有没有谁参加了果蔬汁中马拉硫磷、甲基对硫磷残留量的测定?省局的实验室比对!请问结果多少?
农药的代谢物与残留毒性 上世纪60年代以来的研究进一步发现,除了农药本身以外,它的代谢产物也会出现残留毒性问题,一系列的事件发生引起了对这个问题的重视。美国的Rode L.(1969)报道,在美国加州农场工人进入喷施过对硫磷几天后的柑橘园发生了中毒事件,研究发现是对硫磷的代谢产物对氧磷引起的,这说明有时农药母体化合物的毒性还不如其代谢产物。这一发现不仅促使加强了对农药残留降解的研究,还引发了对农药降解过程中代谢产物毒性的研究。其后,又陆续发现了一些农药的代谢产物具有比母体化合物毒性更大的情况。如乙酰甲胺磷是一种急性毒性不大的农药 (对大鼠的口服半致死剂量LD50为605~1100mg/kg),因此将它列为相对低毒的农药类别。但喷施到植物上后,乙酰甲胺磷会在植物体内代谢成甲胺磷(对大鼠的口服LD50为20~30mg/kg),其毒性提高了20~50倍。茶叶生产上曾对这个问题展开过讨论,即乙酰甲胺磷究竟适不适合在茶树上使用。现在证明尽管乙酰甲胺磷毒性不高,但喷后几天,会出现一个甲胺磷残留的高峰,甲胺磷又是一种高水溶性的化合物,因此存在很高的风险性,不宜在茶叶生产中使用。乐果也是一种相对低毒的有机磷农药(对大鼠的口服LD50为500~600mg/kg),但当喷施到植物上1~2天后,会氧化成为氧乐果(对大鼠的口服LD50为30~50mg/kg),其急性毒性提高 10倍以上。相类似的有涕灭威农药及其在代谢过程中形成的砜和亚砜代谢物,三唑酮代谢形成的烃基三唑酮,杀虫脒代谢形成的4-氯邻甲苯胺,这些代谢物的形成都明显提高了农药的急性毒性或慢性毒性。这就使得在喷施农药后,除了要进行农药母体化合物的残留测定外,还要对其主要的代谢产物,特别是毒性有提高的化合物进行残留测定。 农药杂质与残留毒性 除了农药母体化合物和主要代谢物外,有时农药中含有的杂质也会产生毒性问题。1976年联合国卫生组织和美国援助巴基斯坦时用马拉硫磷杀蚊治疟疾,由于马拉硫磷中含有的杂质马拉氧磷和异马拉硫磷使得数百人中毒,8人死亡。这一事件促使了对农药杂质毒性的研究。上世纪80年代以来我国已经停止生产、销售和使用滴滴涕农药,但在茶叶中还可以检测到滴滴涕农药的残留。经研究发现,这种滴滴涕的残留主要来自三氯杀螨醇农药。由于三氯杀螨醇的化学结构和滴滴涕非常相似,只相差一个氯原子、一个氢原子,因此在三氯杀螨醇加工工艺中,一些环境条件的变化会使产品中出现滴滴涕成分。据对我国三氯杀螨醇产品的成分分析发现,产品中滴滴涕的含量为3%~13%,因此在喷施三氯杀螨醇防治螨类时会出现滴滴涕的残留。正因为如此,1999年农业部颁布了在茶叶生产中禁止使用三氯杀螨醇的决定。此外许多有机磷农药中的氧化物,二硫代氨基甲酸酯类农药(代森锌等)中的乙撑硫脲都是这个问题的实例。 农药残留分析的特点和要求 农药残留分析是应用现代分析技术对各种食品和环境中的微量和痕量的农药母体化合物和代谢物进行定性、定量分析和测定。农药残留分析属于难度较大的分析类别。 1. 农药残留分析属于微量至超微量分析范畴。在上世纪60年代,农药残留分析一般为ppm级,即从1g样品中需要测出微克级的农药物质。但随着科学的发展和对残留测定要求的提高,检测的要求也相应地提高到ppb级(μg/kg,即十亿分之一),甚至ppt级(ng/kg,即一万亿分之一),如果以1g样品计算,最小检出量就需要相应达到纳克(ng,10-9g)级或皮克(pg,l0-12g)级。这就对检测工作提出很高的要求,既要求测定的仪器有非常高的灵敏度,同时还要求检测方法有非常高的精确度。 2. 对样品的前处理要求极高。正因为农药残留分析是一种微量至超微量的分析范畴,所以对前处理的要求就非常高。所谓前处理就是要将样品中的目标物(农药)尽可能完全地提取出来。由于样品中必然含有各种成分,这些成分会对检测过程有很大的干扰,因此要尽最大可能将样品中的其他成分通过纯化而去除,而将提取样品中的目标物质尽可能完全保留,回收率的高低直接关系分析结果的准确度。 3. 由于分析样品都为未知成分样,也就是说样品中含有几种农药及其浓度均为未知,因此对样品的分析就显得非常复杂。
请教牛奶中有机磷农药残留的分析方法,样品如何处理,谢谢!
实验中需要测定河流底泥及河水中草甘膦和百草枯的残留量分析,以前没有这方面的知识,看了好多资料也不知道究竟怎么测定。请教高手:1.采样注意事项2.详细的残留量分析方法(包括样品预处理)。3.如果方便,您也可以推荐检测单位,最好能有详细的联系方式,我把样品送出去做。因为在这里找不到可以测定两种除草剂残留的单位。或者您有测定其他样品中草甘膦和百草枯的残留量分析的方法也可以提供给我,联系方式:ening451@163.com先谢过您的关注和帮助!
有机磷农药是指含有磷原子的有机磷类化合物,在生物体内与胆碱酯酶形成磷酸化胆碱酯酶,使胆碱酯酶活性受到抑制而产生毒性作用的一类农药的总称。有机磷农药大多为磷酸酯类或硫代磷酸酯类,微溶于水,易溶于有机溶剂,对光、热、氧及酸稳定,在碱性溶液中分解、解毒。GC分析时,由于进样口等位置活性位点吸附,经常出现检测结果偏差,使得定量结果不能真实反映样品中有机磷农药残留量,给检测带来一定影响。那么在GC分析有机磷类农药残留时,应注意什么?该如何避免系统中各活性点对农药的吸附?
有机磷(毒死蜱、倍硫磷、喹硫磷、马拉硫磷、杀螟硫磷、甲胺磷、甲基嘧啶磷、甲基毒死蜱、水胺硫磷、敌敌畏、乐果、乙酰甲胺磷),请问这12种有机磷农药残留可以混合在一起分析吗?我今天将它们混合配在一起,可是只出了11个峰,还有一个峰不知道与哪个峰没有分开,反正是少了一个峰。我的色谱条件:RTX1701 30m*0.25mm,0.25um,进样口:250℃ 色谱柱: 80℃(保持1min) 然后以30℃/min升至130℃,再以5℃/min升至250℃并保持8min,总运行时间44.65min,FPD检测器温度280℃
最近让我测蔬菜中的马拉硫磷和乐果残留,实验室以前用固相微萃取测水样中的残留,回收率可达90%,但是蔬菜中我几乎就测不到回收率。大家都用什么前处理方法测有机磷啊?我们实验室条件有限,只有微波萃取仪,超声组织粉碎机,旋转蒸发仪,索式提取器,榨汁机,布式漏斗,丙酮和甲醇,不能固相萃取。请各位高手指点一下适合我们实验室的前处理方法。请说详细些,谢谢。
蔬中的有机磷残留分析,用FPD好还是ECD好?
果蔬中的有机磷残留分析,用FID好还是ECD好?
感觉不错的。[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=83747]FPD四种有机磷的残留分析[/url]
我想知道马拉硫磷有液相分析方法吗?我没有[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]法的林丹内标,试过其他两种效果不好,用毛细柱时间长,并且在N2000工作站响应值很小,请各位指点
我们现在做农残的测试,向分析出以下农药:敌敌畏,甲胺磷,乙酰甲胺磷,敌百虫,甲拌磷,久效磷,乐果,氧化乐果,对硫磷,倍硫磷,透明硫磷,马拉硫磷,毒死蚍蜱,辛硫磷,需要毛细柱分离,主机是岛津德GC-17A ,现有的柱子分离效果不好,怎么选合适的柱子啊?
我今天做有机磷的农药残留,发现毒死蜱,倍硫磷、喹硫磷、马拉硫磷、甲基嘧啶磷这几种有机磷的出峰时间均好接近啊,如果是混标了话,那这几个有机磷岂不是不能分开了,我的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]条件是:[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] 2010plus,色谱柱:Rtx-5 30m*0.25mm, 0.25um 升温程序:80℃保持1min,以20℃/min升至130℃,再以5℃/min升至200℃,最后以15℃/min升至250℃,并保持11min,检测器:FPD,温度250℃,进样口温度220℃。
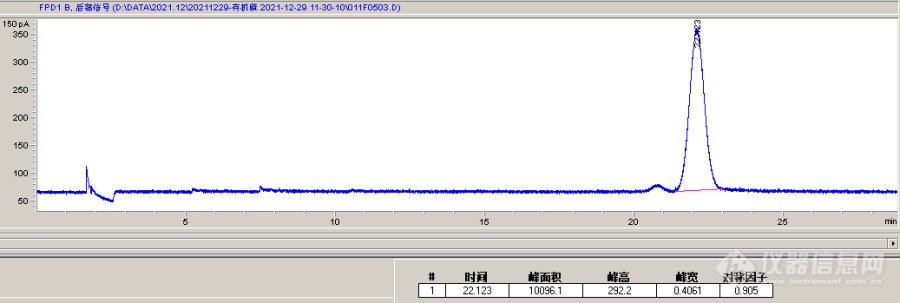
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]为安捷伦7890A,色谱柱为30米长、0.25mm口径、膜厚0.25μm的HP-5MS柱,进样口230℃,不分流进样,柱流量1.0 mL/min,程序升温条件为80℃保持1min,20℃/min升温至130℃,5℃/min升温至200℃,15℃/min升温至250℃保持8min,检测器250℃,氢气流量75mL/min,空气流量100mL/min,尾吹流量30mL/min。分析丙酮标液中的敌敌畏、敌百虫、乐果、甲基对硫磷、马拉硫磷、对硫磷共6种有机磷,进样浓度 2 mg/L,前面的物质出峰都挺高的,峰形也还可以,但马拉硫磷和对硫磷出峰峰形差,峰宽很宽,因此两种物质出峰基本重叠无法分开,试了好多程序升温条件都难以改善,请教各位老师这种情况是什么原因?如何解决?我也试过换一根新的0.32mm的HP-5柱子,情况类似,看论坛里多数人推荐DB-1701的柱子,可是我们实验室只有一根1.0μm膜厚的1701柱,换上去测试出峰太慢了,敌敌畏要12min以后才出峰,马拉硫磷和对硫磷在260℃保持20min后都不见出峰[img]https://simg.instrument.com.cn/bbs/images/brow/em49.gif[/img]图中为2mg/L马拉硫磷单标进样的色谱图,2mg/L对硫磷单标进样的色谱图出峰时间、峰形基本与之类似。[img=,690,232]https://ng1.17img.cn/bbsfiles/images/2021/12/202112301058143478_7214_1916553_3.png!w690x232.jpg[/img]
前言: 近年来,饮水安全越来越受到世界的关注。现代农业生产中农药的广泛应用已经成为水污染的主要来源之一。喷洒后的农药可以分布于空气、水和土壤中,而直接施用于土壤的农药则会流入附近的水体或渗透到地下水中。有机磷农药作为世界上生产和使用最多的农药,正通过各种途径污染水源。目前,水中痕量有机磷农药的分析方法正在不断改进,气相色谱质谱联用仪具有检出限低,定性能力强、定量准确等特点,已广泛应用于水中痕量农药残留的检测。仪器简介GC-MS 6800是天瑞仪器精心打造的一款高性价比气相色谱质谱联用仪,具有完全的自主知识产权,拥有多项专利技术,可广泛应用于环境保护中农药残留的测定。http://www.skyray-instrument.com/cn/images/2015/0420/1.jpg实验原理用有机试剂萃取水中可能残留的有机磷农药,用GCMS进行测定,以保留时间和标准质谱图比对进行定性,以各物质定量离子峰面积及外标法进行定量。在国标GB3838-2002中,对六种有机磷的限量分别为:对硫磷3ug/L,甲基对硫磷2ug/L,马拉硫磷50 ug/L,乐果80 ug/L ,内吸磷30 ug/L,敌敌畏50 ug/L。一、实验部分1.1 试剂及仪器二氯甲烷:色谱纯;无水硫酸钠:分析纯;6种有机磷标准品:各100 ug/mL; 气相色谱质谱联用仪(GCMS6800);旋转蒸发仪;氮吹仪1.2 6种有机磷检测仪器条件色谱柱:DB-5MS(30m*0.25mm*0.25um),柱流量:1.0mL/min进样口温度:270℃,气质接口温度:260℃,离子源:260℃ 柱箱温度:50℃(1min) 20℃/min 200℃(7min)30℃/min 280℃(1min) 进样量:1.0uL(不分流),不分流时间:1.0min,分流比:10:1溶剂切除时间:4min检测器电压:-1000V,电流:100uA扫描方式:SIM扫描 SIM扫描时分段扫描参数设置:扫描起始时间/min持续时间/min扫描离子/amu44109 145 185 2208487 88 93 125 143 170 229122200 233 246 26314293 97 125 173 235http://www.skyray-instrument.com/cn/images/2015/0420/2.jpg图1 6种有机磷(0.5ug/mL)SIM扫描图1.3 实验步骤取水样250mL置于500mL分液漏斗中,加入2g无水硫酸钠溶解后,用20mL二氯甲烷萃取,充分振荡5分钟,静置分层,收集有机相;用20mL二氯甲烷再萃取1次,合并有机相;有机溶剂经盛有2g无水硫酸钠的玻璃漏斗脱水后旋转蒸发至约5mL,氮吹至近干,用正己烷准确定容至2.0mL,过0.22um有机滤膜后测试。二、数据与讨论2.1 精密度和重复性实验取浓度为0.5 μg/mL6种有机磷标准工作溶液,平行进样7针,考察各组分出峰时间及峰面积的相对标准偏差,其相对标准偏差如下表所示:名称保留时间RSD/%峰面积RSD/%敌敌畏0.093.43内吸磷0.065.38乐果0.057.22甲基对硫磷0.086.25马拉硫磷0.094.77对硫磷0.094.39从上表可以看出,六种有机磷的峰面积相对标准偏差<8%,保留时间相对标准偏差<0.1%,结果表明本方法的保留时间重复性和精密度良好。2.2 线性关系和检出限在本方法确定的实验条件下,移取1.0uL 3.4配置好的有机磷标准工作溶液,进样检测,将所得各个组分的峰面积与相应浓度进行线性回归,得各个组分的线性回归方程。同时计算各个组分的检出限,相关结果见下表:物质名称线性方程方法检出限(μg/L)敌敌畏y = 1.6E+05x – 600 R2 = 0.99910.1内吸磷y = 78038x – 3242 R2 = 0.99770.2乐果y = 1.2E+5x – 568 R2 =
谁有环氧乙烷残留量分析—比色分析法德操作步骤和作业指导书啊?
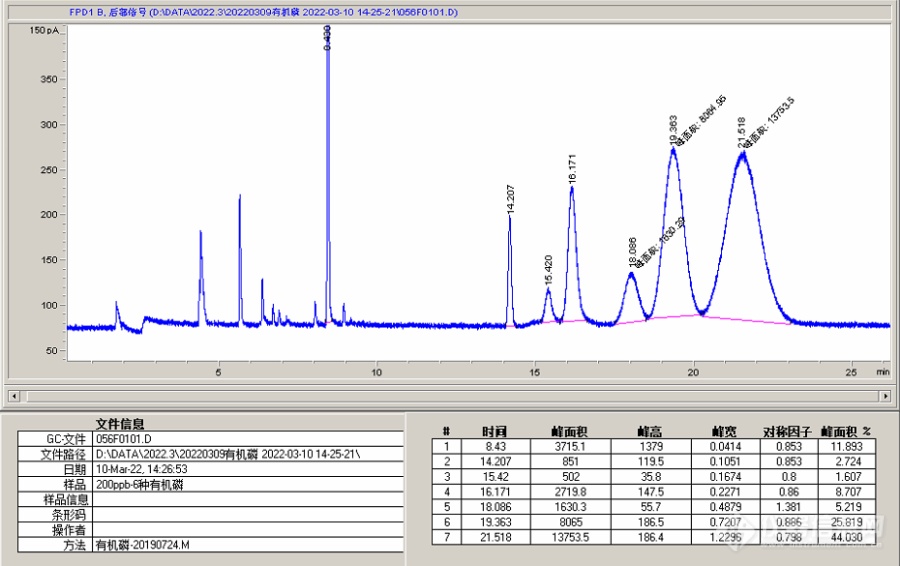
仪器是安捷伦7890A,PFD检测器,色谱柱是DB-1701,30m×0.25mm×0.25μm,柱流量1.5ml/min,进样口220℃,不分流进样,检测器氢气70ml/min,空气100ml/min,尾吹60ml/min,升温程序参考的之前版上高手发过的,80℃保持1min,以20℃/min升温至130℃,再以5℃/min升温至200℃,最后以15℃/min升温至250℃,保持11min。分析6种有机磷混标,包括敌敌畏、乐果、内吸磷、甲基对硫磷、马拉硫磷、对硫磷,除了敌敌畏和内吸磷以外,其他物质峰形都很难看,峰宽矮胖,马拉硫磷与对硫磷也分不开(我在[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]上HP-5MS柱子同样升温程序可以完美分开这两种物质)。也试过其他升温程序和柱流量(1~2mL/min),包括之前换过其他HP-5等柱子,始终出现类似峰形无法分开马拉硫磷与对硫磷,怀疑是否这台[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]的FPD检测器有问题?请教各位高手指点[img=,690,433]https://ng1.17img.cn/bbsfiles/images/2022/03/202203292320019341_7010_1916553_3.png!w690x433.jpg[/img]
在GC 分析时,经常容易出现检测结果偏差,使得定量结果不能真实反映样品中农药有机磷的残留。那么在分析此类有机磷农残时,应注意什么?如何有效的避免活性点对农药的吸附?系统的活性吸附点主要有:1. 进样口衬管和玻璃棉对一些有机磷农药有吸附2. 样品瓶、进样针的吸附3. 色谱柱的吸附解决方案:1. 新换的衬管和玻璃棉,可以再测样前多进几针基质样品或高浓度的标准溶液来饱和,以掩盖活性点2. 增加样品分析保护剂的使用,如山梨醇3.使用惰性衬管和样品瓶,或用硅烷化试剂钝化衬管、进样瓶上的活性位点4. 使用专用农药残留色谱柱检测5. 采用基质空白溶液配制标准溶液来定性定量
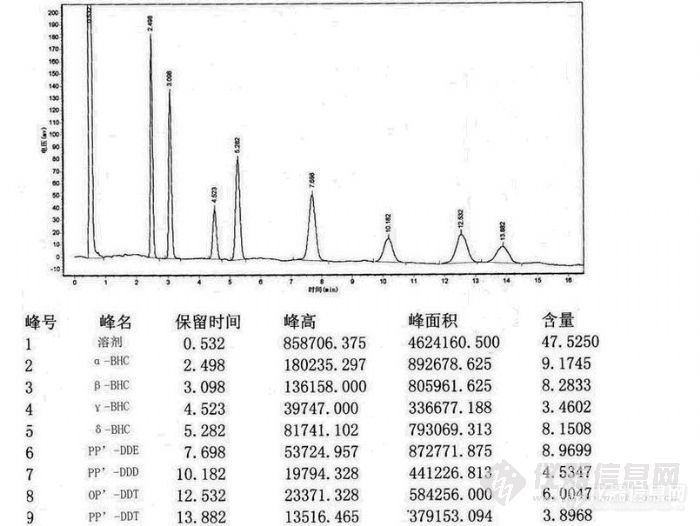
有机氯 有机磷农药残留气相色谱法测定摘要 有机氯、有机磷农药化合物品种多,防治对象和应用范围广,是我国目前使用量最大的农药。粮食、蔬菜、水果、饮料、奶制品、茶叶等食品中农药残留及工业污染给人类的生存环境带来了极大的负担,农产品及农药的滥用所造成的食品中毒事件时有发生。为此南京科捷分析仪器应用研究所采用GC5890气相色谱仪,建立了有机氯类、农药残留的气相色谱(GC)分离、电子捕获检测器(ECD)测定,以及有机磷农药残留的GC分离、火焰光度检测器(FPD)测定方法。提出了用双进样口、双检测器、双分离柱在一台气相色谱仪实现多种有机氯、有机磷等农药残留组分测定的方法,缩短了分析时间,降低了相关实验室应用该方法时的硬件成本。关键词 食品 农产品 有机氯农药、有机磷农药 毛细管气相色谱1.有机磷气相色谱图http://ng1.17img.cn/bbsfiles/images/2011/04/201104211651_290231_2242538_3.jpg2.有机氯气相色谱图http://ng1.17img.cn/bbsfiles/images/2011/04/201104211651_290232_2242538_3.jpg3.方法应用范围本方法可应用在粮食、蔬菜、饮料、奶制品、茶叶、农药残留检测中以及在生产和使用这些农药的过程中的农药检测,残留的农药会将污染延伸到环境水体中,对地表水、地下水造成污染。有机氯农药六六六和滴滴涕以及土壤以及食品等样品中农药残留检测,蔬菜和水果中敌敌畏、甲拌磷、乐果、对氧磷、对硫磷、甲基对硫磷、杀螟硫磷、异柳磷、乙硫磷、喹硫磷、伏杀硫磷、敌百虫、氧乐果、磷胺、甲基嘧啶磷、马拉硫磷、辛硫磷、亚胺硫磷、甲胺磷、二嗪磷、甲基毒死蜱、毒死蜱、倍硫磷、杀扑磷、乙酰甲胺磷、胺丙畏、久效磷、百治磷、苯硫磷、地虫硫磷、速灭磷、皮蝇磷、治螟磷、三唑磷、硫环磷、甲基硫环磷、益棉磷、保棉磷、蝇毒磷、地毒磷、灭菌磷、乙拌磷、除线磷、嘧啶磷、溴硫磷、乙基溴硫磷、丙溴磷、二溴磷、吡菌磷、特丁硫磷、水胺硫膦、灭线磷、伐灭膦、杀虫畏54种有机磷类农药多残留气相色谱的检测方法。食品包括与人关系最密切的8大类:粮食,蔬菜,水果,肉禽,水产,植物没,蛋和乳。本部分适用于蔬菜和水果中上述54种农药残留量的检测。综上所述,本研究建立的方法能满足现代实验室大批量样品检测的需求,实现了快速、简单、便宜、减少污染且能有效去除杂质的目的。4.农药残留专用气相色谱仪配置检测项目有机氯、有机磷农药检测色谱仪器型号GC5890型色谱仪 配有ECD、FPD检测器毛细管色谱柱30*0.32*0.53专用柱两根脱氧管1支色谱工作站N2000(电脑1台自备)氮氢空发生器 HGT300E 1台或高纯氮、氢气、空气钢瓶各一瓶实验单位南京科捷分析仪器应用研究所
蔬菜中有机磷农药多残留分析:方法检出限的对比研究.pdf
农药残留分析技术进展概况本文转摘时未向原作者及杂志申请,如有版权问题请立即与色谱网联系! 农药 第37卷第2期(1998)农药残留分析技术进展概况冯秀琼(南开大学元素有机化学研究所,天津300071)摘 要:本文概述了近几年国外农药残留分析在样品制备及测定中取得的重要进展。关键词:农药 残留分析 在过去几年里,农药残留分析技术在许多领域都取得了重要进步。由于农药品种和用量的不断增加,农药作为对环境和食品的重要污染源之一,越来越受到各国政府和公众的关注。其分析技术的提高也受到高度重视。新的分析方法,特别是多种类型农药的多残留分析方法、同类型农药的多残留分析方法以及新的单个农药在多种试样中的分析方法都取得了重要进展〔1〕。近几年来,在残留分析方法的研究和应用中可看出有如下几点趋势。1 待测组分越来越复杂。除原来农药的几大类型外,新开发的农药包括了各种各样新的结构基团。其中不少化合物,或分子量大,或极性极强,或对热不稳定。除母体化合物外,代谢产物(有活性或有毒性)的分析更加受到重视。代谢产物常留分析技术提出了更高要求。2 由于人类对环境和食品质量要求越来越高,以及检测技术的不断进步,使农药残留检出灵敏度进一步提高。目前,水样一般测到ppb至ppt水平,检测到ppq水平也时有报道。作物、饲料、土壤和其他生物样品一般在较低的ppm到ppb水平。新的食品残留检测下限必须低于最大允许残留量。在定量分析方面,为了提高方法的精密度和准确度,越来越多的方法采用内标法代替过去常用的外标法。3 一向十分费事的样品预处理工作,正向着省时、省力、廉价、减少溶剂、减少对环境的污染、微型化和自动化方向发展。如固相萃取(SPE)、超临界液体萃取(SFE)、微波辅助萃取、样品(matrix)固相分散萃取、自动索氏萃取,在线HPLC萃取等都取得很大进步。尤其是SPE的应用已相当普遍。SPE克服4 了液/液萃取(LLE)及一般柱层析的缺点。与LLE相比,可节省时间和溶剂约90%〔2〕,减少杂质的引入,对操作者更安全,重现性好,可避免LLE中乳化现象的产生。SPE实际上是色谱技术应用的另一种形式。根据柱中填料大体可分为吸附型(如硅胶、大孔吸附树脂等)、分配型(如C8、C18、苯基柱等)和离子交换型。应用中遵守的原理基本上和一般色谱相同5 。对水样和其他液体样品(如牛奶、饮料、血浆、尿等),在选择合适的萃取柱和洗脱液及其他优化条件后,可使萃取、富集、净化一步完成。然后直接进行GC或HPLC分析。Shepherd等〔3〕用C18柱萃取水中莠去津,用1 5小时可处理12个样品。10毫升水样,检出灵敏度可达0 05ppb。Holland等〔4〕用C18柱筛选葡萄酒中74种农药,方法快速,重复6 性也好。对固体样品,SPE主要用于净化。提取仍须先用溶剂萃取。根据需要,净化时可用单柱也可用多个柱串联使用。Schenck等〔5〕用C18和弗罗里硅土SPE柱串联,净化鸡蛋中有机氯及有机磷的乙腈提取液,以GC/ECD/FPD测定,可减少90%的溶剂消耗和85%的废液处理。另一种增强净化效力的方式是用混合型柱。它可利用多种界面效应来分离和纯化分析组分。Mills等〔6〕比较了两种不7 同8 的混合柱在环境及临床化学中的应用。一种是将各种不9 同10 填料经机械混合装入同11 一SPE柱。另一种是将这些带不12 同13 性质官能团的物质,化学键合到同14 一骨架树脂上(copolymerized),将其制成SPE柱。实验表明,极性的均三氮苯类化合物在机械混合的SPE柱上,回收率低于多聚键合型SPE柱。原因可能是由于硅胶粒子的空间隔离,降低了某些活性基团的作用。SPE的另一优点是容易实现预处理的自动化。国外已推出商品化的自动固相萃取装置ASPEC(AutomatedSolid-phaseEx tractionCleanup)。将其与HPLC在线结合可实现许多农药残留的全自动分析。SFE在农药残留分析中应用的报道也在增加。SFE是以超临界流体代替各种溶剂来萃取样品中待测组分的萃取方法。其优点是基本上避免了使用有机溶剂,简单快速、能选择性地萃取待测组分并将干扰成分减到最小程度、减少一般提取方法所占用的玻璃仪器及实验室空间,能实现操作自动化。目前最常用的超临界流体为二氧化碳。为了改进对极性农药的萃取效力,常在二氧化碳中加入少量极性溶剂,如甲醇等。SFE有在线(on-line)和离线(off-line)两种应用形式。在线方式常与GC、超临界流体色谱(SFC)、HPLC或凝胶渗透色谱(GPC)等结合,实现全自动分析过程。离线的SFE,则在萃取不同样品数量和类型以及选择后面的测定方式上有更多的灵活性。Snyder等〔7〕比较了SFE与经典的超声波和索氏提取器对农药的萃取情况。认为三种方法中,SFE精密度最好,比其他两种方法省时省力。Furton〔8〕研究了影响SFE效力的各种因素。指出:不仅温度、压力(密度),而且不同样品种类、萃取容器以及接收体积的变化都明显地影响萃取效力。Lehotay等〔9〕提出了应用Hydromatrix(HMX,一种小粒状硅藻土)到蔬菜水果多残留分析的样品制备中,解决了SFE要求样品的水分含量不能太高,样品用量小(一般1~3克)又必须保证样品的均一性、代表性和方法的准确度等问题。由于SFE需要一定的特殊设备,使目前广泛应用受到限制。但由于它具有许多独特的优点,其应用前景是很光明的。
各位大虾:小弟最近忙着做农药残留(浓缩水果汁中的毒死蜱、马拉硫磷、溴氰菊酯、氯氟氰菊酯)的能力验证,但是实验过程中出现了问题,不知道错在哪里请大家帮助,我按照NY/T761-2004方法作的,最后结果很吓人,马拉硫磷的回收率分别为360%和430%。试验过程如下:称5克样品加入1ug/mL的毒死蜱与马拉硫磷的混标3mL,最后毒死蜱的回收率分别为80%和94%,但是马拉硫磷的回收率分别为360%和430%,而我加入的是混标不可能两种农药加标出错,请问这是什麽原因?请大家帮助我找找原因,只要回帖就送分!!很着急,5号上报结果啊!!
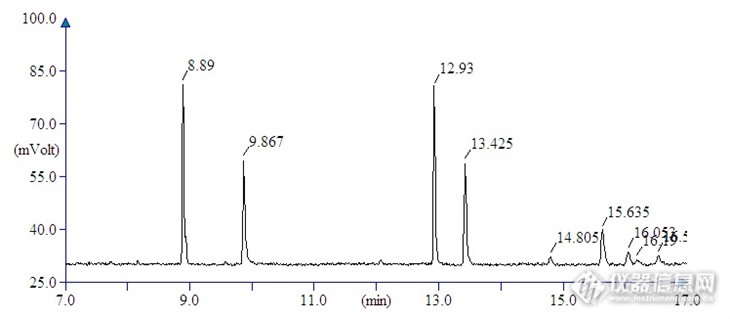
【生活中的仪器分析】活动原创作品:食品安全——果蔬中农药残留及重金属含量检测摘要:通过对固相微萃取(SPME)条件进行优化,建立库尔勒香梨中有机磷农药残留量的快速检测方法。使用顶空-固相微萃取技术提取目标物,采用气相色谱仪检测香梨中9种有机磷农药组分。实验表明在0.05~1.0μg/mL范围内线性回归好,相关系数r大于0.99,样品加标回收率为71.8~93.5%,相对标准偏差为2.14~5.83%。与传统农药残留检测方法相比,具有快速、无溶剂萃取、简便、准确、重现新较好的特点,可作为库尔勒香梨中农药残留快速检测的分析方法。关键词:固相微萃取;有机磷农药;残留量;气相色谱;库尔勒香梨库尔勒香梨我国著名的梨品种,属蔷薇科、梨属中的白梨,已有1500多年的栽培历史,原产地为新疆库尔勒地区,目前栽培面积仍在不断扩大。该品种结果早、品质优,是新疆的名特水果之一,产品销售世界各地,已成为新疆地区支柱产业之一。目前库尔勒香梨主要病虫害有中国梨喀木虱、橄榄片盾、香梨茎蜂、香梨优斑螟、黄化病和腐烂病等,在种植过程中虽然控制农药的使用,但对于农药残留量仍然是检测工作者一直研究的任务。固相微萃取(SPME)技术是20世纪90年代初发明的一种高效、快捷的样品前处理技术,克服了传统样品前处理消耗大量溶剂,操作复杂等缺点,具有萃取、浓缩、进样一体化的优点。近几年随着固相微萃取头材质的不断改进与发展,已逐渐开始应用于基质较为复杂的样品前处理。本文通过优化固相微萃取的试验条件,对库尔勒香梨中添加的有机磷类农药进行萃取,采用气相色谱法检测农药残留量,建立了9种有机磷农药的多残留快速检测方法。1 实验部分1.1 仪器、试剂与材料气相色谱仪(Thermo Fisher赛默飞世尔科技,Trace2000);分析天平(Mettle-Toledo 梅特勒-托利多);匀浆机(IKA仪科);固相微萃取装置(美国SUPELCO公司)、聚二甲基硅氧烷萃取头(SPME-S-01 PDMS上海新拓仪器公司)9种有机磷农药标准品:敌敌畏、甲胺磷、甲拌磷、二嗪磷、乐果、毒死蜱、甲基对硫磷、马拉硫磷、杀螟硫磷(1000 µg/mL,农业部环境质量监督检验测试中心)试验材料:库尔勒香梨种植基地。1.2 标准溶液配制准确配制9种有机磷农药标准品,用丙酮定容,全部配成20 mg/L的标准储备液,吸取以上标准储备液适量,混合后稀释至质量浓度为0.05、0.1、0.2、0.5、1.0 mg/L系列标准溶液。1.3 实验方法1.3.1样品前处理将库尔勒香梨2kg先切成块后等份取出代表部分,在食物破碎机搅碎至浆状。根据试验要求准确称取样品,加入已知量的混合农药标准,均匀混合后备用。1.3.2 SPME[/size
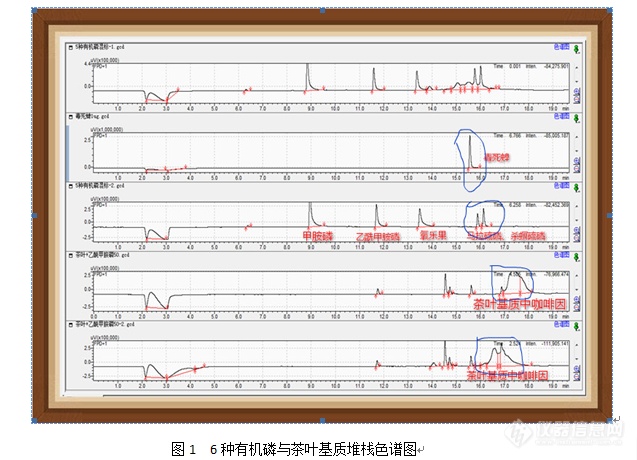
茶叶中咖啡因对有机磷类农药残留测定的影响摘要:茶叶中经常要检测有机磷类农药(甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷),由于茶叶样品基质富含咖啡因的特殊性,本文主要阐述了用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定茶叶中这些有机磷时,咖啡因对这些农药测定的影响,及如何准确地测定这些有机磷农药残留。关键词:茶叶;咖啡因;有机磷农药;[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法;准确定量前言:茶叶是深受人们喜爱的一种饮料这一,最近来,滥用农药的现象也越来越严重,GB 2763-2016[sup][/sup]也对茶叶中各种农药残留的限量作了具体的规定。众所周知,茶叶富含芳香族化合物、多酚和咖啡因[sup][/sup]。咖啡因等化合物会在前处理的萃取过程中与农药残留一起被萃取出来,如果没有有效地去除,将会对目标农药(毒死蜱、马拉硫磷、杀螟硫磷)的准确定量造成影响,同时有机磷类农药(甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷)用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]分析时通常会一起进行净化前处理,一起上机用FPD检测器上机分析。这又给部分农药残留(甲胺磷、乙酰甲胺磷、氧乐果)的准确定量造成了失误。实验仪器:岛津[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] -2010plus(配FPD); 色谱柱:RTX-170130m*0.25mm,0.25um[align=left]仪器条件:进样口温度:220 ℃ 检测器温度:230 ℃[/align]程序升温:[color=black]60℃[/color][color=black]([/color][color=black]1 min[/color][color=black])[/color][sup][color=black]20[b]℃[/b]/min [/color][/sup][color=black]150℃ [sup]15[b]℃[/b]/min[/sup] 230℃ [sup]25[b]℃[/b]/min[/sup] 280℃[/color][color=black]([/color][color=black]5 min[/color][color=black])[/color][color=black][/color]标准品:甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷、[color=red]咖啡因[/color]样品处理(方法一):依SN/T1950-2007[sup][/sup]对茶叶进行处理,同时进行加标实验。提取:取样1.0g(±0.01g)于50mL塑料离心管中,加入1mL饱和氯化钠水溶液浸泡十分钟左右,加入15mL乙酸乙酯先均质,再加入一勺无水硫酸钠和2勺无水硫酸镁均质30s,用15mL乙酸乙酯洗均质头合并提取液,盖上盖子振摇一会,超声5min。把提取液和残渣一起直接过加有2勺无水硫酸镁的漏斗入鸡心瓶中,2×5mL乙酸乙酯洗离心管,振摇,合并提取液于鸡心瓶中。再用20mL乙酸乙酯冲洗漏斗上的残渣合并洗液,35℃旋转蒸发至剩2mL左右,待净化。净化:10mL丙酮+正己烷(1+1, V+V)先活化TPT(10mL,2g)柱子(填料上加1cm左右高的无水硫酸钠)下接15mL玻璃离心管,将上述大约剩2mL左右的乙酸乙酯先吸出直接过活化后的TPT柱,再用丙酮+正己烷(1+1, V+V)2.5 mL×2次洗涤鸡心瓶(必要时超声波),洗液继续过TPT柱子,加丙酮+正己烷(1+1, V+V)继续淋洗柱子共收集12mL淋洗液,35℃左右氮气吹干,丙酮+正己烷(1+1, V+V)定容1mL上机测试。 混标(甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷)与茶叶基质堆栈色谱图,如图1所示:[img=,637,460]https://ng1.17img.cn/bbsfiles/images/2018/10/201810161355391717_7304_2166779_3.png!w637x460.jpg[/img]从图1中可以看出:茶叶基质(空白茶叶)在毒死蜱、马拉硫磷、杀螟硫磷出峰位置附近出现很大的坡(后经确认是茶叶基质中的咖啡因:[color=red]前处理浓缩定容时会产生白色絮状物,此白色絮状物就是茶叶基质中的咖啡因析出产生的[/color]),给茶叶中毒死蜱、马拉硫磷、杀螟硫磷准确定量带来了严重影响。[color=red]且[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]进了茶叶基质中的咖啡因后,要连续进十几针的丙酮空白针,此坡才能消失,才不会对后续的出峰位置的物质定量产生干扰。[/color][color=red][/color]样品处理(方法二):(目标:要去除茶叶基质中的咖咖啡因。)[color=red][/color][align=left]提取:取样2.0g(±0.01g)于50mL塑料离心管中,加入2mL去离子水、20mL丙酮+正己烷(1+1,V+V)溶液和一勺无水硫酸钠,旋紧离心管盖,涡旋1min后超声30min,超声期间每5min振摇一次,4000 r/min离心5min,待净化。[/align]净化: 移取5.0mL上清液至15mL离心管中,35℃下氮气吹干,加入2.5mL正己烷涡旋使样品溶解,[color=red]再加入[/color][color=red]2.5mL[/color][color=red]饱和氯化钠水溶液继续涡旋[/color][color=red]30s[/color]说明:[color=red]用饱和氯化钠水溶液和正己烷分配可去除水溶性杂质及咖啡因)[/color]后2000 r/min离心1min,取出正己烷层,剩余溶液中加入2.5mL正己烷再提出一次。合并正己烷层过经5mL丙酮+正己烷(1+1,V+V)活化上填1cm高无水硫酸钠的Carb/PSA柱(0.5g/ 6mL),用8mL丙酮+正己烷(1+1,V+V)继续洗脱,共收集洗脱液13mL于15mL刻度玻璃离心管中,35℃水浴氮气吹干,用正己烷定容1mL上[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-FPD检测。同时进行加标处理(加标量:加入200ng/mL的有机磷混标1mL,与样品同时同样处理,相当于最终上机浓度为40ng/mL)。从图1中可以看出:茶叶基质(空白茶叶)在毒死蜱、马拉硫磷、杀螟硫磷出峰位置附近出现很大的坡(后经确认是茶叶基质中的咖啡因:[color=red]前处理浓缩定容时会产生白色絮状物,此白色絮状物就是茶叶基质中的咖啡因析出产生的[/color]),给茶叶中毒死蜱、马拉硫磷、杀螟硫磷准确定量带来了严重影响。[color=red]且[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]进了茶叶基质中的咖啡因后,要连续进十几针的丙酮空白针,此坡才能消失,才不会对后续的该出峰位置附近的物质定量产生干扰。[/color][color=red][/color][img=,690,415]https://ng1.17img.cn/bbsfiles/images/2018/10/201810161358058714_1404_2166779_3.png!w690x415.jpg[/img][img=,663,266]https://ng1.17img.cn/bbsfiles/images/2018/10/201810161358110432_4139_2166779_3.png!w663x266.jpg[/img] 表1 用方法二处理各有机磷的回收率从图2及表1可以看出:使用方法二处理([color=red]用饱和氯化钠水溶液和正己烷分配可去除水溶性杂质及咖啡因:茶叶样品基质中的咖啡因大谷峰[/color][color=red]已消失[/color])毒死蜱、马拉硫磷、杀螟硫磷可以准确地给予定量了,然而甲胺磷、乙酰甲胺、氧乐果却也同时会与咖啡因从正己烷层进入饱和的氯化钠层而被洗脱除去。因此用方法二来处理茶叶基质的方法可以很好地去除咖啡因基质,适用于检测毒死蜱、马拉硫磷、杀螟硫磷,不适用于同时要检测:甲胺磷、乙酰甲胺、氧乐果这三种有机磷的净化方法。(必要时)样品处理(方法三):适用于茶叶中甲胺磷,乙酰甲胺磷的净化方法。取干样0.5g于50mL塑料离心管中加2mL水,放置至少30分钟。加入20mL乙酸乙酯和10g无水硫酸钠,均质0.5min,用10mL乙酸乙酯清洗均质头,合并提取液4000r/min离心5分钟,上清液经装有10g无水硫酸钠的漏斗脱水于鸡心瓶中,残渣再用20mL乙酸乙酯涡漩洗涤,4000r/min离心5分钟,上清液并入鸡心瓶中,再用10mL乙酸乙酯冲洗漏斗上的残渣合并洗液35℃浓缩至干。[b]净化:用乙酸乙酯2mL×3次漩涡振荡洗涤鸡心瓶,过经5 mL乙酸乙酯活化过的上填1cm高无水硫酸钠的[color=red]硅胶柱([/color][color=red]LC-Si0.5g / 6mL[/color][color=red]硅胶固相萃取小柱)[/color],待洗涤液流完接近硅胶柱中无水硫酸钠顶端时,再用乙酸乙酯洗, [color=red]弃去前[/color][color=red]9 mL[/color][color=red]洗液,[/color]继续用乙酸乙酯洗并收集15mL于15mL刻度玻璃离心管中,35℃水浴氮气吹干,用1mL乙酸乙酯(色谱纯)定容,上机测试。[/b]说明:[align=left]1)提取剂乙酸乙酯极性较强,能有效地将食品中的甲胺磷提取出来,且样品基质中的共提取杂质相对较少;使用乙腈提取时共提取杂质稍多。使用无水硫酸钠一方面配合均质器研磨,增加分散的均匀度,加强溶剂与样品的接触,提高提取效率,另一方面可以将样品中的水分以结晶水的方式除去,既不对甲胺磷产生吸附,又避免甲胺磷溶于水导致回收率的损失。配合超声波辅助提取,进一步提高提取效率。实验时需先加乙酸乙酯后加无水硫酸钠,以免无水硫酸钠结块导致均质困难。由于本实验对水分的残留较为敏感,提取时要尽可能将水分除干净,否则影响刭PSA填料的吸附性能,净化效果变差; 影响到Lc—si柱的吸附性能,可能改变柱上的洗脱规律,甚至导致实验的失败。可将无水硫酸钠在 650℃焙烧约4 h后备用,必要时增加用量。[/align] 作用机理研究: LC—Si柱/乙酸乙酯选择洗脱净化是本前处理方法的核心步骤。Lc-Si柱是经典的正相同相萃取柱,基于正相原理使杂质吸附于柱上,目标化合物随溶剂洗出,一般使用中等偏弱极性的溶剂洗脱。乙酸乙酯是极性较强的溶剂,在这种介质中,大量中强极性及弱极性杂质均难以保留而与目标化合物一起洗出,导致净化步骤失效。本实验正利用了在乙酸乙酯介质中大量杂质均难以保留的特点,[color=red]使其先于甲胺磷流出[/color][color=red]LC[/color][color=red]—[/color][color=red]Si[/color][color=red]柱,然后甲胺磷在特定阶段流出再与仍然吸附于柱上的强极性杂质分离,达到了良好的净化效果。[/color][color=red][/color]此法适用于各种复杂基质中的甲胺磷、乙酰甲胺磷的检测(如果没有经过[color=red]LC[/color][color=red]—[/color][color=red]Si[/color][color=red]柱净化处理且洗脱液的前[/color][color=red]9mL[/color][color=red]洗脱液要弃去,有的样品基质会再甲胺磷、乙酰甲胺磷的出峰位置如茶叶基质中的咖啡因一样出现很大的坡峰,给甲胺磷、乙酰甲胺磷的定量造成干扰[/color])净化处理。结论:综合所述:茶叶中甲胺磷、乙酰甲胺磷、氧乐果、毒死蜱、马拉硫磷、杀螟硫磷中有机磷的检测要分成两种不同的净化方法进行处理。样品处理(方法一)适用于茶叶中甲胺磷、乙酰甲胺磷、氧乐果的净化处理方法,因为此种净化处理方法会同时萃取出茶叶基质中的咖啡因,茶叶富含咖啡因基质,在[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]中难以消除,会给在咖啡因出峰位置相近的化合物的定量产生干扰(如:毒死蜱、马拉硫磷、杀螟硫磷等)样品处理(方法二)适用于茶叶中毒死蜱、马拉硫磷、杀螟硫磷的测定,如果茶叶样品没有检测甲胺磷、乙酰甲胺磷、氧乐果时,尽量采用此种的净化处理方法,茶叶中的咖啡因用饱和氯化钠水溶液和正己烷液液分配处理后,水溶性杂质、咖啡因及农药(甲胺磷、乙酰甲胺磷、氧乐果、久效磷)会进入饱和氯化钠水溶液层,而其它的农药则留在正己烷层,茶叶样品基质中的咖啡因大谷峰已消失。样品处理(方法三)是遇到个别复杂基质(如有次我们要检测到含茶制品:速溶麦香红茶)时,必要时采用的净化处理方法,过硅胶柱(LC-Si柱)用乙酸乙酯洗脱且弃去前9mL洗脱,收集后面的15mL左右的洗脱液可保证基质中的杂质在前面9mL时洗出弃去,而目标物(甲胺磷、乙酰甲胺磷)被准确定量地收集到。[align=left][/align]
韭菜样品中有大量含硫化合物,干扰选择型检测器(如FPD)的测定。虽然有学者结合凝胶渗透色谱(GPC)和固相萃取(SPE)用于净化并有效地消除了干扰,但该方法费时,费溶剂;也有学者将样品用微波预处理,显著地降低了干扰,尽管比前一方法简便,但该方法包括两个浓缩步骤,因而也需要较长的样品处理时间。 气相色谱-质谱(GC-MS)有一定的选择性,但不能消除韭菜中含硫化合物的干扰。有人用气相色谱-离子阱质谱检测蔬菜中多种农药残留,但离子阱质谱不适于定量分析。有学者用GC/MS/MS分析绿叶蔬菜中的多种农药残留,但韭菜不同于一般的绿叶蔬菜。 本文旨在介绍一种检测含硫蔬菜中的有机磷农药(敌敌畏、甲胺磷、乙酰甲胺磷、二嗪磷、异稻瘟净、久效磷、甲基毒死蜱、乐果、甲基嘧啶磷、毒死蜱、甲基对硫磷、马拉硫磷、杀螟硫磷、倍硫磷、对硫磷、毒虫畏、喹硫磷、丙硫磷、丙溴磷)残留的方法,通过修改的QuEChERS方法进行样品前处理,GC-MS/MS检测。本方法具有快速、简便、准确、灵敏等优点。本文首次用复杂的样品验证GC-MS/MS的选择性。 方法提要:称取样品3 g→微波加热(640 W,10 s)→冰水浴冷却→加水3mL,乙腈(含0.1%醋酸)15 mL,混匀→加无水硫酸镁4 g,醋酸钠2 g,混匀→离心→移取上清液4 mL→加入100 mg PSA,40 mg GCB ,600 mg MgSO4,混匀→离心→取上清液2 mL,N2吹干→加1mL正己烷定容→GC-MS/MS分析。 方法线性范围:0.002-0.20 ug/mL,R2:0.9897-0.9997 方法回收率:81.0-109.4%.添加浓度为:10、50、100 ppb 方法精密度:RSD(n=3):1.3%-10.4%. 方法检测限LOD:0.07–1.5 ug/kg;方法定量限:LOQ:0.25 to 5 lg/kg。 LOD比气相色谱-离子阱质谱方法(文献方法)低10倍甚至更多。 尽管不经微波处理时本底要高些,但仍然有良好的选择性,基本无干扰。但较脏的样品污染进样口和色谱柱并降低它们的寿命,因此建议使用微波处理。
随着兽药的种类和应用规模剧增,人们对兽药残留问题的日益关注以及国际间贸易等原因,使兽药残留分析对象、样本数量和测定难度大大增加,迫切需要发展简便、快速、灵敏,并能同时处理测定大批量样品的兽药残留分析技术。 传统的波谱、色谱等理化分析手段难以适应兽药残留分析的要求。 近年来在兽药残留分析领域所取得的重要进展或发展趋势主要有以下方面: ①样品分离纯化技术(提取和净化方法)的简单化、微型化和自动化,提高了提取或净化效率及自动化水平。如固相萃取法(SPE)、固相微萃取(SPME),超临界流体萃取(SFE)、微波萃取法(MAE)、免疫亲和色谱(IAC)技术、基质固相分散(MSPD)技术,凝胶渗透色谱(GPC)净化,分子印迹技术等。②在定量分析上的新技术包括毛细管电泳、超临界流体色谱、液相色谱-质谱联用技术、免疫分析技术和生物传感器等。下面将这些研究进展作一综述。一、样品分离纯化技术 动物性食品中兽药残留的特点是样品中残留物水平很低,样品基质复杂,干扰物质多,不易从样品中分离、纯化残留物。因此兽药残留分析是复杂生物样品基质中痕量组分的分析技术,样品的分离纯化是兽药残留分析中最费时和劳动强度最大的步骤。传统的样品制备技术如液- 液分配等仍在广泛使用,同时一些新的样品分离纯化技术也不断被引入到兽药残留分析中。免疫亲和色谱技术、分子印迹技术、基质固相分散技术、超临界流体萃取(SFE)等是残留分析中最有效的分离纯化方法,目前是兽药残留分析领域中的研究热点。
梁贵平 侯震 摘 要:采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,以p,p'-DDE为内标物,对马拉硫磷水胺硫磷悬乳剂进行测定.两种有效成分马拉硫磷、水胺硫磷在同一条件下测定,方法回收率均在99.0%~100.6%之间,变异系数均小于0.40%.方法简便、快速、准确且重现性好.
做农残的应该都深有体会,复杂基质样品中的甲胺磷残留分析是个相当棘手的问题。相对来说葱还算不太复杂的样品,最可怕的是熏硫处理过的干香菇、调味粉,简直是无解了,还有大蒜,真是头疼。。。。主要还是因为前处理目前没有什么好的办法。正头疼中,一天突发奇想,哈哈,搞了一个前处理方法,很好用,速度很快,成本也不高,跟大家分享。主要有2个优势:1、含硫基干扰物质(挥发性硫化物)如葱蒜类样品、熏硫产品也可以用GC-FPD做了,没干扰。2、通吃各种不同基质样品,验证了黑胡椒粉、茶叶、干香菇、小麦、葱姜蒜、韭菜、烤鳗、黄鱼、菠菜、苹果等基质,目前暂时没有发现不能做的样品基质。这点在农残检测中很少见。 其它的看附件啦,下个月在分析化学刊登出来,解释得比较详细了,包括[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](GC)和液相色谱-串联质谱(UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS)方法:正相硅胶/选择洗脱-[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法、液相色谱-质谱法检测食品中甲胺磷残留及其作用机理研究。大家试一下,有什么问题可以跟帖,互相交流,呵呵[img]http://bbs.instrument.com.cn/images/affix.gif[/img][url=http://bbs.instrument.com.cn/download.asp?ID=195647]食品中甲胺磷残留分析方法.pdf[/url]
用NY761-2008对蔬菜中农药残留进行检测,气相条件是FPD检测器,色谱柱是DB1701,如果有蔬菜超标,要求用不同色谱柱检测,那么用什么型号的柱子呢?检测19种农药:甲胺磷、氧乐果、甲拌磷、对硫磷、甲基对硫磷、甲基异柳磷、水胺硫磷、乐果、敌敌畏、毒死蜱、乙酰甲胺磷、三唑磷、丙溴磷、杀螟硫磷、二嗪磷、马拉硫磷、亚胺硫磷、伏杀硫磷、辛硫磷
用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检测茶叶中有机磷类农药残留,你们都是如何前处理的,如何去除咖啡因对茶叶有机磷特别是马拉硫磷,毒死蜱、杀螟硫磷的影响







 我要推广仪器
我要推广仪器
 下载APP
下载APP