顺式氯氰菊酯残留分析(WHO)[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=57997]顺式氯氰菊酯残留分析(WHO)以及毒理资料[/url]
有谁参加了这次玉米中的溴氰菊酯、氯氰菊酯残留检测的比对,可以交流一下吗?我们做的初步结果为1号样为100、200ppb,2号样为50、100ppb,但是我们的回收率较高,约为130%,有朋友能交流吗?
蜂王浆样品用水溶解后经正已烷-二氯甲烷(1:1,V/V)提取,无水乙醇破除乳化,样液经离心后,用反相固相萃取柱富集与净化,用乙腈洗脱后,氮吹至近干,用80%乙腈水溶液溶解,经0.22μm滤膜过滤后,采用超高效液相色谱梯度洗脱与紫外检测器分析蜂王浆中的氟胺氰菊酯与氟氯苯氰菊酯残留。以XTerra Shield RP18柱(2.1mm×50mm,1.7μm)为色谱柱,以水和乙腈为流动相,两种菊酯残留在4min中内实现了较好的分离,方法快速准确,灵敏度高,是一种较好的定性和定量方法。所研究的提取净化方法及色谱分离条件能有效排除蜂王浆中的杂质干扰,添加回收率在75.7%~82.8%之间,方法检出限为10μg/kg。
[b][color=#444444]食品中的氯氰菊酯、氰戊菊酯残留量检测需用何种色谱柱?[/color][color=#444444]现GB/T 14929.4—1994和 SN 0217—93的检测方法所要求的色谱柱类型不一样,不知要采用何种色谱柱比较[/color][/b]
出口蔬菜中氯菊酯、溴氰菊酯残留量检验方法 中华人民共和国进出口商品检验行业标准 SN 0217-93 Method for the determination of permethrin, cypermethrin, fenvalerate, deltamethrin residues in vegetables for export 1 主题内容与适用范围 本标准规定了出口蔬菜中氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯残留量检验的抽样、制样和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定法。 本标准适用于出口花菜、蘑菇中氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯残留量的检验。2 抽样和制样 2.1 检验批 以不超过 1 000 件为一检验批。 同一检验批的商品应具有相同的特征,如包装、标记、产地、规格、等级等。 2.2 抽样数量 批量,件 最低抽样数,件 1~25 1 26~100 5 101~250 10 251~1 000 152.3 抽样方法 按 2.2条规定的件数随机抽取,逐件开启。每件至少取 500g作为原始样品,原始样 品总量不得少于 4kg,加封后,标明标记并及时送实验室。 2.4 试样制备 分取出部分有代表性样品,取可食部分切碎,用四分法缩分出 1kg左右。置高速组织 捣碎机中,捣碎成果酱状,均分成二份,装入洁净容器内,密封,标明标记。2.5 试样保存 将试样于-18℃以下冷冻保存。 注:在抽样和制样的操作过程中,必须防止样品受到污染和发生残留物含量的变化。 3 测定方法 3.1 方法提要 蔬菜中残留的氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯,采用丙酮提取,提取液用正已烷萃取,硅胶柱净化,二氯甲烷洗脱。净化液用配有电子俘获检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测定, 外标法定量。 3.2 试剂和材料 除另有规定外,试剂均为分析纯,水为蒸馏水或相适应的去离子水。 3.2.1 丙酮:重蒸馏。 3.2.2 正已烷:重蒸时收集 68~69℃馏分。 3.2.3 苯:重蒸馏。 3.2.4 二氯甲烷:重蒸馏。 3.2.5 无水硫酸钠: 650℃灼烧 4 h,冷却后贮于密封瓶中备用。 3.2.6 2%(m/m)硫酸钠水溶液:称取 2 g无水硫酸钠,溶于 100 mL水中。 3.2.7 硅胶:Merck Kieselgel60,70~230目。在130℃烘箱中烘 10h,贮于密封瓶中备用。 3.2.8 氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯标准品:纯度 ≥99%。 3.2.9 氯菊酯、氯氰菊酯、氰戊菊酯、溴氰菊酯标准溶液:分别准确称取适量的农药标 准品,用少量苯溶解,然后用正已烷各配制成浓度为 0.100 mg/mL的储备液。根据需要再 稀释配制成适用浓度的混合标准工作液。 3.3 仪器和设备3.3.1 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]并配备电子俘获检测器。 3.3.2 振荡器。3.3.3 离心管:具塞 50 mL。 3.3.4 微型层析柱:15 cm×0.5 cm(内径),带有 10 mL储液斗。3.3.5 空气流(或氮气)浓缩装置。 3.3.6 刻度试管:10 mL,具磨口塞。3.3.7 无水硫酸钠柱:6 cm×1.8 cm(内径),内装 5 cm高的无水硫酸钠。3.3.8 微量注射器:10μL。3.3.9 脱脂棉:用正已烷回流 2 h,取出挥发至干,保存在清洁容器中备用。3.4 测定步骤 3.4.1 提取 称取约20g试样,精确至0.1g,置于250mL锥形瓶中。加入80mL丙酮,振荡40min,过滤。用丙酮洗涤残渣。将滤液收集在100 mL容量瓶中并以丙酮稀释定容。3.4.2 净化 准确吸取5mL提取液于具塞离心管中,加入20mL硫酸钠水溶液(2%),用正已烷对丙酮-水溶液相萃取二次(每次 10 mL)。合并正已烷萃取液,并通过无水硫酸钠柱脱水,以少量正已烷洗涤柱。将合并的流出液浓缩至约1mL。于微型层析柱的下端填入少量脱脂棉,依次装入0.5cm高的无水硫酸钠、用正已烷拌 湿的1g硅胶和1cm高的无水硫酸钠。用5mL正已烷预淋层析柱,弃去流出液。待液面下降至上层无水硫酸钠表面时,将上述浓缩液倒入柱内,并用5mL正已烷-二氯甲烷(4+1) 洗涤器皿,倒入柱内,弃去流出液。继用二氯甲烷洗脱层析柱,收集洗脱液10mL于收集管 内并加入2mL正已烷。浓缩以除尽二氯甲烷(温度控制于40℃),再以正已烷定容为10mL,供[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定。3.4.3 测定 3.4.3.1 色谱条件 a. 色谱柱:石英毛细管柱,5 m×0.53 mm(内径), 2.65 μm (膜厚度)。固定 相为:HP-1[100%dimethyl-polysiloxane (Gum)]。b. 氮气:纯度≥99.99% ,13.3 mL/min。c. 尾吹气:氮气,20 mL/min。d. 柱温:245℃。 e. 进样口温度: 260℃。 f. 检测器温度:280℃。g. 进样方式:柱头进样,不分流。 3.4.3.2 色谱测定 根据试样中被测农药含量情况,选定峰高相近的标准工作液,标准工作液和待测样液中农药的响应值均应在仪器检测的线性范围内。对标准工作液与样液应体积参插进样测定。 在上述色谱条件下,被测农药的保留时间为:氯菊酯约 2.5 min,氯氰菊酯约 3.4.3.3 min, 氰戊菊酯约3.4.3.4 min,溴氰菊酯约 5.7 min。 3.4.4 空白试验:除不称取试样外,均按上述测定步骤进行。3.5 结果的计算和表述 用色谱数据处理机或按下式计算试样中农药的残留含量: Acs X=━━━━━ Asc 式中:X- 试样中被测农药的含量,mg/kg;A-样液中被测农药的色谱峰面积(或峰高),mm**2(mm);As-混合标准工作液中被测农药的色谱峰面积(或峰高),mm**2(mm);Cs-混合标准工作液中被测农药的浓度,μg/mL; c-最终样液所代表的试样浓度,g/mL。 注:计算结果需将空白值扣除。 4 测定低限、回收率 4.1 测定低限 本方法的测定低限:氯菊酯为 0.020 mg/kg,氯氰菊酯为0.040 mg/kg,氰戊菊酯为 0.020 mg/kg,溴氰菊酯为 0.020 mg/kg。4.2 回收率 回收率的实验数据:氯菊酯浓度在0.020~1.00mg/kg范围内,回收率为93.4%~113.8% 氯氰菊酯浓度在 0.040~2.00 mg/kg 范围内,回收率为 91.7%~105.7% ; 氰戊菊酯浓度在0.020~1.00mg/kg范围内,回收率为86.7%~98.6%;溴氰菊酯浓度在 0.020~1.00 mg/kg范围内,回收率为 93.0%~114.2%。 附加说明:本标准由中华人民共和国国家进出口商品检验局提出。 本标准由中华人民共和国上海进出口商品检验局负责起草。本标准主要起草人陈余英、习娟华、朱坚。中华人民共和国国家进出口商品检验局 1993-12-28 批准 1994-05-01实施
请问: 高效氯氟氰菊酯 和氯氟氰菊酯 的区别 还有 它们的残留量分析 有没有参考标准呢
江苏出入境检验检疫局动植物与食品检测中心2015年能力验证计划目录 实施机构:江苏出入境检验检疫局动植物与食品检测中心通讯地址:南京市中华路99号 邮编:210001网站:http://www.apfic.com/和http://www.foodmate.net监督电话:025-52345193计划编号计划名称测试项目对应的实验室领域代码对应的PT子领域推荐的测试/测量方法实施时间实施机构及联络信息预计费用APFIC PTP-001 2015果蔬汁中氯氰菊酯、氰戊菊酯和毒死蜱农药残留量的测定氯氰菊酯,氰戊菊酯和毒死蜱227.02食品与农产品中残留物GB/T 5009.146-2003植物性食品中有机氯和拟除虫菊酯类农药多种残留量的测定GB/T 5009.145-2003 植物性食品中有机磷和氨基甲酸酯类农药多种残留的测定2015.8~2015.12联系人:赵增运、沈伟健电话:025-52345193,传真:025-52345180E-mail:shenwj18@jsciq.gov.cn1000元
请各位高人指点,现在刚开始做溴氰菊酯在水产动物中的残留代谢规律研究(从毒理学角度去研究),查到有国标“拟除虫菊酯多组分测定方法”,想请问有没有单残留测定的国标,或者其他成熟的色谱条件?谢谢大家,务必请给帮忙指点!

我要检测硫丹(包括:1-硫丹,2-硫丹,硫丹硫酸盐),甲氰菊酯,氯菊酯,氟氯氰菊酯,这6种物质,由于我不清楚这6种物质在我的气相上的大致出峰时间,所以我决定用全扫描的方式试下,程序升温的方法:40度保持1min;然后以30度/min升至130度,再以5度/min升至250度,最后以10度/min升至300度,并保持5min,后运行5min,以去除残留于色谱柱中的杂质。色谱柱:HP-5ms;溶剂延迟时间:5min,从5min开始至35min,SCAN从50到500,我的TIC图是这样的:http://ng1.17img.cn/bbsfiles/images/2016/07/201607140743_600414_2166779_3.jpg局部放大:http://ng1.17img.cn/bbsfiles/images/2016/07/201607140743_600415_2166779_3.jpg对着一个峰右键双击,看其碎片:http://ng1.17img.cn/bbsfiles/images/2016/07/201607140744_600416_2166779_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607140744_600417_2166779_3.jpg
各位,我是农药残留的生手,向各位求教:溴氰菊酯在水中和土壤中的残留检测方法以及标准,溴氰菊酯通过呼吸道的急性毒性和对鱼类的毒性试验方面有关的文献,可以在哪里查得到?急用,谢谢了!
按NY/T761-2008做蔬菜中菊酯类农药残留(三唑酮、联苯菊酯、甲氰菊酯、三氟氯氰菊酯、氟氯氰菊酯、氯氰菊酯、溴氰菊酯、氰戊菊酯),回收率普遍达到180%,请教各位高手有什么原因造成回收率这么高?
谁做过毒死蜱和氯氰菊酯55%乳油混合制剂的农药残留分析?有没有具体的分析方法?
GB 29705-2013 食品安全国家标准 水产品中氯氰菊酯、氰戊菊酯、溴氰菊酯多残留的测定 气相色谱法
PONY谱尼测试最新了解到,厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。PONY谱尼测试在农药残留方面具有丰富的经验,可以依据日本肯定列表进行检测。对于这三种农药,企业应该引起重视,PONY谱尼测试将鼎力帮助企业进行检测,顺利出口。
有参加ACAS-PT243(2016)果蔬汁中毒死蜱、氯氰菊酯、多效唑、哒螨灵农药残留能力验证的吗?加群交流一下啊595456862
各位大虾:小弟最近忙着做农药残留(浓缩水果汁中的毒死蜱、马拉硫磷、溴氰菊酯、氯氟氰菊酯)的能力验证,但是实验过程中出现了问题,不知道错在哪里请大家帮助,我按照NY/T761-2004方法作的,最后结果很吓人,马拉硫磷的回收率分别为360%和430%。试验过程如下:称5克样品加入1ug/mL的毒死蜱与马拉硫磷的混标3mL,最后毒死蜱的回收率分别为80%和94%,但是马拉硫磷的回收率分别为360%和430%,而我加入的是混标不可能两种农药加标出错,请问这是什麽原因?请大家帮助我找找原因,只要回帖就送分!!很着急,5号上报结果啊!!
最近我们做蔬菜中菊酯类农药残留(六六六、滴滴涕、氯菊酯、氟氯氰菊酯、氟氰戊菊酯、氰戊菊酯、溴氰菊酯),样品均为未检出,标准曲线的系列浓度为:各农药浓度均为100ng/mL、200ng/mL、400ng/mL、800ng/mL、1000ng/mL,各化合物的线性关系是不错,可是就是样品的农药残留均为未检出,我们也有做加标回收率,问题是加标回收的样品:氯菊酯、氟氯氰菊酯、氟氰戊菊酯、氰戊菊酯、溴氰菊酯这几个菊酯类物质均未出峰,也就是回收率为0啊,我们蔬菜样品的前处理是按照NY/T761-2008来的:称取25.0克蔬菜样品,加入50.0mL乙腈提取,加6克氯化钠使乙腈层与水层分层,取10.0mL乙腈层用旋转蒸发仪于40℃水浴中减压旋转至近干,用正已烷:丙酮=90:10溶解,过艾捷尔的Carb/NH2复合柱(1000mg/6mL)净化除色素,氮吹,用色谱纯正已烷1.0mL定容。可是为什么回收率会为0呢,每进10个样品我们也有插入一个1000ng/mL的标准溶液,标准溶液的测定数据正常啊,为什么就是加标回收的样品不正常?
厚生劳动省医药食品局发布食安发0928第2号:部分修改食品、添加剂等的规格标准(2009年厚生劳动省告示第422号),设定农药氯虫酰胺、氰氟虫腙以及甲基碘在食品中的残留标准。根据此通知,厚生劳动省将如下记修改部分食品、添加剂等的规格标准(昭和34年厚生省告示第370号)。第1 修改的摘要根据食品卫生法(昭和22年法律第233号。以下简称“法”。)第11条第1项的规定,设定农药氯虫酰胺、氰氟虫腙以及甲基碘等在食品中的残留标准。第2 实施• 适用日期由公布之日起开始实施第3 应用须知1、此次,在设定了氯虫酰胺标准值的食品中,桃、西瓜以及香瓜是包括果皮的。2、此次,设定了标准值的氰氟虫腙是指:将氰氟虫腙(E-同分异构体)、氰氟虫腙(Z-同分异构体)以及作为氰氟虫腙代谢物的p-[m-(三氟甲基) 苯甲酰甲基] 苯甲腈换算为氰氟虫腙之后的和。第4 其它以残留标准值(根据“法”设定)以及农药取缔法(昭和23年法律第82号)为依据,在农林水产省将氯虫酰胺、氰氟虫腙以及甲基碘注册为农药。关于氯虫酰胺、氰氟虫腙以及甲基碘的检验法将在日后通知。
[color=black][font=Times New Roman][/font][/color][color=#f10b00][size=2][font=宋体]维权声明:本文为[b][color=#d15502]lonpine[/color][/b]原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[/font][/size][/color][color=black][font=宋体][b]摘要:[/b][/font][/color][color=black][font=宋体]本文结合[/font][/color][font=Times New Roman]NY/T 761-2004[/font][font=宋体]农药残留分析方法,[/font][color=black][font=宋体]把[/font][/color][font=宋体]氯氰菊酯残留量测定[/font][color=black][font=宋体]不确定度分为称量、提取、净化、色谱测定等分量,用校正因子来表征测定过程中的重复性不确定度,并加以评定。结果表明,当测定结果为[/font][/color][font=Times New Roman]0.1026 mg/kg[/font][color=black][font=宋体]时,扩展不确定度为[/font][/color][font=Times New Roman]0.0020 mg/kg[/font][font=宋体]([/font][font=Times New Roman]k= 2[/font][font=宋体])。其中,色谱[color=black]定量重复性误差及同一样品的重复性误差所引入的不确定度是影响[/color][/font][font=宋体]残留量测定不确定度的较大因素。[/font][b][color=black][font=宋体]关键词:[/font][/color][/b][font=宋体]氯氰菊酯[/font][font=Times New Roman] [/font][font=宋体]残留量[/font][font=Times New Roman] [/font][color=black][font=宋体]不确定度[/font][/color][color=black][/color]
茶叶农药残留检测方法一般都是参照食品中农药残留检测方法,相对前处理过程较复杂,想请教各位能否有相对较简单的前处理方法,如用固相萃取小柱净化(主要检有机磷、有机氯、菊酯类农药残留)等等!
据美国环保署(EPA)消息,11月7日美国环保署发布公告,将溴氰菊酯(Deltamethrin)在鱼体内的最大残留限量修订为0.01ppm。 本规定自发布之日起生效。征求意见截止日期为2015年1月6日。 具体修订内容如下:产品名称限量要求(ppm)淡水有鳍鱼0.01淡水养殖有鳍鱼0.01其他海水有鳍鱼0.01海水有鳍金枪鱼0.01
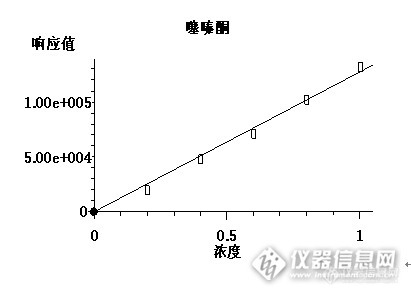
茶叶中硫丹、甲氰菊酯、噻嗪酮类农药残留的检测1、仪器: 安捷伦5975C/7890GC-MS;电子分析天平,箱式电阻炉,涡旋混合器;固相萃取装置;氮吹仪;安谱 GraphiCarb 固相萃取小柱(57084;250mg/3mL)。2试剂:硫丹、甲氰菊酯、噻嗪酮混合标准溶液;无水硫酸钠、色谱纯乙腈、甲苯;茶叶样品硫丹、甲氰菊酯、噻嗪酮混合标准使用液的配制:(α-硫丹、β-硫丹,硫丹硫酸盐、甲氰菊酯、噻嗪酮)0、0.2、0.4、0.6、0.8、1.0ug/mL。http://ng1.17img.cn/bbsfiles/images/2014/12/201412270910_529372_2266096_3.png3、样品准备将1g粉碎茶叶加到50mL离心试管中,加入3mL水润湿,浸泡10min,加15mL色谱纯乙腈,涡旋2min,静置,过滤,滤液待用,再用15mL色谱纯乙腈重复提取一次,过滤,合并滤液,滤液用50度的水浴中氮吹浓缩至2mL左右待净化。净化 柱子上端装入约1cm高的于650度灼烧过的无水硫酸钠层,以吸附除去多余的水分。a 活化: 4mL 乙腈:甲苯=3:1活化,流出液弃去;b 上样:将待净化的样液加入小柱(安谱 GraphiCarb 固相萃取小柱(57084;250mg/3mL)),收集流出液;用6mL 乙腈:甲苯=3:1分两次洗涤,合并流出液。c 重新溶解:于50度的水浴中氮吹浓缩至近干,加2mL农残级的正已烷溶解定容,GC-MS 检测。4、仪器条件色谱柱:HP-5MS; 30m*0.25mm*0.25um ;柱温:初始温度80 ºC,维持1min,以30 ºC/min 升温至200 ºC,并保持1min,再以5 ºC/min 升温至280 ºC,保持3 min;流速:1.0mL/min ;载气:高纯氦气;进样量:1μL;进样方式:不分流;进样口温度:230 ºC;接口温度:[
有参加CNAS下半年的茶叶中溴氰菊酯、甲基毒死蜱、甲氰菊酯、硫丹农药残留量的测定能力验证的吗?有做预实验的吗?都是用什么标准检测的?
请问如果要建立一种测脐橙中有机氯和拟除虫菊酯的农药残留的方法,该如何建立啊?我是想按照761中提取,净化想比较弗罗里硅土,中性氧化铝,活性炭,或者弗罗里硅土,中性氧化铝混合,,但是里面的用量都要进行实验比较。。。这段时间比较了活性炭对回收率的影响,用量是0.1,0.2,0.3,0.4,0.5,发现没有很明显的差别,0.4克,稍微好点,,但是重复性很差。。所以很难得出结论。。(这个实验加了样品的25克的) 还有一个实验后来是不用样品,想比较填料对回收率的影响,只是加农药和溶剂(1+1ML)作为上样液,柱子分别比较装了弗罗里硅土(2,3,4,5克),中性氧化铝(1,2,3,4克),活性炭(0.2,0.3,0.4,0.5克)以及弗罗里硅土+中性氧化铝(2:2,2:1,2:3,3:1,3:2),但是实验结果测出来他们对回收率都没有影响,,几乎都一样?怎么会这样,,我实验设计哪里有问题,请大家指点,,不胜感激。。该怎么设计呢?
能力验证名称:茶叶中溴氰菊酯、甲基毒死蜱、甲氰菊酯、硫丹农药残留量的测定编号:CNAS T0616有效期:具体实施时间:2011.6-2011.9组织方:中国测试技术研究院下个星期样品送到
要用液相测土壤中农残含量。有机磷类:辛硫磷(提取剂:丙酮;定容:正己烷)菊酯类:功夫(高效氯氟氰菊酯)(提取剂:石油醚+丙酮;定容:丙酮)流动相怎么选择?原因?波长怎么选择?根据?其他的条件都怎么确定?新手,不懂,望指教
请各位帮忙,急需GB/T17332-1998 食品中有机氯和拟除虫菊酯类农药多种残留测定,谢谢
http://www.instrument.com.cn/bbs/shtml/20080521/1270090/——第1部分:蔬菜和水果中54种有机磷类农药多残留的测定;——第2部分:蔬菜和水果中41种有机氯和拟除虫菊酯类农药多残留的测定;——第3部分:蔬菜和水果中10种氨基甲酸酯类农药及其代谢物多残留的测定。本标准代替NY/T 761-2004《蔬菜和水果中有机磷、有机氯、拟除虫菊酯和氨基甲酸酯类农药多残留检测方法》。二〇〇八年四月三十日农业部公告发布
出口粮谷中溴氰菊酯残留量检验方法 中华人民共和国进出口商品检验行业标准 SN 0219-93 Method for the determination of deltamethrin residues in cereals for export 1 主题内容与适用范围 本标准规定了出口粮谷中溴氰菊酯残留量检验的抽样、制样和[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定方法。 本标准适用于出口玉米中溴氰菊酯残留量的检验。 2 抽样和制样 2.1 检验批 以不超过 200 t为一检验批。 同一检验批的商品应具有相同的特征,如包装、标记、产地、规格、等级等。 2.2 抽样数量 2.2.1 袋装商品:按式(1)计算抽样袋数。 __ a=√ N …………………………………(1) 式中:a-抽样袋数: N-全批袋数。 注: a值取整数,小数部分向前进位为整数。 2.2.2 散积商品:粮堆高度不超过2m。按粮堆面积划区设点。以 50m**2 为一个取样区,每区设中心及四角(距边线 1 m处)5个点。每增加一个取样区,增设 3个点。 2.3 抽样工具 2.3.1 金属单管取样器:全长 55cm(包括手柄),直径 2.5cm,沟槽长度应超过袋对角线长度的一半。 2.3.2 金属双套管取样器:长度 1m(包括手柄),直径 2.5cm 。 2.3.3 取样铲。 2.3.4 分样板。 2.3.5 样品筒(袋):可密封。 2.3.6 分样布或适用铺垫物。 2.4 抽样方法 2.4.1 袋装抽样 2.4.1.1 倒包抽样:从堆垛的各部位随机抽取 2.2.1条规定的应抽样件数的10%(每一批一般不少于3包),将袋口缝线全部拆开,平置于分样布或其他洁净的铺垫物上,双手紧握袋底两角提起约成 45°倾角,倒拖1m以上,使袋内货物全部倒出。检查货物的外观、气味,有无发霉、变质等,并查看袋内和袋间品质是否均匀。确认情况正常后,用取样铲随机在各部位抽取样品,每袋抽取样品数量应基本一致,即将样品倒入盛样器内。2.4.1.2 袋内抽样:按2.2.1 条规定的应抽样件数的90%,在垛堆四周上、中、下各层以曲线形走向,随机抽取。将取样器(2.3.1条)管槽朝下,从每袋一角依斜对角方向插入袋内,然后将管槽旋转朝上,抽出取样器,立即将样品倒入盛样容器内。每袋抽取样品数量应与2.4.1.1条基本一致。每批总样品量应不少于 4kg。 2.4.2 散积抽样:按 2.2.2 条规定的取样点,逐点将取样器(2.3.2条)插入粮堆至相应深度扦取代表性样品。从各点扦取的样品量应基本一致。总样品量应不少于 4kg。 2.4.3 大样缩分 袋装样品:集中袋内和倒包抽样所取全部样品,倒于分样布上,用分样板按四分法缩分样品至不少于 2kg,加封后标明标记并及时送实验室。散积样品:将扦取的全部样品倒于分样布上,以下按上述同样方法进行。 2.5 试样制备 将样品缩分至 1 kg,全部磨碎并通过 20目筛,混匀,均分成两份,装入洁净的容器 内,密封,标明标记。2.6 试样保存 将试样于-5℃以下避光保存。 注:在抽样和制样的操作过程中,必须防止样品受到污染或发生残留物含量的变化。3 测定方法3.1 方法提要 试样用丙酮提取后,再用石油醚反提取,提取液经弗罗里硅土柱净化,脱水、浓缩、定容后,用配有电子俘获检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测定,外标法定量。3.2 试剂和材料 3.2.1 无水硫酸钠:分析纯,经 650℃ 灼烧 4h,置干燥器内备用。3.2.2 弗罗里硅土(100 ~200目): 650℃灼烧 4h。使用前一天在 130℃下烘1h,冷却后加2%(m/m)的水脱活,贮于干燥器中。3.2.3 石油醚:分析纯,重蒸馏,收集 65~75℃馏分。3.2.4 丙酮:分析纯,重蒸馏后备用。 3.2.5 乙醚:分析纯。3.2.6 溴氰菊酯标准品:纯度≥99%。3.2.7 溴氰菊酯标准溶液:准确称取适量的溴氰菊酯标准品,用少量苯溶解,再用石油醚配成浓度为 1.00 mg/mL 的标准储备溶液,根据需要再配成适当浓度的标准工作液。3.3 仪器和设备3.3.1 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],配有电子俘获检测器。3.3.2 净化柱: 200mm×15mm(id)玻璃柱。柱底填约 0.5 cm高的脱脂棉和 2cm高的无水硫酸钠,装入 5g弗罗里硅土,顶端加入2cm高的无水硫酸钠。使用前用 20 mL石油醚淋洗。3.3.3 旋转蒸发器。3.3.4 无水硫酸钠柱:80mm×40mm(id)筒形漏斗,底部垫 0.5cm高的玻璃棉,再装 5cm 高的无水硫酸钠。3.3.5 微量注射器:10μL ,100μL。 3.3.6 超声波水浴。3.3.7 离心机:4 000r/min。3.4 测定步骤3.4.1 提取 称取试样约 2g(精确到 0.01g)于离心管中,加入 5ml丙酮,于超声波水浴上提取5min。于4000r/min离心1min,取出上清液。残渣再用5mL丙酮按上述同样步骤提取,合并提取液于心形瓶中。加入石油醚和蒸馏水各20mL,于超声波水浴中提取5min。取出上层 石油醚提取液。再加入石油醚20mL,同样提取一次,合并石油醚提取液于容器中。经无水硫酸钠柱脱水,并用少量石油醚分数次洗涤无水硫酸钠柱和容器。流出液收集于心形 瓶内。流出液在 70℃水浴中减压旋转浓缩后,转移至刻度试管内。用少量石油醚洗涤心形瓶,并入刻度试管,稀至 10 mL。 3.4.2 净化 将上述 10mL提取液移入净化柱(3.3.2条)中,弃去流出液。用100mL乙醚-石油醚混 合液(3+7)洗脱,收集全部洗脱液。在 70℃ 水浴中通氮气浓缩,用石油醚定容至5mL, 供测定。 3.4.3 测定 3.4.3.1 色谱条件a. 色谱柱:玻璃柱 2m×3 mm(id),填充物为 2%(m/m)OV-1涂于 Chromosorb W HP(80~100目)。b. 色谱柱温度:250℃。c. 进样口温度:280℃。d. 检测器温度:300℃。e. 氮气:纯度 ≥99.99%,70 mL/min。 3.4.3.2 色谱测定 根据样液中溴氰菊酯含量情况,选定峰高相近的标准工作溶液。标准工作溶液和样液中溴氰菊酯响应值均应在仪器检测线性范围内。取标准工作溶液和样液等体积参插进行 测定。在上述色谱条件下,溴氰菊酯保留时间约为 5.8min 。 3.4.4 空白试验 除不加试样外,均按上述测定步骤进行。 3.5 结果计算和表述 用色谱数据处理机或按式(2)计算试样中溴氰菊酯残留量: h c V X=───── ……………………………………(2) hs m 式中: X-试样中溴氰菊酯残留量,mg/kg; h-样液中溴氰菊酯的峰高,mm; hs-标准工作溶液中溴氰菊酯的峰高,mm; c-标准工作溶液中溴氰菊酯的浓度,μg/mL; V-最终样液的体积,mL; m-最终样液所相当的样品量,g。 注:计算结果需扣除空白值。4 测定低限、回收率4.1 测定低限 本方法的测定低限为 0.1 mg/kg。 4.2 回收率 回收率的实验数据:溴氰菊酯添加浓度在 0.02~2 mg/kg范围内,回收率为85.0%~97.5%。附加说明: 本标准由中华人民共和国国家进出口商品检验局提出。 本标准由中华人民共和国陕西进出口商品检验局负责起草。 本标准主要起草人庄金侍、康定民。中华人民共和国国家进出口商品检验局 1993-12-28批准 1994-05-01 实施
我们买的 农药标准品是100μg/ml的1ml,气相用的是安捷伦的。公司要求测茶叶是否低农,请问我该怎么配各个标样的浓度呢?具体的种类是哒螨灵、氟氯氰菊酯、甲氰菊酯、硫丹、氯氟氰菊酯、氯菊酯、氯氰菊酯、溴氰菊酯、滴滴涕、六六六、三氯杀螨醇。如果按照GB/T2763里的最大残留限量配最大浓度的话,氯菊酯和氯氰菊酯的量就不够用了,请教一下,该怎么办







 我要推广仪器
我要推广仪器
 下载APP
下载APP