本人[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]新手,要建立一种中药成分 [url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相[/color][/url]鉴定方法;使用仪器是waters,鉴定的物质是皂苷类,打算用一种方法测定6种不同皂苷含量流动相为乙腈A-水B,流速0.6ml/min,柱温25℃,波长203nm,进样量20ul梯度洗脱 0~12min 20%~35%A 12~32min 35%~40%A ;32~36min 40%A;36~37min 40~20%A 37~42 20%A。目前标品目标峰分离良好,测量样品无法与目标峰对应,无法完全分离,且杂质影响较大。请问大神们,像这种情况应该如何优化
第一次接触HPLC-ELSD,现打算做皂苷类含量测定,请问需要注意什么?氮气流速、气压条件如何定的?诸位多帮忙,急啊[em0808]
主成分甾体皂苷除杂如色素,甾醇,脂类有什么方法啊
流动相选用乙腈和水,梯度洗脱,建立测量皂苷的方法,从结果来看标品都能分开,样品与目标峰对应不上,而且分不开,这种问题该如何解决?
流动相选用乙腈和水,梯度洗脱,建立测量皂苷的方法,从结果来看标品都能分开,样品与目标峰对应不上,而且分不开。这种问题该如何解决?
齐墩果酸五环三萜类极性很小,人参皂苷类极性较大,但是两者都同属三萜类,都是末端吸收203~210nm,按道理说可以采用紫外检测器同时检测。这几天走了几个梯度条件都不理想http://emuch.net/bbs/images/smilies/cry.gif,我的思路是这样的:先采用中国药典2010版人参项下条件走60min(主要是乙腈-水,水比例较高80~45),然后梯度增加乙腈的比例,走至100min,但是齐墩果酸还是走不出来,请教各位能不能把保留时间限定在100min内,把上述两类成分分离呢?http://emuch.net/bbs/images/smilies/work.gif
安捷伦1100,乙腈-水梯度洗脱,皂苷类成分,在开始的时候出现倒峰是怎么回事啊
藻类制品的铅检测中要以干重计,如何烘干吗?望老师不吝赐教
[b]Q:[b][b][b][/b][/b]西洋参中人参皂苷的检测,样品前处理步骤是?[/b]A:标准品溶液:人参皂苷Rg1:0.1mg/ml (溶剂70%甲醇水) 人参皂苷Re :0.35 mg/ml(溶剂70%甲醇水) 人参皂苷Rb1:1 mg/ml(溶剂70%甲醇水).===============================================================【活动内容】1、每个工作日上午10:00左右发布一个关于应用数据库的应用问答题,版友根据题目给出自己理解的答案。2、每个工作日下午15:10公布参考答案。【活动奖励】幸运奖:抽奖软件,当天随机抽取3个或5个回答正确的版友ID号(最后一个ID号,截止至下午15:00),每人奖励[color=#ff0000]2钻石币[/color](抽奖人数≤10,抽取3个版友;抽奖人数>10,抽取5个版友);中奖名单:yifan1117(注册ID:yifan1117)捌道巴拉巴巴巴(注册ID:v3082413)PAEs(注册ID:v2911392)WUYUWUQIU(注册ID:wulin321)dahua1981(注册ID:dahua1981)[img=,690,388]https://ng1.17img.cn/bbsfiles/images/2018/10/201810171513425489_3187_1610895_3.png!w690x388.jpg[/img][img=,690,388]https://ng1.17img.cn/bbsfiles/images/2018/10/201810171513451756_1050_1610895_3.png!w690x388.jpg[/img]积分奖励:所有回答正确的版友奖励[color=#ff0000]10个积分[/color](幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次[/b][align=left][color=#ff0000][b]PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。[/b][/color][/align][align=left][color=#ff0000][b] 下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。[/b][/color][/align][align=center]=======================================================================[/align]方法:2015药典基质:标准品应用编号:102441色谱柱:[url=http://www.dikma.com.cn/product/details-861.html]Diamonsil Plus C18-A 5μm 250 x 4.6mm[/url]样品前处理:标准品溶液:人参皂苷Rg1:0.1mg/ml (溶剂70%甲醇水) 人参皂苷Re :0.35 mg/ml(溶剂70%甲醇水) 人参皂苷Rb1:1 mg/ml(溶剂70%甲醇水).色谱条件:色谱柱:Diamonsil 5μm C18 Plus-A 250*4.6mm #99406流动相:A:乙腈 B:0.1%磷酸水流速:1.0 mL/min柱温:40℃进样量:10 uL波长:203nm[img]http://www.dikma.com.cn/UploadImage/edit/images/23.jpg[/img]文章出处:天津应用实验室关键字:西洋参中人参皂苷的检测、2015药典、Diamonsil C18 Plus-A图谱:[img]http://www.dikma.com.cn/UploadImage/edit/images/45.jpg[/img]
寻求流化床造粒,实验室用,要求与样品接触部分是有机类材料,不能有不锈钢含铁类物质。拜托各位,发布内容有效日期17年12月5日-17年12月30日
完全按照药典标准做的三七总皂苷,可是对照品和样品的Re均不出峰,其他几个峰都出得很好
独圣活血片中三七皂苷R1和人参皂苷Rg1、Re的分析2010版药典解决方法独圣活血片中三七皂苷R1、人参皂苷Rg1、Re的分析本方法来源于2010版药典P940中“独圣活血片”的分析l 前言虽然药典中只要求分析三七皂苷R1和人参皂苷Rg1,但实际上是需要有效分离人参皂苷Rg1和人参皂苷Re的,因为作为独圣活血片原料的三七中或多或少总会含有Re,如果不将Rg1和Re分开,将会对Rg1的定量产生极大的干扰; l 样品分子结构中文名英文名结构式三七皂苷R1Notoginsenoside R1见附页人参皂苷Rg1Ginsenoside Rg1见附页人参皂苷ReGinsenoside Re见附页 l 样品来源记录
用药典方法测定柴胡药材中柴胡皂苷a、d的含量,刚开始预实验,对照品和样品的峰型都挺好,后来平衡好色谱柱,直接批量进针,问题就出现了,无论是对照还是样品,柴胡皂苷a的峰高变低,峰型变胖,保留时间也提前了4分钟,但是柴胡皂苷d没有什么变化,求助大家,问题在哪?而且每个样品进3针,第1针与第2、3针的柴胡皂苷a在保留时间上也有0.5分钟的差别...

作者:http://d.g.wanfangdata.com.cn/Images/head_pic.gif雷灼雨 http://d.g.wanfangdata.com.cn/Images/head_pic.gif邹丽 作者单位:重庆市药品检验所,重庆,401121 四川省食品药品检验所,四川成都,摘要:建立测定血塞通片中三七皂苷R1、人参皂苷Rg1、人参皂苷Rb1含量的反相高效液相色谱(RP-HPLC)法.方法 色谱柱为Diamonsil C18柱(250 mm×4.6 mm,5μm),柱温30℃,采用乙腈-水线性梯度洗脱,检测波长203 nm,流速1.0 mL/min....http://ng1.17img.cn/bbsfiles/images/2012/08/201208201721_384768_2379123_3.jpg
香草醛-冰醋酸显色针对的苷类包括黄酮苷、蒽醌苷、皂苷吗?因为我做的UV里面苷类成份特别多,而我只想测黄芪总苷,但阴性有干扰,现在解决不了,谁能帮我解决一下,谢谢
小弟在测甾体皂苷,用的是已腈-磷酸水(酸万分之一),梯度洗脱,DAD检测。待测样品用普通分析纯的甲醇溶解,微孔滤膜过滤,进样分析。做了好几次,发现皂苷的峰不明显,并且更要人命的是,谱图中有很多溶剂峰,强度很大很高,现在才意识到,以前都当成皂苷的吸收峰,因为不像溶剂峰,都是末端吸收,没办法区分。今天,在进对照品时,发现对照品进入液相后,有好几个末端吸收峰,发现问题比较严重,对照品纯度没问题的。我怀疑:1、普通分析甲醇溶解对照品和样品后,才导致出现谱图中不同位置出现大小不一样的溶剂峰。我马上做了空白对照,确实甲醇单独进入液相分析,有很多溶剂峰,末端吸收,真是无语。2、考虑到溶剂问题,我又做了已腈空白对照,发现还是有溶剂峰,只不过少了很多,只有两个看起来比较高的峰。一个出峰在10分钟,一个在很靠后。接下来,我想做下面尝试:1、用流动相来溶解样品,就是已腈-磷酸水,同时,先做溶剂空白。2、考虑用已腈溶解样品紫外检测,感觉皂苷的峰不灵敏,但是比较实用,所以还是选择用紫外检测。对以上问题,请各位朋友多多指导。十分感谢!!![em09508]
人参(Panaxginseng C. A. Mey)五加科、人参属多年生草本植物。人参皂苷,人参中的活性成分,是一种固醇类化合物,三萜皂苷。因为人参皂苷影响了多重的代谢通路,所以其效能也是复杂的,而且各种人参皂苷的效能是难以分离出来的。人参皂苷都具有相似的基本结构,都含有由30个碳原子排列成四个环的甾烷类固醇核。人参皂苷成分:如Rb1、Rb2、Rb3、Rc、Rd、Rg3、Rh2及糖苷基PD等。目前测定人参皂苷含量的标准有国标(GB/T 22996-2008)和行标(NY/T 1842-2010),本文仅仅对上述两个标准的适用范围、样品处理、检测方法等进行比较。1、 标准的适用范围标准号:GB/T 22996-2008标准名称:人参中多种人参皂甙含量的测定 液相色谱-紫外检测法标准规定了人参中人参皂甙Re、Rg1、Rf、Rb1、Rc、Rb2含量的液相色谱-紫外检测方法,适用于生晒人参中人参皂甙Re、Rg1、Rf、Rb1、Rc、Rb2含量的测定。标准号:NY/T 1842-2010 标准名称:人参皂苷的测定标准规定了测定人参中9种人参皂苷的高效液相色谱方法。适用于人参中人参皂苷Rbl、Rb2、Rb3、Rc、Rd、Re、Rgl、RgZ和Rf的测定。注意:在GB/T22996-2008中,测定的是6种人参皂苷,而NY/T1842-2010测定的是9种。并且,包含了国标中的6种。
三萜皂苷与甾体皂苷在薄层色谱上如何能分开?(甾体皂苷含量低于三萜皂苷)本人用硫酸乙醇显色出现的点都是紫红色的,没有墨绿色,并且表面看上去点都是分开的,会不会是甾体皂苷显示的颜色被三萜皂苷的盖住了。忘高人指点一下为谢!
请教大家一个问题,我做的是藻类的一些挥发半挥发性物质的检测(烯醛类),因为一些物质买不到标样,也难以合成,所以想说能不能自己从样品中分离出来。 我看过一般是用制备液相色谱分离一些生物活性物质,不知道是否可以分离半挥发性的物质。 此外,看到一种制备[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url],但是国内用的人好像不多,有没有对这方面了解的版友呢?
【作者】 杨秀伟; 桂方晋; 宋燕; 张尉清; 田建明; 李龙云;【Author】 YANG Xiuwei1,GUI Fangjin1,SONG Yan1,ZHANG Weiqing1,TIAN Jianming2,LI Longyun2 (1.School of Pharmaceutical Sciences,Peking University Health Science Center,Beijing 100191,China;2.Jilin Institute of Chinese Materia Medica,Changchun 130021,China)【机构】 北京大学医学部药学院; 吉林省中医药科学院;【摘要】 目的:研究静脉给予大鼠人参皂苷Rg2后,其在胆汁、粪便和尿液中的排泄。方法:采用反相高效液相色谱(HPLC)-紫外检测器(UVD)法测定大鼠胆汁、粪便和尿液中的人参皂苷Rg2;Dikma Diamonsil TMC18色谱柱(4.6 mm×250 mm,5μm),以甲醇-4%磷酸水溶液(65∶35)为流动相,检测波长为203 nm。结果:HPLC-UVD测定方法的标准曲线线性关系、样品回收率和日内、日间精密度均符合生物样品分析要求。给大鼠静脉注射人参皂苷Rg2后,5.5 h内胆汁中原形人参皂苷Rg2累积排泄量为给予剂量的27.2%,24 h内粪便中原形人参皂苷Rg2累积排泄量为给予剂量的22.6%;尿液中未检出人参皂苷Rg2。结论:静脉给予大鼠人参皂苷Rg2,原形药物主要通过胆汁和粪便途径排出体外。
从药材中提取纯化皂苷时,会涉及到总皂苷含量的估算。然而在紫外检测器中,酚类物质的响应高于皂苷。用峰面积估算就不合理了。其他检测器可以吗?例如质谱检测器、蒸发光散射检测器?

【作者中文名】赵春芳; 刘金平; 赵岩; 李平亚;【作者英文名】ZHAO Chun-fang1; LIU Jin-ping2; ZHAO Yan2; LI Ping-ya2(1.Pharmaceutical Academy of Jilin University; Changchun 130021; China; 2.Institute of Frontier Medical Science of Jilin University; China);【作者单位】吉林大学药学院药物分析; 吉林大学再生医学科学研究所; 吉林大学再生医学科学研究所 吉林长春; 吉林长春;【摘要】目的:研究大鼠舌下静脉给药伪人参皂苷GQ后,其在胆汁、粪和尿中的排泄情况。方法:采用高效液相-蒸发光散射色谱(HPLC-ELSD)法测定大鼠胆汁、粪和尿中伪人参皂苷GQ,Diamonsil C18色谱柱(4.6 mm×250mm,5μm),以甲醇-水(24∶7)为流动相,流速1.0 mL.min-1,检测温度为50℃,灵敏度为10,以氮气为载气,压力为303 975 Pa。结果:HPLC-ELSD测定方法的标准曲线线性关系、样品回收率和日内、日间精密度均符合要求。伪人参皂苷GQ大鼠舌下静脉给药后,主要以胆汁排泄为主,占总药量的41.60%;其次为粪排泄,占总药量的9.97%;尿液中仅检出少量伪人参皂苷GQ。结论:大鼠胆汁、粪和尿中主要以伪人参皂苷GQ原形药物排泄。http://ng1.17img.cn/bbsfiles/images/2012/08/201208131711_383575_2379123_3.jpg

全自动固相萃取-比色法测定保健食品中总皂苷 作者:赵晓冬http://ng1.17img.cn/bbsfiles/images/2015/11/201511261708_575182_2904170_3.jpg目的:建立保健食品中总皂苷的含量测定方法。方法:采用Sepline全自动固相萃取装置对保健食品中的总皂苷富集,通过70%乙醇回收,以香草醛-高氯酸为显色剂,采用比色法测定。结果:比色法测定的线性范围为0.038~0.268mg,相关系数 r=0.9997; 在不同保健食品中的平均加样回收率均在98.3%~102.4%,RSD〈2.2% (n=6)。结论:本法简便、准确、灵敏度高、重复性好,可用于不同类型保健食品中总皂苷的含量测定。 目前保健食品中的总皂甙测定方法尚无国家标准,大多都是按照《保健食品检验与评价技术规范 (2003年版) 》“保健食品中总皂甙的测定”方法检测,该方法采用人参皂苷 Re为对照,用香草醛比色法测定总皂苷。其显色体系为:香草醛-冰醋酸-高氯酸,但在实际操作中和据相关文献报道中,但采用该方法检测结果误差较大,尤其是不同实验室间测定结果差异更大,不利于产品的质量控制。有文献报道,卫生部食品卫生监督所2000年组织全国22个省、市、自治区省级疾病预防控制中心对这一方法进行比对试验,结果显示:离散度较大,有的浓度水平还出现离群值,各实验室间的RSD达80%,因此需要对测定方法加以改进。本文采用莱伯泰科公司Sepline全自动固相萃取提取装置 具有洗脱液流速恒定可控、大批量产品自动化处理,保证试验系统的密闭性等优点可保证结果的平行性和准确性。在对大孔树脂进行品牌筛选的基础上,定制统一规格固相萃取柱、对总皂苷测定前处理过程进行了优化,整套方法简单,数据结果离散性小,平行性得到提高。1材料与方法1.1 仪器 UV-2450 紫外可见分光光度计(日本,岛津公司),CP2250分析天平,AS10200ADT超声波清洗仪,TLL-DCⅡ型氮吹仪,Sepline全自动固相萃取系统(北京莱伯泰科有限公司),固相萃取柱(底层有砂芯隔板,内装XAD-2大孔树脂2.7g,Alumina-N中性氧化铝,6ml,北京莱伯泰科有限公司)。1.2 试药 人参皂苷Re对照品(中国食品药品检定研究院,含量92.7%),冰醋酸、香草醛、高氯酸、乙醇均为分析纯,水为纯化水。1.3 供试品 上普牌西洋参含片 (厦门上普药业有限公司,批号14040201);天信减肥茶( 福建安溪馨隆保健品有限公司,批号: 20140504 ) ;辅助降血脂片(哈尔滨久盛医药科技开发有限公司,批号:20140714)珍迪牌黄芪阿胶口服液(南昌健民营养补品厂,批号: 20130701)2方法与结果2.1 标准溶液制备精密称取20mg人参皂苷Re对照品置于100mL量瓶中,用甲醇溶解并稀释至刻度,摇匀。吸取10mL人参皂苷Re标准溶液(0.2mg/ml)放蒸发皿中,放在水浴挥干(低于60℃),准确加入10ml水溶解,作为标准溶液2.2 样品溶液制备 精密称取适量样品,置于50mL容量瓶中,加水适量溶解,超声30min,放置至室温,用水稀释至刻度,摇匀, 3000r·min-1离心5min,0.45um微孔滤膜过滤。若为非乙醇类液体试样,先经0.45um微孔滤膜过滤,取精密过滤后样品1mL (根据试样含总皂苷量定) 进行固相萃取。2.3 样品溶液吸附及洗脱 取样品溶液上清液置于收集管中(大于2ml), 放置于Sepline全自动固相萃取系统中,按以下步骤进行吸附及洗脱(详见图1)。2.3.1 活化:在固相萃取柱中,以2.5 ml·min-1 流速冲洗70%乙醇25ml、然后以2.5 ml·min-1 流速冲洗水25ml。2.3.2 上样:以1ml·min-1 流速加1ml样品溶液于固相萃取柱中。2.3.3 洗脱:以0.8ml·min-1 流速, 依次用25ml水,25ml70%乙醇进行洗脱,收集70%乙醇洗脱液,在收集结束时氮气吹扫管理残留洗脱液50s。2.3.4 挥干:将收集的洗脱液转入50ml的离心管中,在60℃下氮气吹干。https://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXHUdyj5UNWFsWqjnVFOfw77OyyjdE8AtsH0V1ibNylQgZibv2FialaXicibA/0?wx_fmt=jpeg2.4 显色与测定 在上述已挥干的洗脱液中准确加入0.2ml5%香草醛冰乙酸溶液,使残渣都溶解,再加0.8ml 高氯酸,混匀后60℃水浴上加热 10min,冰浴冷却后,准确加入冰乙酸 5.0ml,摇匀后,以 1cm 比色池于 560nm 波长处与标准管一起进行比色测定。2.5 线性关系考察 精密吸取“2.1”项标准溶液0、0.2、0.5、0.8、1.0、1.2、、1.4人参皂苷Re标准溶液(0.2mg/ml),按 “2. 3”“2.4”项下方法进行前处理并测定。以吸光度值为横坐标, 以人参皂苷Re含量 (mg) 为纵坐标进行回归计算,回归方程为y =0.3521x+0.0018,r=0.9997,结果表明,总皂苷( 以人参皂苷Re计) 在0.038~0.268mg 范围内线性关系良好。2.6 加样回收率试验 采用加样回收率法,精密称取4类保健品适量,分别按样品量的100%的比例各6份加入对照品 ,按“2.3”项下和“2.4”项下进行测定,并分别计算回收率(见表1-表5),结果各品种平均加样回收率均在98.3%~102. 4% ,RSD<2.2% ,并与传统方法(手填层析柱、人工进行柱洗脱)进行比较,表明该方法准确度良好,且离散度低于传统方法。https://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXpKdklF1UoeMzvU6eQJEseF6P0shyEyUViacyO2M0iaBmcQmLVtq4YUIQ/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXcxdobibU0cPzwap5qsdShaQtxlrmMbDxOG6AKyWSTxYHbwu085loQog/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXudGTrHbh0Eu8R6zY3PNo4iaHwJMh58wAxhK00QPk76bje6KXTQ6LWAA/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXO3ae9XyfEiaaUU6Qkibib1WgWDiaAVh9AQEFhV6tJ8oJxfsibuHW7tMWHUw/0?wx_fmt=jpeghttps://mmbiz.qlogo.cn/mmbiz/1M6WGftQWKcMCtAjjEeTO31icqxEfIgZXfVUKQfmpzF7ootVP2rp4VREoKIGLKDM54tMwEK2GyZ8DazgJL6tfug/0?wx_fmt=jpeg2.7 稳定性试验 将显色后样品分别放置 0、0.5、1、1.5、2、2.5、3h, 测定吸光值。结果表明该显色体系在1 h内吸光度基本不变,1h后吸光度有所降低,故在显色后1 h内测定。2.8 样品含量测定 取4类保健食品 ,按“2.3”项下和“2.4”项下进行测定,于 560nm 波长处测定吸光度值, 根据所得到的标准曲线计算样品中总皂苷的含量(见表6) 。[align

地表水体的富营养化引起藻类及其它浮游生物迅速繁殖,导致溶解氧下降,水质恶化,鱼类和其它生物大量死亡,甚至引发供水危机。色素是藻类光合作用时吸收、转化和传递光能的主要物质,叶绿素以多种形式存在于藻类植物中,大约占有机物干重的1-2%,是估算其生物量的重要指标,快速准确测定水体中叶绿素a浓度对于评价水体营养状态和水质管理具有重要意义。 叶绿素测定方法有很多,大致分为萃取测定、荧光活体测定及水华遥感监测。萃取分析利用有机溶剂萃取植物细胞叶绿素进行光谱光度测定,根据分析技术的不同分为分光光度法、荧光法和高效液相色谱法。萃取分析只要操作准确,提取完全,可以得到准确和重复性好的结果,但是过程繁琐,不方便用于连续监测。 AOA在线藻类分析仪利用叶绿素荧光特性进行分类定量分析,活体测量,无需样品预处理,仪器操作简单,可以快速地获得大量叶绿素数据,广泛运用在在线监测。为保证在线分析仪的有效运行,需要用标准样品来校准、性能考核和日常数据质量控制,但国内还未开发叶绿素的标准样品和质控样品,已知叶绿素含量(用萃取方法测定)的浮游植物悬浮液被认为是最好的标样替代品。当水体发生水华,生物多样性下降,往往是一种藻类成为绝对优势,可作为纯种藻类标准样品,本文将几个代表性的比对实验整理分析,探讨误差原因。[b]1.比对方法[/b]1.1清洗AOA在线藻类分析仪蠕动泵、测量室及其底盖,检查纯水透光率值。1.2水样不经任何处理,测量叶绿素浓度值。1.3剩余水样酌量加入1%碳酸镁悬浊液(按1升水量加入1ml),防止酸化。1.4带回实验室,尽快分析,实验室分析方法依据《无水乙醇热浴超声法测定淡水中的叶绿素a》[sup][/sup]。[b]2实验2.1微囊藻水样2.1.1样品来源[/b] 2014年8-10月某水库出现铜绿微囊藻水华,采集水华“浓密”处水样作为标准储备液,分别取0、2、5、10、15、20、25、50、100ml,用纯水稀释成500ml,检查两种方法叶绿素a和藻密度线性。取水库水华严重区、进水口、湖心和出水口四个水样,做实际水样比对分析。[img=,690,690]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011537059882_3331_3247383_3.jpg!w690x690.jpg[/img][b]2.1.2实验结果[img=,690,361]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011538312332_220_3247383_3.png!w690x361.jpg[/img][/b]表1微囊藻稀释水样比对结果[b][img=,622,352]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011539397062_938_3247383_3.png!w622x352.jpg[/img][/b]图2微囊藻水样线性比对[img=,690,215]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011611492722_7678_3247383_3.png!w690x215.jpg[/img]表2含微囊藻实际水样比对表1、表2和图2表明,两种测量方法的结果与藻类数量之间存在非常显著的相关性,但AOA在线分析仪测量结果低于分光光度法,浓度越高,差异性越大。[b]2.1.3结果分析[/b]2.1.3.1分光光度法误差分析 萃取后叶绿素,对光照和氧气很敏感,极易造成不可逆的分解,因此,节时高效是分光光度法的质量保证,而温度是质控措施的关键。在超声波萃取过程,提高温度加快叶绿素溶出速度,同时,叶绿素降解的速度也是加快的,最佳超声波萃取条件:60℃萃取25分钟,2小时内完成测定。另外,实验室遮光也是必要。[img=,690,690]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011541253150_3659_3247383_3.jpg!w690x690.jpg[/img]2.1.3.2AOA荧光法误差来源 AOA在线藻类分析仪原理:叶绿素具有荧光特性,一定的激励光,任何一种藻产生的荧光强度与其所含叶绿素a成正比,几种不同藻混合体产生的荧光强度等于各自荧光的总和。采用325纳米、450纳米、525纳米、570纳米、590纳米和610纳米作为激励光,其中325纳米是用来补偿黄色物质(有机物),测定685nm的光强,计算总叶绿素a值和不同藻的浓度。(1)水样原始性状发生变化暗室环境下,激励光照到藻上,能量分成三部分:光合作用、叶绿素自发荧光和热能辐射。如果藻的活性强,光合作用消耗的能量就越多,产生荧光的强度越小,反之,如果藻的活性弱,荧光强度就强。荧光能量和光合作用的能量是相互竞争的,叶绿素荧光常常被认为光合作用无效指标的依据。比对实验的样品,从采样经实验室样品处理,到水质自动监测站仪器分析,剩余样品送回实验室做分光光度法比对,再紧凑也需要3-4天完成,运输、保存过程,叶绿素在活体内也和其它物质处于不断更新变化中,可能衰老、也可能分解破坏,比对实验要求的同步水平也不可能实现,这是比对误差不可忽视的部分。(2)微囊藻特殊的细胞结构使水样分布不均匀微囊藻群体有胶鞘和伪空泡,可以自由漂浮在水中,水样在实验室静置一天后,出现明显的分层,测量过程难以保证均匀分布。[img=,690,690]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011542423667_4598_3247383_3.jpg!w690x690.jpg[/img](3)工作温度与校准温度不一致参考各类荧光法仪器说明书,浮游生物悬浊液的荧光反应受温度的影响很大,有些仪器的温度补偿2%⁄ ℃,但这种经验补偿不能保证准确的现场测量,因为每一种浮游植物的荧光强度随温度变化程度不同。这台AOA藻类分析仪安装测试在冬季,这次比对实验在9月份进行,温差亦有影响。(4)浊度的影响 实验室分光光度法经过0.8um滤纸过滤,比色扣除750nm吸光度值,只要操作正确,浊度的影响很小。水中的悬浮颗粒物对激励光和产生的荧光有反射、散射和阻挡的作用,造成激励光和荧光产量的下降,YSI3026传感器检测结果:1NTU的 浊度影响大约是0.03ug/L叶绿素。 除色素以外的细胞结构(细胞壁、胶被、储藏物质)以溶解有机碳为主要成分,对紫外光和蓝光具有强烈的吸收,也可视为影响测定的悬浮物,AOA在线藻类分析仪称之为黄色物质,用325纳米补偿。铜绿微囊藻细胞壁分为两层,内层是纤维素,外有胶鞘,有相当的厚度,互相溶合形成多细胞群体。图5显示黄色物质与叶绿素含量相关,AOA的测量结果反映了铜绿微囊藻细胞群体特征。分光光度法和AOA扣除浊度方法各有不同,是否有可比性,有待进一步了解。[img=,586,305]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011543564741_5446_3247383_3.png!w586x305.jpg[/img]图5(5)水样镜检微囊藻绝对优势,视野内不见其它藻类,AOA分析结果有少量绿藻、硅藻,其它的比对实验也出现藻类识别错误。[b]2.2薄甲藻水样[img=,690,920]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011545089042_4958_3247383_3.jpg!w690x920.jpg[/img][/b]图6[b]2.2.1实验结果[/b][img=,690,289]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011547036798_1735_3247383_3.png!w690x289.jpg[/img]表3薄甲藻水样比对结果2.2.2结果分析2.2.2.1镜检视野内为单一薄甲藻,与AOA定性结果相差较大。从图7可知,绿藻和硅甲藻的光谱图很相似,从光谱图识别绿藻和硅甲藻是困难的,AOA的藻类分类和藻密度估量值仅能作为参考,要将监测数据做水体生态评估的依据,必须结合实验室镜检和萃取分析方法。[img=,690,496]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011547417547_4700_3247383_3.png!w690x496.jpg[/img]图72.2.2.2薄甲藻AOA测量值约为光度法的1/3,薄甲藻有鞭毛,可以自由游动,离开有利的生活条件则很快失去活性,沉入水底不再活动,且细胞内容物溶出。不能保证水样原始性状,是叶绿素a比对测定误差的主要来源。[img=,690,690]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011548482013_3683_3247383_3.jpg!w690x690.jpg[/img][color=#423B3B]2.2.2[/color][color=#423B3B].3[/color][color=#423B3B]薄甲藻细胞内容物溶出后,显微镜照片清楚看到细胞壁很薄,AOA测量结果印证黄色物质影响极小。[/color][b]2.3小球藻[img=,690,920]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011550015246_3694_3247383_3.jpg!w690x920.jpg[/img]图9 小球藻[/b]垃圾渗滤液水样,实验室放置几周,变绿,镜检结果:小球藻绝对优势,少量硅藻。[b]2.3.1实验结果[/b][img=,690,367]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011551332128_4472_3247383_3.png!w690x367.jpg[/img]表4小球藻水样比对[b]2.3.2结果分析[img=,537,303]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011552368052_2948_3247383_3.png!w537x303.jpg[/img][/b]图10小球藻水样线性分析2.3.2.1藻类分类与实验室镜检结果基本吻合;2.3.2.2低浓度出现少量蓝藻,因为仪器刚做完微囊藻样品,检测室可能有少量残留,比对实验之前彻底清洗检测室很有必要;2.3.2.3分光光度法相关系数小于AOA,并非方法不可行,而是叶绿素萃取太依赖分析人员的操作,稍有疏忽就会造成萃取损失,相比之下仪器要稳定可靠些。2.3.2.4小球藻叶绿素a测量结果AOA/分光光度法比值0.7-1.5,高于铜绿微囊藻和薄甲藻。两种方法测量结果比较接近的原因是因为小球藻不会运动,活体样品保持比较稳定的悬浮液状态,而AOA测量结果高于分光光度法的误差,一方面来自叶绿素和藻密度系数的确定,一方面来自萃取的损失。[img=,601,407]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011554177672_7731_3247383_3.png!w601x407.jpg[/img]图11AOA仪器校正界面 荧光测定仪校准即确定叶绿素和藻密度相关系数,可以给活体叶绿素传感器提供的最好标准物是浮游植物悬浮液,此悬浮液亦有部分用萃取方法测定叶绿素含量,并且应该从监测地点获得,这样的标准物质产生的荧光可以尽可能接近现场的生物体。荧光测定同时受浊度、温度、细胞活性、不同藻的种类、大小、形状、和叶绿素种类的影响,这些都大大限制活体测量的准确度。2.4不同水体样品分析[img=,690,267]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011555277271_2806_3247383_3.png!w690x267.jpg[/img]表5不同水体样品分析2.4.1与单一藻类样品结果一致,绿藻含量高的水样AOA测量值偏高,微囊藻水样上浮分层、甲藻和裸藻活性降低下沉造成水样不均匀分布,是比对结果偏低的一个因素。而在线监测时,水样中的藻类足够鲜活,吻合度会好些。2.4.2按照AOA提供的分类方法(图12),绿藻和蓝藻分类结果与镜检匹配,单一种类薄甲藻检测结果显示为绿藻、隐藻、蓝藻、极少量甲藻,镜检中数量可观的硅藻、裸藻在AOA测量结果中未见。[img=,600,350]http://ng1.17img.cn/bbsfiles/images/2018/07/201807011602453540_1118_3247383_3.jpg!w600x350.jpg[/img]图122.4.3水库水质良好,杂质少,其它水体有机杂质含量相对高,AOA黄色物质测量结果与水体洁净程度吻合。[b]小结[/b]:萃取分析耗时长,不方便用于连续监测,需要有经验、高效率的分析人员才能得到准确、重复性好的结果。活体荧光法简便易操作,但准确度受更多因素的限制。两种方法不可互相替代,而应该取长补短,互为参照。参考文献 王丽娟,章岳蓬,郭步平等. 无水乙醇热浴超声法测定淡水中的叶绿素a.当代环保,2010,2(4):14-17.
藻类干制品样品到样时,没有明确的脱水率,测重金属元素时还需要测样品水分吗?还是直接检测出报数?望老师不吝赐教
问题:启脾口服液中人参皂苷Rg1、人参皂苷Re的检测:对照品中人参皂苷Rg1、人参皂苷Re的分离度是多少?答案:2.467获奖名单:吕梁山(ID:shih20j07)dahua1981(ID:dahua1981)m3071659(ID:m3071659)http://ng1.17img.cn/bbsfiles/images/2016/02/201602231542_584931_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602231543_584932_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602231543_584933_708_3.jpg【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。启脾口服液中人参皂苷Rg1、人参皂苷Re的检测样品制备 制备方法1. 对照品:取人参皂苷Rg1对照品、人参皂苷Re对照品适量,精密称定,加甲醇制成每1 mL各含0.25 mg的混合溶液,摇匀,即得。2. 供试品:精密量取本品50 mL,加三氯甲烷振摇提取3次,每次30 mL,弃去三氯甲烷提取液,水液加水饱和正丁醇振摇提取5次(50 mL、30 mL、30 mL、20 mL、20 mL),合并正丁醇提取液,加氨试液洗涤4次,每次50 mL,弃去氨试液,再加正丁醇饱和的水轻轻振摇洗涤2次,每次50 mL,弃去水洗液,正丁醇液回收溶剂至干,残渣加甲醇溶解并转移至5 mL量瓶中,加甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。分析条件 色谱柱Diamonsil C18(2) 250 × 4.6 mm,5 μm (Cat#:99603) 流动相A:水 B:乙腈 梯度流速1.0 mL/min 柱温35 ℃ 检测器UV 203 nm 进样量5 μL 色谱图对照品http://ng1.17img.cn/bbsfiles/images/2016/02/201602231201_584890_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 52.603 387347 6585 18460.678 1.019 -- 2 56.668 262702 4169 16820.782 0.971 2.467 *药典要求理论板数按人参皂苷Re峰计算应不低于2500 供试品http://ng1.17img.cn/bbsfiles/images/2016/02/201602231202_584891_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 52.664 117855 2108 18875.714 0.982 -- 2 56.654 291453 4502 18266.289 1.019 2.486 *药典要求理论板数按人参皂苷Re峰计算应不低于2500本品种同时使用了Leapsil C18色谱柱,在药典规定条件下进行人参皂苷Rg1[/sub
用原子荧光测定贝类的镉,其消化样品不赶尽硝酸对测贝类的中镉有什么影响呢?另外选择什么样的仪器条件为最佳呢?谢谢

问题:颈痛颗粒中三七皂苷、人参皂苷的检测使用了哪几款液相色谱柱?答案:Platisil ODS、Leapsil C18、Spursil C18-EP【活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币mengzhaocheng(ID:mengzhaocheng)999youran(注册ID:999youran)WUYUWUQIU(注册ID:wulin321)http://ng1.17img.cn/bbsfiles/images/2016/01/201601291556_583923_1610895_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601291556_583924_1610895_3.png积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。=======================================================================颈痛颗粒中三七皂苷、人参皂苷的检测样品制备制备方法1. 对照品:取人参皂苷Rg1对照品、人参皂苷Rb1对照品和三七皂苷R1对照品适量,精密称定,加甲醇制成每 1 mL含人参皂苷Rg1 0.5 mg、人参皂苷Rb1 0.5 mg、三七皂苷R1 0.1 mg的混合溶液,即得。2. 供试品:取装量差异项下的本品适量,研细,取约1.3 g,精密称定,加乙醚40 mL,加热回流30分钟,滤过,弃去乙醚液,药渣及滤纸挥尽乙醚,再精密加入甲醇40 mL,称定重量,加热回流1小时,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,精密量取续滤液20 mL,回收溶剂至干,残渣加水10 mL使溶解,用水饱和的正丁醇振摇提取5次,每次10 mL,合并正丁醇提取液,用2%碳酸钠溶液洗涤2次,每次20 mL,再用正丁醇饱和的水洗涤2次,每次20 mL,取正丁醇液回收溶剂至干,残渣用甲醇溶解,转移至10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得。分析条件色谱柱Platisil ODS 250 x 4.6 mm,5 μm (Cat#:99503)流动相A:水 B:乙腈 梯度流速1.0 mL/min柱温30 ℃检测器UV 203 nm进样量10 μL色谱图对照品 http://ng1.17img.cn/bbsfiles/images/2016/01/201601290942_583886_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 36.408 264983 25847 265850.102 1.018 -- 2 38.915 1775914 178004 318998.439 0.998 8.983 3 54.506 1316933 127848 597818.242 0.870 55.926 *药典要求理论板数按三七皂苷 R1峰计算应不低于3000供试品http://ng1.17img.cn/bbsfiles/images/2016/01/201601290943_583887_1610895_3.jpg 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 36.344 250230 25147 274216.739 0.996 --
最近用甲醇-水结晶的方法分了一些皂苷,感觉效果还不错,做皂苷的同志们可以尝试一下哦。
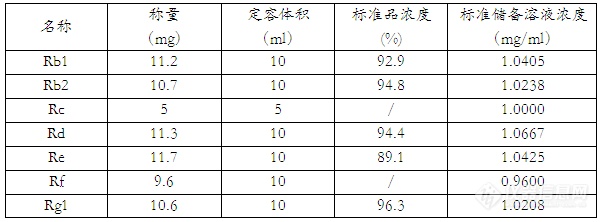
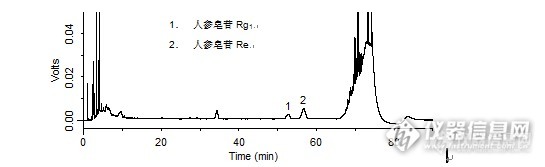
一、实验目的及原理:参照药典中人参皂苷的检测,使用超高效液相色谱,对于人参中7种皂苷成分进行检测,大大提高了检测速度,并拓展了检测的皂苷种类。人参中的皂苷成分通过超声波萃取,高校液相色谱柱进行分离,采用紫外检测器进行检测,外标法定量。二、实验仪器及试剂:仪器:UPLC(waters H-Class),检测器:TUV紫外检测器、超声波仪、超纯水机、离心机、涡旋振荡仪、50ml离心管、20ml胖肚移液管试剂:人参皂苷标准品Rb1、Rb2、Rd、Re、Rf、Rg1购自食品药品检定院, 人参皂苷标准品Rc购自百灵威、乙腈(HPLC)、甲醇(HPLC)三、色谱条件:色谱柱:BEHC18 2.1mm×50 mm×1.7um 柱温:35℃ 样品温度:25℃进样体积:5µL 流速:0.4ml/min 波长:203nm 采样速率:20点/秒流动相配比:时间水乙腈曲线类型08218/1082186185050618.1821862082186四、实验步骤4.1、标准储备溶液配制:称取人参皂苷标准品,用甲醇溶解定容,具体见下表:http://ng1.17img.cn/bbsfiles/images/2012/10/201210241734_399095_1854832_3.jpg4.2、样品处理称取样品约0.4g(精确到0.0001g)于50ml离心管中,加入20.0ml超纯水,涡旋15s,超声波萃取30min,5000转/min离心5min,过0.22um水膜,待测。五、计算5.1样品中人参皂苷浓度的计算: X=C*100/m式中:X——样品中人参皂苷的浓度(mg/g) m——样品称量(g) C——待测溶液中人参皂苷的浓度(ug/mL)六、标准曲线与线性相关系数取标准储备溶液混合,用水稀释至约5、10、20、50、100ug/ml,进行2次平行测定,得到相应的峰面积平均值。以浓度为横坐标,以峰面积为纵坐标,绘制标准曲线,结果如下:http://ng1.17img.cn/bbsfiles/images/2012/10/201210241728_399091_1854832_3.jpg七、样品测定及回收率实验(以6年红参体为样本)http://ng1.17img.cn/bbsfiles/images/2012/10/201210241730_399092_1854832_3.jpg八、对照与样品测定的色谱图http://ng1.17img.cn/bbsfiles/images/2012/10/201210241731_399093_1854832_3.jpg九、结果与讨论本实验尝试多个色谱条件, Re、Rg1组分性质非常接近较难分离,选择梯度洗脱的方式加快其他组分的分离,相对于传统液相方法需要至少一个小时的进样时间,UPLC的分离时间在20min,具有较大的优势。







 我要推广仪器
我要推广仪器
 下载APP
下载APP