Digital WB在基因治疗眼部疾病细胞和类器官模型中应用
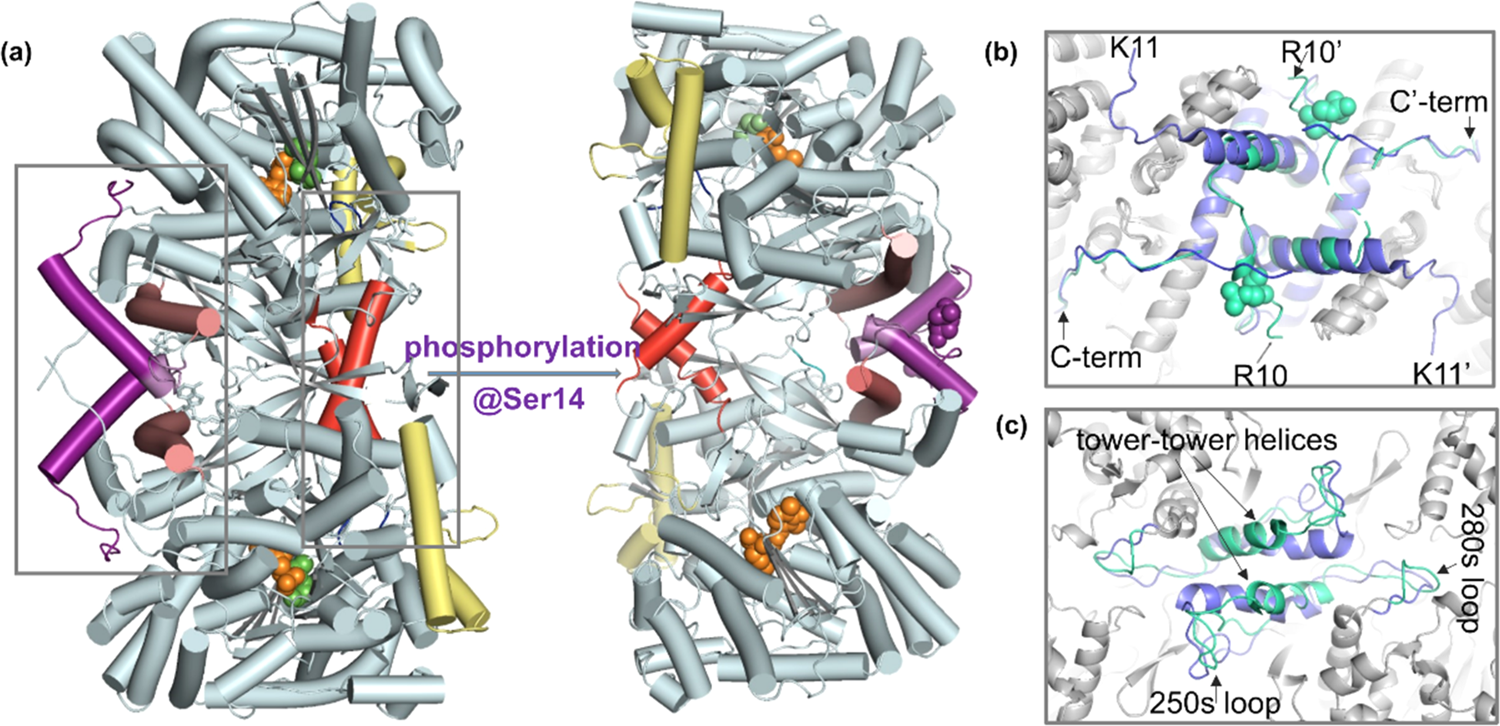
遗传性视网膜营养不良(Inherited retinal dystrophies, IRDs)是可导致进行性视网膜退化的遗传缺陷性罕见疾病,常见的IRD相关基因缺陷超过200种。近几年,眼科领域的基因治疗临床试验项目数量激增,包括基因替换、基因编辑和基因沉默多个技术方面。2017年美国FDA首次批准了视网膜Voretigene Neparvovec基因疗法(Luxturna, Spark Therapeutics),用于治疗RPE65.1双等位基因突变引起的罕见眼科疾病,称为Leber先天性黑蒙。这个里程碑意义的决定为眼科疾病基因疗法打开了大门。目前大部分临床研究疗法目标是通过导入正常功能基因,从而恢复缺陷基因编码蛋白质的正常表达。在非临床研究和临床研究中,检测转基因目的蛋白表达是基因疗法开发的一个关键方面。 目前,有多种技术可实现目的蛋白表达定量检测包括配体结合法(Ligand binding assay,LBA)如酶联免疫吸附方法(ELISA)、液相色谱-质谱(LC-MS)、流式细胞术、蛋白质免疫印迹(Western Blot)和组织染色技术。每种技术都有各自优势和局限,如目的蛋白为分泌性表达,可采用ELISA方法检测细胞培养上清液或体液系统中目标蛋白含量;如目的蛋白不能分泌表达,可采用Western Blot或质谱方法;如需要检测细胞膜蛋白,可采用流式细胞术;如要确定蛋白质在细胞和组织内分布,可采用免疫荧光检测。 在体内和体外模型中研究基因治疗产物与治疗靶点的相关作用机制和效应,选择生物相关性模型来检测目的基因表达和生物学活性非常重要。对于眼部疾病可探索选择临床前研究模型如细胞系模型、人诱导多能干细胞(hiPSC)衍生的视网膜类器官疾病模型、啮齿动物和非人灵长类动物等,根据生物学相关性和测定时间可在不同阶段综合选择特异性评估模型。眼部疾病细胞模型案例1:iPSC衍生视网膜色素上皮细胞(RPE)中低丰度大分子量蛋白质表达检测 从三名Stargardt病人皮肤活检样本产生多个iPS细胞系,这些患者都携带一个致病性ABCA4基因变异。采用RNA-Sep和Digital WB分析正常对照和患者细胞衍生的RPE。这个细胞模型与活检组织相比,可用于评估难以检测的非表达变异体,患者来源的细胞可能更密切地反映患者体内发生的剪接和编辑事件,可用于病人药物敏感性研究,指导临床试验。采用全自动Digital WB技术分析pABCA4蛋白质表达,制备了20 μg 总蛋白 dRPE 细胞匀浆,阳性和阴性对照分别是20 μg野生型和 ABCA4 敲除小鼠视网膜匀浆。参考下图,小鼠视网膜(Mouse ret)在野生型(WT)中pABCA4表达丰度很高,敲除(KO)小鼠没有表达。人类对照(NHDF)具有比WT小鼠视网膜更高表观分子量,同时有更高的表达丰度。与对照相比,所有患者细胞系(H、J和S)中均可检测到pABCA4 ,但这些低丰度pABCA4蛋白可能被降解,作为截短蛋白或降解产品形式存在(除S2外)。与mRNA表达谱结果一致,S2细胞系具有相对正常的pABCA4表达水平和修饰后成熟膜蛋白的分子量。本研究利用了Digital WB对低丰度和大分子量蛋白质分析检测能力。案例2:眼角膜内皮细胞信号通路中多重蛋白质表达检测 本研究采用人源和鼠源细胞,分别是敲低了SLC4A11表达水平的原代人角膜内皮细胞(primary human corneal endothelial cells, pHCEnC),即SLC4A11 (SLC4A11 KD pHCEnC);还有Slc4a11+/+和Slc4a11-/-鼠角膜内皮细胞系(murine corneal endothelial cells, MCEnC),即 Slc4a11-/- MCEnC和Slc4a11+/+ MCEnC。比较转录组学分析揭示了SLC4A11 KD pHCEnC和Slc4a11-/- MCEnC中细胞代谢和离子转运功能抑制以及线粒体功能障碍,导致ATP生产减少。AMPK-p53/ULK1通路激活也表明线粒体功能障碍和线粒体自噬。稳态 ATP 水平降低和随后 AMPK-p53 通路激活提供了代谢功能缺陷和转录组改变之间的联系,以及 ATP 不足以维持 Na+/K+-ATPase角膜内皮泵的证据,这是 SLC4A11 相关角膜内皮营养不良特征性水肿的原因。所以SLC4A11缺陷角膜内皮中分子作用导致内皮功能障碍,是先天性遗传性角膜内皮营养不良 (congenital hereditary endothelial dystrophy, CHED) 和Fuchs 角膜内皮营养不良的主要特征。 下图结果表明SLC4A11缺陷角膜内皮中AMPK-p53 通路激活,采用Digital WB检测信号通路中各蛋白质表达水平。图B说明与 scRNA pHCEnC 对照相比,SLC4A11 KD pHCEnC 中 p53 Ser15 磷酸化水平增加,表明p53转录翻译后激活。图C在Slc4a11-/- MCEnC晚期传代中观察到相似结果(p53 Ser18磷酸化增加,对应于人p53 Ser15)。图C和D结果表明在Slc4a11-/- MCEnC 早期和晚期传代中总 p53 水平增加,代表p53转录激活。进一步研究磷酸化和p53转录激活的激酶,根据报道AMPK介导 Ser15(小鼠中Ser18)磷酸化和p53转录激活,图B和C实验结果也说明AMPKα的Thr172磷酸化增加,AMPKβ1的Ser182磷酸化没有变化。图E和F,与 scRNA pHCEnC 相比,AMPK 另一种下游底物 Unc-51 样自噬激活激酶 1 (ULK1) 在SLC4A11 KD pHCEnC中磷酸化水平(Ser555)增加。综合这些结果表明,ATP水平下降导致AMPK及其下游底物p53 和 ULK1 激活,分别导致转录组改变和线粒体自噬增加。同样,鉴于 SLC4A11 在预防氧化损伤中的作用,SLC4A11 缺失导致线粒体 ROS 产生增加,随后线粒体功能障碍和线粒体自噬增加。此发病机制支持使用Slc4a11-/-小鼠作为SLC4A11相关角膜内皮营养不良的模型,评估各种治疗方法的转化潜力。 基于Digital WB技术的全自动蛋白质表达分析系统Jess可实现化学发光和荧光两种检测模式,是多重蛋白质表达分析有力工具。2022年,ProteinSimple发布了Stellar全自动双色荧光蛋白质表达检测方案,特别适合同步分析细胞信号通路磷酸化蛋白和总蛋白表达,将细胞信号通路研究工具带到一个新高度。iPSC衍生视网膜类器官模型案例1:Digital WB检测iPSC衍生的视网膜类器官中视紫红质表达含量 美国NIH研究人员利用成纤维细胞重编程获得诱导多能干细胞(iPSC),再分化产生视网膜类器官。通过转录组学分析,确定了视网膜类器官发育过程中调节信号,在体外生成了更成熟视网膜,可促进疾病建模和基因治疗研究。本研究采用Digital WB技术揭示了不同培养条件下类器官培养物种视紫红质(Rhodopsin)表达差异。下图结果表明,DHA处理的类器官在32天时视紫红质表达增加了30%,而亚油酸(LA)处理类器官视紫红质表达降低,这表明DHA处理的类器官中视紫红质表达增加不是脂肪酸添加带来的。案例2:AAV基因治疗的RetGC-GUCY2D视网膜类器官疾病模型 Leber先天性黑蒙可由多种不同突变基因导致包括RPE65、CEP29、GUCY2D和CRX等。其中Leber先天性黑蒙1型由GUCY2D基因突变导致,可导致严重视力损害或失明。GUCY2D基因正常拷贝编码了一种鸟苷酸环化酶(RetGC),其是感光器生理学中关键酶之一,视网膜中光敏杆状细胞和视锥细胞使用该酶将光转换为电化学信号。 英国MeiraGTx公司研究人员利用CRISPR/CAS9 技术生成 RetGC 敲除 (RetGC KO) 视网膜类器官,iPSC衍生视网膜类器官分化后,将RetGC KO 视网膜类器官与同一细胞系的野生型类器官进行对比研究。总共设计了四种 AAV 载体来测试RetGC 蛋白在光感受器中的恢复情况,所有载体采用AAV7递送。CMV 和视紫红质激酶 (RK) 两个启动子,并评估了WoodChuck肝炎病毒翻译后调控元件 (WPRE) 影响。采用Digital WB检测6组类器官中RetGC蛋白表达水平。实验结果揭示,与非转导样本组比,所有载体设计均以不同效率产生RetGC蛋白。加入WPRE似乎显示出效力降低趋势,通过其他量化指标验证了这个趋势。 Digital WB相比传统Western blot,只需要几十分之一样本量就可实现类器官等珍贵样本中蛋白质定量检测,而且重复性更高和速度更快,非常适合眼部疾病类器官模型的转基因目的蛋白及相关通路蛋白表达分析。“全自动Digital WB技术是眼部疾病蛋白质表达定量的重要工具 Jess全自动数字化蛋白质表达定量分析系统 (Digital WB) 是Bio-Techne集团旗下蛋白质分析品牌ProteinSimple所有。系统利用毛细管电泳免疫学分析技术,可从微量样品中自动吸取、分离、捕获蛋白质,并通过化学发光或荧光检测目的蛋白含量。针对眼部疾病基因治疗应用技术优势Digital WB技术适合眼科基因治疗体外和体内各种模型中转基因目的蛋白表达定量分析,用于视网膜细胞系、iPSC衍生视网膜色素上皮细胞(RPE)和类器官、小鼠动物模型和非人灵长类动物模型的关键蛋白质分析。适合于基因治疗研发的不同阶段对转基因目的蛋白及相关信号通路蛋白检测需求。满足类器官和视网膜微量样本蛋白质分析需求,Digital WB技术样本量需求是传统Western Blot几十分之一,只需要3 μL样本量就可实现多重蛋白质表达检测,特别适合眼部疾病微量珍贵样本蛋白质分析。Digital WB精准定量检测,传统Western Blot只能满足样本半定量需求,重复性比较差。基因治疗某些目的蛋白表达与临床治疗效果相关联,可作为替代生物标志物,建立量效关系。要求目的蛋白分析检测标准需要提高,要求技术需要经过严格验证,Digital WB可满足这些需求。符合基因治疗产业对自动化标准化和效率的需求,面对行业激烈竞争,需要提升研发效率。Digital WB实现了全自动化和标准化,软件符合FDA 21 CFR Part 11合规性需求。系统3个小时完成一批次蛋白质分析,比传统Western Blot快4倍,大大提高了实验效率,同时减少人力成本。 Digital WB自动化程度高、重复性好、灵敏度高和具有较宽动态检测范围,这些特点满足眼部疾病基因治疗项目不同阶段的目的蛋白定量需求。Digital WB已被国内外知名基因治疗机构采用如Biogen, Sarepta Therapeutics, MeiraGTx,ATGC, Spark Therapeutics,Regenxbio,CRISPR Therapeutics, Editas Medicine, Bluebird bio,杭州嘉因生物、中国食品药品检定研究院等,必将在基因治疗研发阶段、非临床研究和临床研究阶段发挥更大的作用。扫描下方二维码,获取更多关于Digital WB资料参考文献: Gordon, Kathleen Del Medico, Amy Sander, Ian Kumar, Arvind Hamad, Bashar (2019). Gene therapies in ophthalmic disease. Nature Reviews Drug Discovery.MacLaren, R.E. A 2020 vision of ocular gene therapy. Gene Ther 28, 217–219 (2021).基因治疗产品非临床研究与评价技术指导原则(试行)Thilo M. Buck and Jan Wijnholds. Recombinant Adeno-Associated Viral Vectors (rAAV)-Vector Elements in Ocular Gene Therapy Clinical Trials and Transgene Expression and Bioactivity Assays. Int. J. Mol. Sci. 2020, 21, 4197.Annemieke Aartsma-Rus, Jennifer Morgan, et at. Report of a TREAT-NMD/World Duchenne Organisation Meeting on Dystrophin Quantification Methodology. Journal of Neuromuscular Diseases. 6 (2019) 147–159Beekman C, Janson AA, Baghat A, van Deutekom JC, Datson NA (2018) Use of capillary Western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS ONE 13(4): e0195850.Matynia A, Wang J, Kim S, Li Y, Dimashkie A, Jiang Z, Hu J, Strom SP, Radu RA, Chen R, Gorin MB. Assessing variant causality and severity using retinal pigment epithelial cells derived from Stargardt disease patients. Transl Vis Sci Technol. 2022 11(3):33Zhang W, Frausto R, Chung DD, et al. Energy shortage in human and mouse models of SLC4A11-Associated corneal endothelial dystrophies. Invest Ophthalmol Vis Sci. 2020 61(8):39Brooks et al., Improved Retinal Organoid Differentiation by Modulating Signaling Pathways Revealed by Comparative Transcriptome Analyses with Development In Vivo, Stem Cell Reports (2019)Arifa Naeem, etc. RetGC-GUCY2D retinal organoid disease model for AAV gene therapy development. MeiraGTx LTd



























 我要推广仪器
我要推广仪器
 下载APP
下载APP