整合结构质谱法和计算模拟法探究糖原磷酸化酶中磷酸化介导的蛋白变构调控和构象动态性
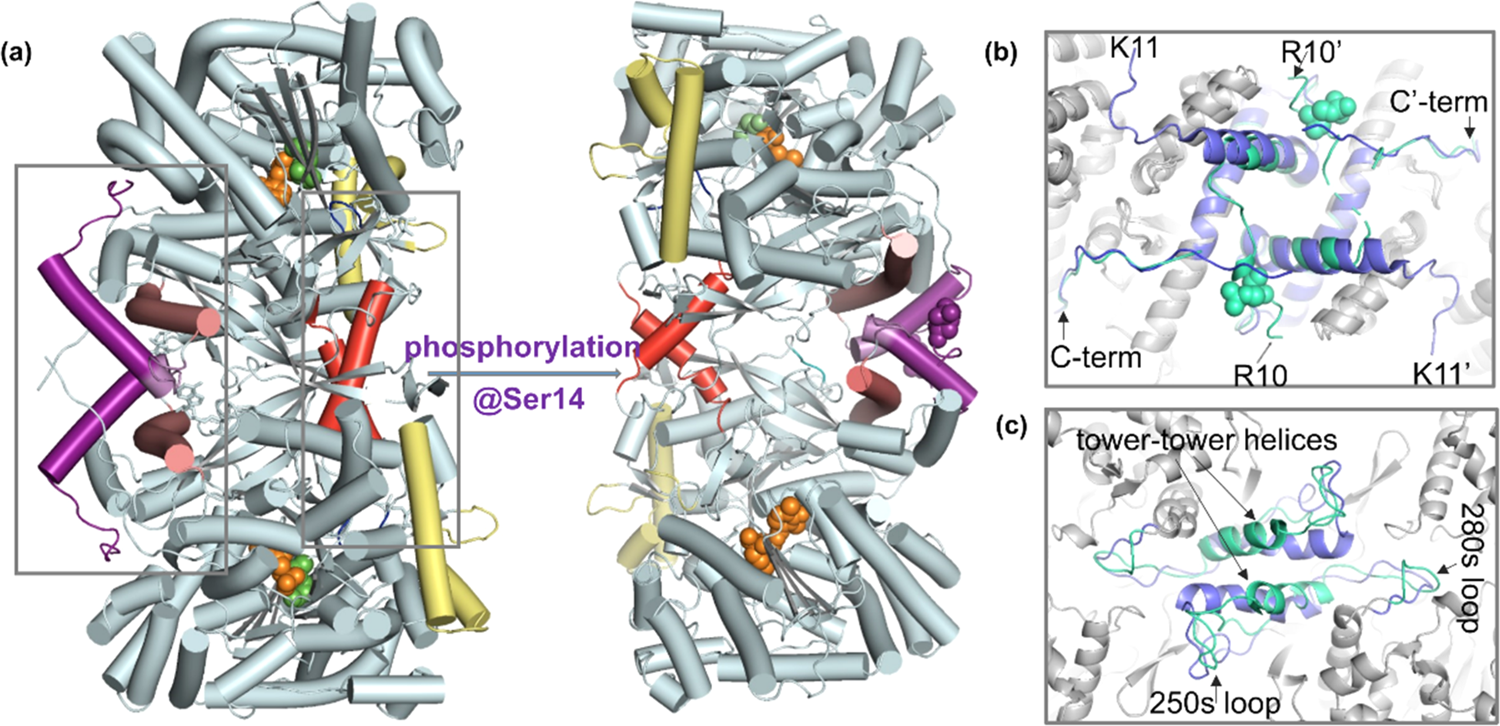
大家好,本周为大家介绍一篇本课题组发表在ACS Chem. Biol.上的文章,Insights into Phosphorylation-Induced Protein Allostery and Conformational Dynamics of Glycogen Phosphorylase via Integrative Structural Mass Spectrometry and In Silico Modeling1。变构调节在自然界中广泛存在,可以用于调控细胞过程。糖原磷酸化酶(GP)是第一个被鉴定出的与变构调节相关的磷酸化蛋白。GP是一个分子量约196kD的同源二聚体蛋白,是糖代谢中重要的组分,也是2型糖尿病及癌症的靶点。AMP结合以及Ser14的磷酸化介导了GP的变构调节,使其构象从非活化的T-state GPb(未磷酸化状态)转变为活化的R-state GPa(磷酸化状态)。即使目前X-射线晶体学法解析出了GP的原子级蛋白结构,但受限于较大分子量,其结构动态性的检测较为困难,因此与GP变构调节相关的结构动态变化过程仍较为模糊。核磁共振(NMR)谱及分子动力学(MD)模拟等是探究蛋白质结构动态性的常用方法,但NMR分析存在分子量上限,且样品消耗量大,MD模拟的时间尺度和力场准确度有限。质谱(MS)法具有快速、灵敏的特点,是蛋白质结构、动态性以及构象变化分析中强有力的一款技术。氢氘交换质谱(HDX-MS)通过监测蛋白骨架酰胺氢原子与溶液中氘的交换来反映蛋白质构象动态性,因此适用于探究由配体、蛋白结合或共价修饰引起的蛋白质构象变化。同时,多个软件实现了由HDX-MS数据计算保护因子(PFs)和吉布斯自由能,从而提取残基水平的蛋白动态性信息。此外,在先前的工作中2, 3,我们整合了native MS和top-down方法(native top-down,nTD-MS技术),成功实现了多个蛋白复合物的一级序列到高阶结构等多方面信息的检测(包括测序、翻译后修饰、配体结合、结构稳定性、朝向等)。整合多种结构质谱法(整合结构质谱法)可以有效填补传统生物物理法中结构到动态性联系中的空缺,更好地表征变构调控现象。本文整合了HDX-MS、nTD-MS、PF分析、MD模拟以及变构信号分析检测了磷酸化介导的GP变构调控的结构和动态性基础,为GP的变构调控过程提供了见解。根据X-射线晶体学结构报道(图1a),T-state GPb转变为R-state GPa时,二聚体界面中N-末端尾部、α2、cap’(图1b)以及tower-tower helices区(图1c)发生了明显的结构重排,导致催化位点开放,从而底物磷酸吡哆醛(PLP)可以结合。尽管有晶体学报道,但与变构调控关联的构象动态性仍有待探寻。图1.(a)磷酸化介导T-state GPb(PDB:8GPB)向R-state GPa(PDB:1GPA)的构象转变;亚基相互作用界面:(b)C端区域和(c)tower-tower helices,GPb为蓝色,GPa为绿色。首先我们通过nTD-MS进行了检测。如图2a、b,谱图中观察到了GPb的单体和二聚体信号,其中二聚体为主要形式;GPa除了单体和二聚体外,谱图中还存在少量四聚体,但仍以二聚体为主要形式。当增加sampling cone(SC)电压时,GPb、GPa保留了其二聚体形式(图2c、d)。随后我们选择离子(29+)并在trap池中进行了碎裂(图2e、f、g、h),谱图低质荷比区GPa的碎片相对峰强度较GPb高,说明GP的二聚体互作界面较为稳定,且GPb亚基结构较GPa稳定。nTD-MS不仅能够探究GPb、GPa的结构差异,也能够为接下来的HDX-MS实验做好前期样品质量检查工作。图2.不同活化条件下GPb、GPa的nTD-MS谱图。(a、b)SC=40V;(c、d)SC=150V;(e、f)SC=150V、trap=100eV;(g,h)SC=150V、trap=200eV。左侧为GPb,右侧为GPa。随后我们进行了HDX-MS实验。图3a中展示了五个时间点的HDX heat map。图3b为通过PyHDX软件计算产生的PF值。其中N-端(1-22)以及tower helix前的loop区域(256-261)的氘代值较高、PF值较低,说明这些区域较为柔性或是结构较为无序。此外我们发现,tower-tower helices(262-276)区域的氘代值较低、PF值较高,表明helices的旋转可能是由前端可塑性铰链区触发的,而非helices本身的变形和重塑引起的,这些发现在晶体结构数据中均有吻合之处。除这两个区域外,GPa和GPb基本保持了稳定的整体结构。而从1μs原子级MD模拟计算得到的均方根波动(RMSF)和溶剂可及表面(SASA)中我们也发现(图3c),这两个区域数据与HDX-MS信息有所吻合,但MD模拟中部分区域未和HDX-MS相吻合的区域可能跟序列覆盖不足相关。图3. (a、d)GPb和GPa在不同标记时间下的氘代热图并映射到结构中(PDB: 1GPA)。(b、e)基于HDX-MS数据计算得到的PF值并映射到晶体结构中。(c、f)MD模拟中RMSF和SASA值并映射到结构中。从氘代差异图(图4a)中可以看出,4个区域呈氘代降低趋势(红色方框),多个区域呈氘代上升趋势(蓝色方框)(GPa-GPb)。而PF差的变化趋势与氘代变化趋势基本一致(图4b)。由数据可知,N-端和tower-tower helices的变化说明磷酸化介导的变构稳定了这两个区域,α1-cap-α2区域的动态性轻微下降。除此之外多个区域(尤其是tower-tower helices序列后的区域)均表现为PF值下降,说明相比于GPb,GPa催化位点附近的区域动态性增强了。接下来我们根据HDX kinetic plot特征将其进行了分类,并详细讨论了所属区域的变化。图4.(a)GPa-GPb HDX-MS的氘代差异图。(b)GPb到GPa PF的变化。 首先是N-端和C-端的变化(图5)。N-端残基1-22表现氘代下降,这说明N-端具有一定可塑性。受N-端区域磷酸化和结构变化影响,C-端区域也产生了一定的变化。此外,残基30-50(cap区)和残基111-117(α4back-loop)区表现氘代下降,而103-109(α4front)表现氘代上升。根据晶体结构推测,cap区和α4back-loop的氘代变化受N-末端变化影响,原有的残基相互作用被打破,形成新的残基间相互作用,同时这两个区域也经历了结构重排,因此表现出较明显的氘代变化。残基88-99(β2-α3)和残基125-141(β3-L-α6)氘代上升。总的来说,磷酸化使得cap′/α2界面互作增强了,同时磷酸化基团和精氨酸残基的静电相互作用是cap区产生变化的主要原因,而α1和α2起到锚定作用,其相对位置基本保持不变。图5.GPb(a)和GPa(b)的N-端和C-端区域的局部结构和HDX动力学曲线(c)。 此外,tower-tower helices(α7,残基262-278)区的变化同样值得关注(图6)。250s loop是表面暴露区域,未与其他区域发生接触,其氘代下降可能是因为自身结构的收缩。而肽段262-267和268-274氘代下降提示该区域可能发生了低周转率或强互作的结合反应。280s loop区氘代值上升。这些变化均说明,tower-tower helix的角度的改变不仅影响了二聚体界面结构,而且还影响了其靠近催化位点的周围区域。因此我们结合晶体结构推测,磷酸化和N-端相对位置的改变,使250s loop自身结构收缩,从而打破了Tyr262' -Pro281和Tyr262-Tyr280′之间的相互作用,导致两个亚基的tower helices发生相对滑动,倾斜角度增加。图6.GPb(a)和GPa(b)tower helix区域的局部结构和HDX动力学曲线(c)。 最后是催化位点、PLP结合位点和糖原存储位点的变化情况(图7)。催化位点周围多数区域均表现氘代上升趋势。我们推测,随着Pro281、Ile165和Asn133间的相互作用被打破,Arg569与Ile165、Pro281、Asn133间的互作也随之打破,因此催化位点和PLP结合位点周围的残基溶剂可及性上升,局部区域结构变得更为灵活,催化位点开放并转变为活化构象。糖原储存位点位于GP表面,距离催化位点30Å,除了α23(残基699−708)外,HDX-MS在糖原存储区没有观察到明显的变化。图7.GPb(a)和GPa(b)的催化位点和PLP(橙色)结合位点的局部结构和HDX动力学曲线(c)。结合以上所有数据,我们对磷酸化调节的动态机制进行了推测(流程图1)。磷酸化后,N-端尾部残基与acidic patch的互作被打破,也导致N-端尾部的有序化以及C-端尾部的无序化以及伴随的其他结构变化。通过在pSer14和Arg69和Arg43′之间形成新的盐桥,N-端残基被重定位,随之带来的是Asp838和His36′间的盐桥断裂。随着三级和四级结构的转变,250s loop收缩并发挥类似“门环”的作用,当其收缩时,Tyr262′-Pro281与Tyr262-Tyr280′之间的相互作用、276-279区与162-164区之间的氢键也被打破,导致tower helix发生相对滑动,tower-tower helices之间的作用被打破,同时将结构变化传递到催化位点。最后,280s loop和催化位点以及PLP结合位点附近的残基松动,通往催化位点和底物磷酸盐识别位点的通道打开,酶得以活化。流程图1.GP变构调节过程中,被打破(蓝色)或新形成的(红色)关键残基相互作用。 本文整合nTD-MS、HDX-MS、PF分析和MD模拟检测了GP磷酸化变构调节过程的结构和动态基础,通过该整合结构手段揭示了GP构象柔性、局部动态性以及长程变构调控构象变化中值得关注的信息。各个方法具有各自的优势,但也在一定层面存在局限,我们期待将HDX-MS信息与计算模拟信息进行更深度的整合以实现二者对蛋白质结构更精确的分析。撰稿:罗宇翔编辑:李惠琳原文:Insights into Phosphorylation-Induced Protein Allostery and Conformational Dynamics of Glycogen Phosphorylase via Integrative Structural Mass Spectrometry and In Silico Modeling李惠琳课题组网址:https://www.x-mol.com/groups/li_huilin参考文献1. Huang, J. Chu, X. Luo, Y. Wang, Y. Zhang, Y. Zhang, Y. Li, H., Insights into Phosphorylation-Induced Protein Allostery and Conformational Dynamics ofGlycogen Phosphorylase via Integrative Structural Mass Spectrometry and In Silico Modeling. ACS Chem. Biol. 2022.2. Li, H. Nguyen, H. H. Ogorzalek Loo, R. R. Campuzano, I. D. G. Loo, J. A., An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem. 2018, 10 (2), 139-148.3. Li, H. Wongkongkathep, P. Van Orden, S. L. Ogorzalek Loo, R. R. Loo, J. A., Revealing ligand binding sites and quantifying subunit variants of noncovalent protein complexes in a single native top-down FTICR MS experiment. J. Am. Soc. Mass Spectrom. 2014, 25 (12), 2060-8.





























 我要推广仪器
我要推广仪器
 下载APP
下载APP