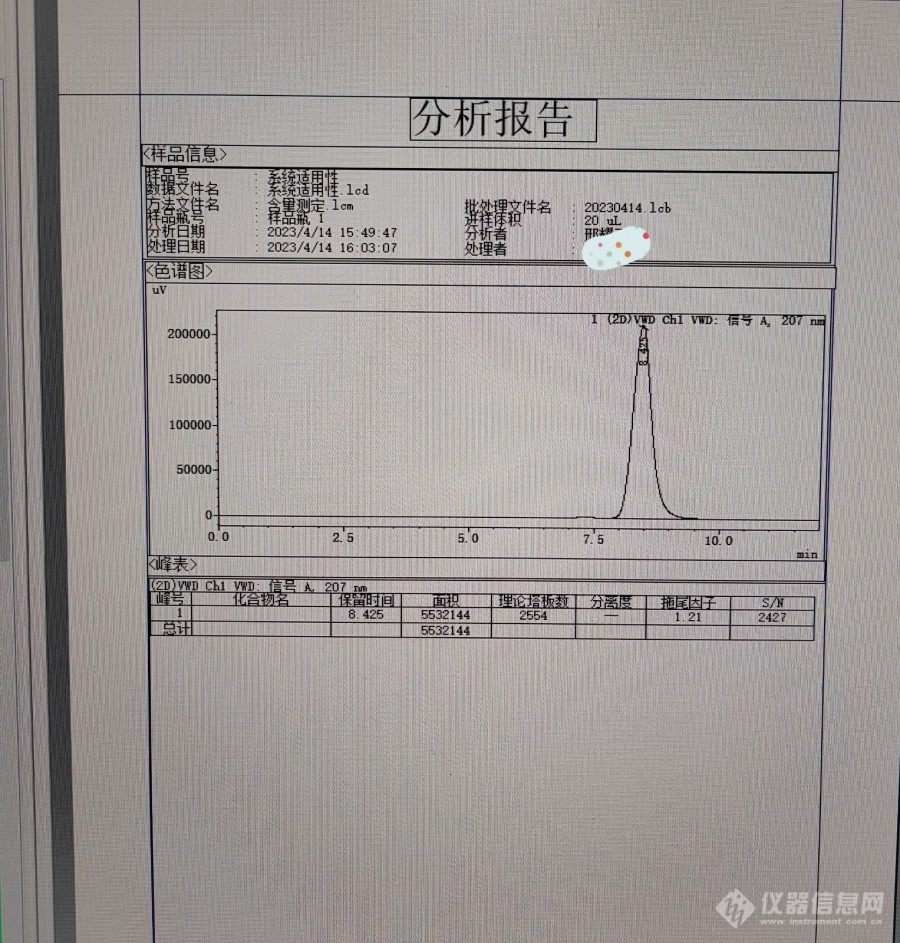
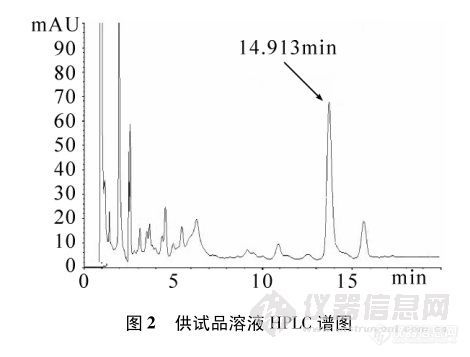
[b][font=宋体][/font]1.[font=宋体]样品简介[/font][/b][font=宋体]利巴韦林颗粒主要用于治疗呼吸道合胞病毒引起的病毒性肺炎与支气管炎,皮肤疱疹病毒感染等。该药主要化学成分为利巴韦林,利巴韦林是广谱强效的抗病毒药物。[color=#333333][back=white]本文主要简介使用[/back][/color][/font]HPLC[font=宋体]法测定利巴韦林颗粒含量。[/font][b]2.[font=宋体]仪器设备与试剂[/font]2.1[font=宋体]仪器设备[/font][/b][font=宋体]岛津[/font]LC-2030C[font=宋体]高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url][/font][font=宋体]赛多利斯[/font]CPA225D[font=宋体]分析天平[/font][font=宋体]色谱柱:[/font]Agilent Hi-Plex H[font=宋体]([/font]7.7mm×100mm[font=宋体])[/font][b]2.2[font=宋体]对照品和试剂[/font]2.2.1[font=宋体]对照品[/font][/b][font=宋体]利巴韦林(中国食品药品检定研究院;[/font]140629-202204[font=宋体];[/font]99.8[font=宋体]%)[/font][b]2.2.2[font=宋体]试剂[/font][/b][font=宋体]超纯水(临用新制);稀硫酸(配制批号:[/font]20230701[font=宋体])。[/font][b]3[font=宋体]色谱条件、样品制备及检测方法[/font]3.1[font=宋体]色谱条件及系统适用性要求[/font][/b][font=宋体]用磺化交联的苯乙烯[/font]-[font=宋体]二乙烯基共聚物的氢型阳离子交换树脂为填充剂;以水(用稀硫酸调节[/font]pH[font=宋体]值至[/font]2.5±0.1[font=宋体])为流动相;检测波长为[/font]207nm[font=宋体];进样体积[/font]20μl[font=宋体]。[/font][font=宋体]系统适用性要求:理论板数按利巴韦林峰计算不低于[/font]2000[font=宋体]。[/font][b]3.2[font=宋体]溶液制备[/font][/b][font=宋体]对照品溶液:取利巴韦林对照品,精密称定,加流动相溶解并定量稀释制成每[/font]1ml[font=宋体]中约含[/font]50μg[font=宋体]的溶液。[/font][font=宋体]供试品溶液:取装量差异项下的内容物适量,混合均匀,精密称取适量(约相当于利巴韦林[/font]0.1g[font=宋体]),加流动相溶解并定量稀释制成每[/font]1ml[font=宋体]中约含利巴韦林[/font]50μg[font=宋体]的溶液,滤过,取续滤液。[/font][b]3.3[font=宋体]检测方法[/font][/b][font=宋体]精密量取供试品溶液与对照品溶液,分别注入[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]。按外标法以峰面积计算。[/font][b]3.4[font=宋体]图谱数据[/font]3.4.1[font=宋体]对照品图谱[/font][/b][font='Calibri',sans-serif][img=,605,634]https://ng1.17img.cn/bbsfiles/images/2023/08/202308182050061104_4837_6126746_3.jpg!w690x722.jpg[/img][/font][b]3.4.2[font=宋体]供试品图谱[/font][/b][img=,605,638]https://ng1.17img.cn/bbsfiles/images/2023/08/202308182050574121_6408_6126746_3.jpg!w690x728.jpg[/img][b]4.[font=宋体]结果与讨论[/font][/b][font=宋体]由系统适用性图谱知,利巴韦林峰保留时间为[/font]8.425[font=宋体],理论塔板数[/font]2554[font=宋体](大于[/font]2000[font=宋体]),符合要求。[/font][font=宋体]因此,[/font]Agilent Hi-Plex H[font=宋体]([/font]7.7mm×100mm[font=宋体])色谱柱能满足利巴韦林颗粒含量分析要求。[/font][font=宋体][/font]
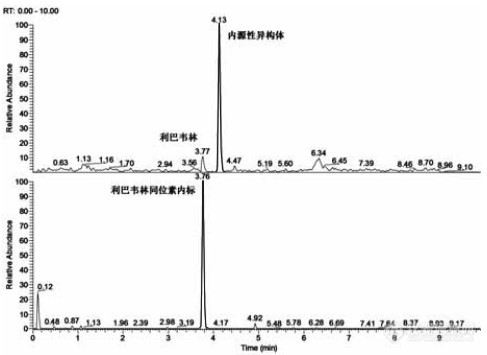
[align=center]利用SPE-HPLC/MS技术检测鸡肉中利巴韦林的含量[/align] 利巴韦林是一种合成的核苷类抗病毒药,是一种前体药物,当微生物遗传载体类似于嘌呤RNA的核苷酸时,它会干扰病毒复制所需的RNA的代谢,抑制病毒的RNA和DNA合成。体外细胞培养试验表明,利巴韦林对呼吸道合胞病毒(RSV)具有选择性抑制作用。无论是基层社区医院还是三级医院,无论是综合医院还是儿童专科医院,无论是普通感冒还是轮状病毒肠炎,还是普通型手足口病,都可能被开利巴韦林。虽然利巴韦林的体外试验对呼吸道合胞病毒具有选择性抑制作用,但其治疗腺病毒、呼吸道合胞病毒、副流感病毒或流感病毒感染临床效果目前并不确定。国外只有北美国家对于极少数重症呼吸道合胞病毒肺炎才偶尔应用。 抗病毒药物在动物源食品中乱用的现象层出迭起,各类抗病毒药物标准品为有效的抗病毒药物的质量掌握和有害残留检测提供主要作用。本文以鸡肉中的抗病毒药物(利巴韦林)作为研究对象,创立而且优化了鸡肉中残留药物的高效液相色谱-质谱分析方法, 使用200 mg的3mL HyperCarb 固相萃取柱,取2 g鸡肉,加入15mL水,涡旋混匀,超声提取10 min,6000 rpm离心10 min,取3 mL上清液进行固相萃取。SPE柱净化分析物过程:活化:1 mL乙腈,1mL甲醇。上样:3 mL上清液用1mL/min流速上样。洗脱:500 mL 20%乙腈-水溶液,洗脱两次,合并洗脱液,直接进样。分析方法采用HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS方法。流动相选择A:水溶液(5mM乙酸铵),B:乙腈。梯度洗脱程序:0-1min,90%A;1-2min,90%-40%A;2-5min,40%A;5-6min,40-90%;6-8min,90%A。流速:0.2 mL/min。柱温:25 ℃。进样量:10 mL。MS条件:电喷雾电离源(HESI),正离子模式选择反应监控(SRM)扫描模式。喷雾电压:3700V,毛细管温度:350℃。离子对参数:利巴韦林:245.1→96.2,245.1→113.2,碰撞能量:43V,14V,透镜电压:80V,80V;利巴韦林同位素内标:249.9→95.9,249.9→112.9,碰撞能量:43V,14V透镜电压:64V,64V;结果:鸡肉空白添加1 ppb的利巴韦林和同位素的HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS图。见下图。添加50 ppb的加样回收率为121.8%,重现性良好。[align=center][img=,487,355]https://ng1.17img.cn/bbsfiles/images/2019/10/201910101335284818_7713_3255306_3.jpg!w487x355.jpg[/img][/align]
具体要求见中国药典2005年二部260页,利巴韦林【含量测定】
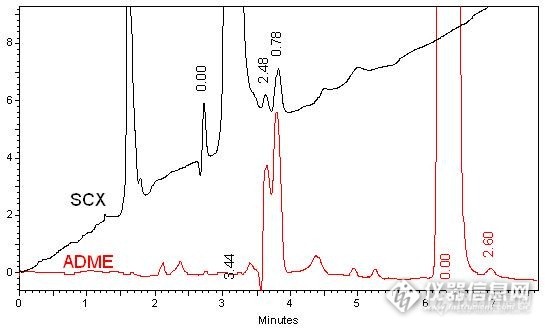
利巴韦林作为一种广谱强效的抗病毒药物,目前广泛应用于病毒性疾病的防治,常用剂型有注射剂、片剂、口服液、气雾剂等。因利巴韦林本身的强极性特点,在中国药典中使用聚合物基材的阳离子交换柱进行含量和有关物质的分析,在国外的一些标准中使用100%水相下的C18柱进行分析。资生堂实验室通过使用新推出的ADME色谱柱,应用其对强极性化合物特有的保留能力,对利巴韦林样品进行了实验,得到了下面的分析结果,供大家参详。首先对比了传统SCX的分析方式,参考中国药典中含量测定下系统适应性试验,【液相条件】色谱柱:CAPCELL PAK SCX S5;4.6mm i.d.×250mm CAPCELL PAK ADME S5;4.6mm i.d.×250mm流动相:ADME: 2%CH3OH SCX: H2O(adjusted pH=2.5 with H2SO4)流速:1.0 mL / min温度:35°C检测:ADME: UV 207 nm SCX: UV 254nm进样量:20 µl样品浓度:50 μg/ mL实验谱图如下http://ng1.17img.cn/bbsfiles/images/2014/12/201412091042_526276_2222981_3.jpg然后,参照药典中有关物质的分析方法,对比实验如下,【液相条件】色谱柱:CAPCELL PAK SCX S5;4.6mm i.d.×250mm CAPCELL PAK ADME S5;4.6mm i.d.×250mm流动相:ADME: 2%CH3OH SCX: H2O(adjusted pH=2.5 with H2SO4)流速:1.0 mL / min温度:35°C检测:ADME: UV 207 nm SCX: UV 254nm进样量:20 µl样品浓度:400 μg/ mL实验谱图如下http://ng1.17img.cn/bbsfiles/images/2014/12/201412091050_526277_2222981_3.jpg局部放大图如下http://ng1.17img.cn/bbsfiles/images/2014/12/201412091052_526278_2222981_3.jpg
求助肠溶阿司匹林片中游离水杨酸的含量测定!谢谢大家了!
据香港文汇报报道,[B]美国食品及药物管理局(FDA)日前发信警告雀巢食品公司,指公司一款婴儿奶粉的钙及磷含量低于法例规定。[/B] 警告信中指出,他们在今年5月巡查公司在威斯康星州的生产厂房后,测试GoodStart系列其中一款婴儿奶粉,发现样本的钙及磷的含量都低于法例规定和奶粉罐上的标示。 美国规定每100千卡路里的婴儿奶粉,钙含量不得低于60毫克,而雀巢该款奶粉样本的钙含量为58.6毫克,罐上标示的钙含量则为64毫克。另外奶粉样本每100千卡路里的磷含量为29.4毫克,低于法例要求的30毫克,而罐上标示的含量则为36毫克。 当局给予雀巢15个工作天,订出解决方法并制定防止问题重现的方案,但没有提及受影响奶粉的数量。 雀巢后来则发表声明,公司自行进行的测试显示奶粉的钙及磷含量均达法例要求,正与当局合作了解两个测试存在分歧的原因。
我用铂金埃尔默的ICP-oes做六氟磷酸锂中金属的含量,因为里面有锂金属的影响,选择什么方法比较合适,我们现在用的是往标样中添加锂来做校正,但是磷也是会有影响的吧,求比较合理的分析方法。
各位达人: 请问有什么比较实用的方法来测磷铁中磷和铁的含量,如何样品前处理。ps:磷铁为电炉制黄磷的一种废渣。其中磷的含量有20%左右,铁的有60%左右。请大家多多指教!
大家对磷青铜中磷含量与锡含量测定 都用什么方法?我用EDS测的跟ICP测的相差很大。而磷又不能用ICP测。有什么好方法?
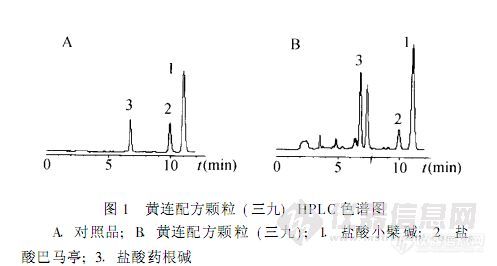
【作者中文名】雷鹏; 刘韶; 李新中; 徐平声;【作者英文名】Lei Peng; Liu Shao; Li Xin-zhong; Xu Ping-sheng(Department of Pharmacy of Xiangya Hospital; Central South University; Changsha 410008);【作者单位】中南大学湘雅医院药剂科; 中南大学湘雅医院药剂科 长沙;【摘要】目的 考察不同厂家黄连配方颗粒中盐酸小檗碱、盐酸巴马亭和盐酸药根碱含量。方法 用高效液相色谱法测定盐酸小檗碱、盐酸巴马亭和盐酸药根碱含量。色谱柱为Diamonsil C18 (4 6 mm×250 mm, 5μm); 流动相为0 2 mol·L-1磷酸二氢钠(用磷酸调节pH至3 0) 乙腈(70∶30); 流速为1 mL·min-1; 柱温为30 ℃; 检测波长为345 nm。结果 不同厂家黄连配方颗粒中盐酸小檗碱含量为3 .67~72. 53 mg·包-1, 盐酸巴马亭含量为0 .60~28. 70 mg·包-1, 盐酸药根碱含量为5. 40~26 .54 mg·包-1。结论 不同厂家产品盐酸小檗碱、盐酸巴马亭和盐酸药根碱含量差异显著。http://ng1.17img.cn/bbsfiles/images/2012/08/201208061421_381879_2379123_3.jpg
本人接到样品是高含量的钯碳,加水合肼 .最后加王水,测试出来的结果含量严重偏低,不知道是不是前处理有问题还是哪里出了问题,麻烦高人指教,多谢!
哪位大神有测试邻苯和总铅含量测试记录表分享一下啊邻苯[back=transparent][font=&]CPSC-CH-C1001-09.4[/font][/back]可迁移元素[font=&][back=transparent]ASTM F963-17 8.3[/back][/font][font=&]总铅: [/font][font=&]CPSC-CH-E1003-09.1[/font]
不知道哪位朋友知道三溴氧磷的含量测定方法,没错的话应该可以用滴定的方法吧?谢过各位了!
按照GB28142-2011方法测定吡虫啉可湿性粉剂,Aginent1100,ZORBAX SB-C18 4.6*250mm,5um,检测波长260nm,甲醇/水=40/60,柱温35摄氏度,0.8ml/min,初入职场,由于工作需要称取四个样做平行测定,每个样进两针,不知是什么原因四个样品中总有两个样品的两针之间相差较大,经过测定吡虫啉含量在30%左右,但一个样品瓶连续两针可能相差0.9%-1.5%之间。称样量使被测样品及标准样品中吡虫啉含量大致处于0.05g,定容至100ml进样量5ul时峰面积处于10000左右,之后选择进样3ul,再之后选择稀释10倍进样量10ul,此时峰面积在2200左右,但还是有一个样品小瓶中两针之间相差1%左右,柱子也换了一次,实在不知道是哪里的原因,望高手指点,谢谢!
有没有谁做过ICP测定硅片硼磷硅玻璃层中硼磷含量,大家来讨论一下,如何处理样品才能保证测量精确?
本人接到样品是高含量的钯碳,加硝酸 高氯酸,最后加王水,有红色沉淀,测试出来的2个结果平行性差,含量严重偏低,不知道是不是前处理有问题还是哪里出了问题,麻烦高人指教,多谢!
PET(固体)磷含量测定,磷含量7500ppm左右,Fe含量20ppm,Sb含量160-200,Ti含量0-2000ppm,标准曲线0,0.5,1,5,10mg/L,ICP参数1300w,等离子气体15,辅助气0.2,雾化0.8,样品提升1.5ml/min,取样0.1g,5ml硫酸加热碳化,过氧化氢消解至透明后继续加热至过氧化氢消失,冷却后用up水定容100ml,铁,锑,钛含量双样极差较大,P含量双样检测极差很小,但是磷含量的测定值在定容后直接检测和存放1-3小时后检测明显随着存放时间变长而减小。消解液磷存在水解可能?这个不是太懂,所以又重新制备消解液并加入10%硝酸5ml再定容,检测发现没有随着时间而变小,但是双样重复性不太好,磷含量极差100-200ppm,并且磷含量测定值要小于不加硝酸200-400ppm。其他微量元素重复性也很差。觉得标曲对于低浓度元素误差太大,且器皿会有残留,消解和定容玻璃器皿浸泡10%硝酸24h,另建标曲0,0.05,0.1,0.5,1mg/L用于测定Fe,Sb,Ti元素,标曲0,0.5,1,5,10mg/L再次复测,重复上述两种处理的消解液。1,消解液不加硝酸,双样重复性良好,低浓度曲线测Fe含量在10ppm左右,高浓度曲线Fe含量接近零(双样平均值为0或者0.1-0.2),但磷含量第一组定容后直接测定7500ppm,第二组2h后测定7300ppm,Sb,Ti,Fe元素比较稳定;2,消解液加硝酸,双样重复性较差,Fe含量在3-40ppm,磷含量第一组7300左右,第二组2h后测定7300左右,Sb,Ti元素比较稳定。有几个疑问需要大佬们指点:1,磷含量为什么会减小,是消解过程损失还是在消解液中损失了?2,酸化消解液为什么重复性变差了?3,由于磷含量较高,在前后样品检测时该怎么防止上个样品残留影响呢,我目前解决办法是增加排空时间和后个溶液冲洗时间不知道对不对4,还有一些添加剂磷含量6%-14%,我是该减小称样量还是进行稀释后测定。跪拜
请,本版的高手能够指点一下.我们用ICP测定高含量的硅铝磷复合氧化物.其中硅10%,铝40%,磷50%;我测定后将各结果相加只有90%左右请问,怎样设计实验.本人将拿出200分奖励!谢谢!希望大家踊跃参与[em09507]
利巴韦林柱,真的只能用于利巴韦林的分析吗?我用作别的品种可不可以?
【作者】:董芳,万丽,吕芳,王云红,陈曙【摘要】:目的:建立冬瓜子中胡芦巴碱的含量测定方法。方法:采用 Diamonsil C18柱( 250 × 4. 6 mm,5 μm) ; 流动相 甲醇 - 水 - 0. 05% 十二烷基磺酸钠溶液( 5: 15: 80) ; 检测波长 265 nm,流动相流速 1. 0 mL·min - 1 。结果:胡芦巴碱在 25. 2 ~201. 6 μg·mL - 1 呈良好线性关系( r = 0. 9998) ,该方法平均回收率为 99. 76% ,RSD 0. 99% 。结论:所建方法准确、简便、为科学地评价冬瓜子药材的质量提供了依据。【作者单位】: 成都中医药大学药学院【关键词】:冬瓜子; 胡芦巴碱; 高效液相色谱法http://ng1.17img.cn/bbsfiles/images/2012/07/201207311327_380848_1838299_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207311327_380849_1838299_3.jpg
今天做阿司匹林肠溶片的释放度,两批结果都高于含量,分别是:130%,150%多???请教高手分析一下原因了。水我是烧开脱气的!含量的结果分别是104%,102%符合规定。
为什么我公司处理过的水里总磷含量总是超标呢?环保局老是找俺们的茬,我现在想找一家公司给我们测一下,市场上那一家好呢?大家给推荐一下?
方法联用测定铬铁中的铬、硅、磷含量摘要:建立了一种同时测定铬铁中主量、次量元素的方法。样品经溶解后,用电感耦合等离子体发射光谱法测定铬铁中的硅、磷含量,用自动电位滴定法测定铬铁中的铬含量。该方法用于两种标准物质GBW01425a和GSB03-1058-1999的实际分析,Cr、Si、P的测定值与标准值吻合,RSD(n=11)为0.09%~3.92%;Cr、Si、P 的回收率为100.45%~105.88%。与现行国标方法相比,分析周期短,适用于大宗铬铁进出口检验的要求。关键词:铬铁 铬 硅 磷 电感耦合等离子体发射光谱法 自动电位滴定法铬铁是铬和铁组成的铁合金,由于它具有质硬、耐磨、耐高温、抗腐蚀等特性,在冶金产业、耐火材料和化学产业中得到了广泛的应用。在冶金产业中,铬铁矿主要用来生产铬铁合金和金属铬。铬铁合金作为钢的添加料生产多种高强度、抗腐蚀、耐磨、耐高温、耐氧化的特种钢,如不锈钢、耐酸钢、耐热钢、滚珠轴承钢、弹簧钢、工具钢等。在耐火材料中,铬铁矿用来制造铬砖、铬镁砖和其他特殊耐火材料。铬铁矿在化学产业主要用来生产重铬酸钠,进而制取其他铬化合物,用于颜料、纺织、电镀、制革等产业,还可制作催化剂和触媒剂等。随着中国经济的发展,汽车、道路、建筑、房地产等市场的持续发展,对钢铁的需求量将继续增大,因而对铬铁的需求也会持续增大。铬、硅和磷的含量是评价铬铁质量的重要指标。现有的对铬铁中铬、硅、磷含量的检测方法,多采用对不同元素逐一样品前处理,再测定,而且测定方法较为复杂,不利于口岸的快速通关。例如现行国标方法GB/T 4699.2-2008采用滴定法测定铬含量,方法1过硫酸铵氧化滴定法使用试剂较多,操作步骤繁琐,方法2电位滴定法前处理步骤复杂,且使用的电位滴定仪自动化程度较差;GB/T 5687.2-2007测定硅含量的高氯酸氧化法及GB/T4699.3-2007测定磷含量的两种方法的操作步骤都较繁琐,不易掌握;由于铬铁是一种重要的战略物资,在当前的贸易形势下,建立对铬铁合金的铬、硅、磷含量的快速检测方法有利于维护我国的贸易利益。 电感耦合等离子体原子发射光谱法具有灵敏度高,干扰少、线性范围宽并能连续测定多种元素等优点,自动电位滴定仪以不需要指示剂,自动判断滴定终点等当点,人为误差因素小等优点在食品、化工、矿产、环保及其它领域得到了广泛的应用。笔者也曾用电位滴定法测定了铬铁里的铬含量。本方法旨在采用样品一次前处理,引入ICP-AES(电感耦合等离子体发射光谱),对硅、磷含量进行测定,用自动电位滴定仪法通过对铬含量进行测定,实现快速测定铬铁的铬、硅、磷含量,精密度和准确度均满足国标要求,方法快速,准确,容易操作,尤其适合口岸大批检验的要求。1 实验部分1.1 仪器及工作条件美国热电ICAP6300 型光谱仪,光室通氩气12h以上,发生器功率1150w;辅助器流量1.0L/min;雾化器流量0.5L/min;分析泵速100r/m;观测高度17mm;短波积分时间15s,长波积分时间10s;进样清洗时间45,波长范围及背景扣除见表1。瑞士梅特勒公司DL55自动电位滴定仪,搅拌速度50%,搅拌时间20s,电极DM140,滴定剂添加模式:动态添加,终点识别模式:等当点控制,阈值:3000,等当点个数:1个,dE(set):2.0mV, dV(min):0.01mL, dV(max): 0.2mL。
测定磷的含量时,采用钼锑抗分光光度法。如果硝化实验最后不中和,那得出的实验结果和真实有什么区别吗?
氧瓶燃烧法测磷元素的含量,步骤如下所示!为什么我测出的实际含量都偏低好多呢?磷不能用氧瓶燃烧做?磷酸三苯酯MW:326.29理论P含量9.49%用氧瓶燃烧法燃烧吸收液为10ml10%过氧化氢溶液,重量及响应值如下表 重量mg0.590.9131.2341.9822.51响应面积0.3730.70091.35272.25992.9036y = 1.3391x - 0.4181R2 = 0.994用30mg/L响应面积1.5574磷酸根标准溶液单点法标定重量mg1.5791.4891.234响应面积1.99131.59451.3527单点法测得磷酸根含量38.36mg/L30.71mg/L26.06mg/L转换成磷重量ug12.5ug10.1ug8.5ug磷百分含量7.9%6.7%6.9%
纺织品纤维含量检测写为:成分分析和成份分析都不算错吧?
各位大神好,小弟最近检测猪肉、鸡肉中的利巴韦林出现了很多问题。样品加标总是出不来,具体表现为:1、加标值异常的高,而且离子比与标准品的离子比对不上。时间也与标准品相差较远2、样品中会有一个非常大的干扰峰,无法判断是否含有目标物。3、我是用sn 4519的那个方法做的亲各位大神帮帮忙,看看问题出在哪里,本人实在是没辙了
各位大虾: 小弟又有事请教了!哈哈 今天我们主管问我LiCoO2中钴含量的测定方法,理论钴含量为59%,这么高含量用ICP由于存在自吸收不好测啊,请问怎么进行样品前处理?如果用化学分析法又怎么测呢? 希望大家不吝赐教啊,谢谢
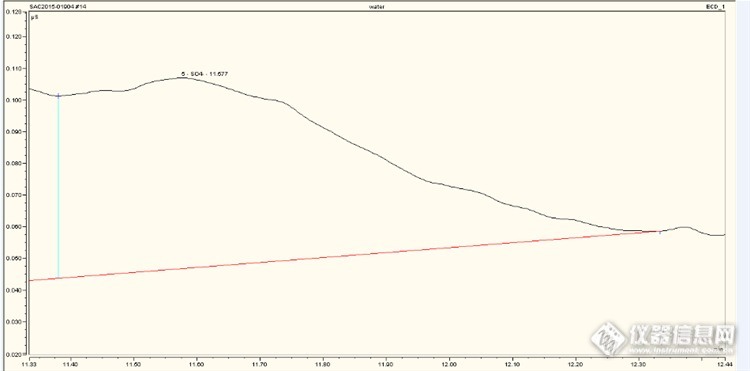
纯磷(赤磷)含量大于99.9%,准备测试下里面的微量杂质硫,用碳硫仪测试没有合适的标样,氧弹然后后IC测试磷酸根的干扰很大,硫酸根的峰很难分辨出,用ICP测试的话需要碱溶,太高的盐雾化器有不满足测试,大家有没测试过,有什么好的方法可以检测的。附图:离子色谱测试谱图http://ng1.17img.cn/bbsfiles/images/2015/04/201504141236_541953_2042772_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/04/201504141236_541954_2042772_3.png
按药典测定甘油磷酸钠中磷含量,用钼酸铵,对苯二酚,乙酸钠,配制好后,显蓝色,720nm波长检测,吸光度一直降低,原来0.629,一个小时后,0.518,第二天,颜色是淡淡的蓝色了,这是为什么?







 我要推广仪器
我要推广仪器
 下载APP
下载APP