我在注射用头孢唑肟中N-甲基匹咯烷含量测定中发现,N-甲基匹咯烷峰前面有一未知峰干扰,降低柱温和流速不能增加分离度,相反增加流速、提高柱温对分离度有一定的帮助,但改善不明显,十分令我头疼。
大家好,我最近在做头孢地嗪钠的含量测定,发现头孢地嗪峰拖尾十分严重,拖尾因子达到了2,试了很多柱子都这样。请问有谁做个这个品种的含量测定嘛,请帮帮我!
用Sephadex G-10做头孢他啶,个别能分离,大多数不能分离,之所以这样,头孢他啶里有头孢他腚聚合物,但是我一直不没搞清楚这聚合物含量是多少呢,它分子结构是什么样呢?还有,我用的是G-10,10mmX300mm的柱子?望各位大虾多指教和发表言论![em09]
请问一下中国药典 05版第二部 头孢氨苄的有关物质和含量测定是怎样计算取样量的 大家能细细说一下吗本人很笨 数学学的不太好

样品制备制备方法:【有关物质】取本品适量,加流动相A溶解并稀释至每1ml中含1.0mg的溶液,滤过,取续滤液作为供试品溶液;精密量取1ml,置100ml量瓶中,用流动相A稀释至刻度,摇匀,作为对照溶液。取7-氨基去乙酰氧基头孢烷酸对照品和α-苯苷氨酸对照品各约10mg,精密称定,置同一100ml 量瓶中,加pH7.0磷酸盐缓冲液约20mL超声使溶解,再用流动相A稀释至刻度,摇匀。精密量取2 ml,置20 ml量瓶中,用流动相A稀释至刻度,摇匀,作为杂质对照品溶液。【含量测定】系统适应性试验: 取供试品溶液适量,在80℃水浴中加热60min,取20μl,测定,头孢氨苄峰与相邻杂质峰的分离度应符合要求。含量测定法:取装量差异项下的内容物,混合均匀,精密称取适量(相当于头孢氨苄0.1g),置100 ml量瓶中,加流动相适量,充分振摇,使头孢氨苄溶解,再用流动稀释至刻度,摇匀,滤过,精密量取续滤液10mL,置50mL量瓶中,用流动相稀释至刻度,摇匀,取20μl,注入液相色谱仪。分析条件【有关物质】色谱柱:Spursil C18,150×4.6 mm,5um,Cat#:(82001)流动相:流动相A为0.2mol/L磷酸二氢钠溶液(用氢氧化钠调pH至5),流动相B为甲醇洗脱方式线性梯度流速:1mL/min柱温:30 ℃检测器:UV 220nm进样量:20 μL【含量测定】色谱柱:Spursil C18,150×4.6 mm,5um[/fon
2005《中国药典》头孢氨苄含量测定项下: 色谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;以水-甲醇-3.86%醋酸钠溶液-4%醋酸溶液(742:240:15:3)为流动相;检测波长为254nm。头孢氨苄干混悬剂、头孢氨苄片、头孢氨苄胶囊和头孢氨苄颗粒含量测定方法同头孢氨苄。 文献报道的方法(HPLC法),试验条件除药典的水-甲醇-3.86%醋酸钠溶液-4%醋酸溶液系统外,其它的如: 周嘉等用HPLC法测定头孢氨苄胶囊的含量。仪器:岛津LC-10AD 液相色谱仪。色谱柱为Spherisorb C18柱(5μm,4.6mm×250mm);流动相为0.025mol/L磷酸溶液(用三乙胺调pH3.0±0.1)-乙腈(88:12);流速为0.9ml/min;检测波长为240nm;柱温为室温。 王建宁等用HPLC法测定头孢氨苄片的含量。仪器:日立L-6200A高效液相色谱仪。色谱柱为C18(250mm×4.6mm,5μm);流动相:0.04mol/L磷酸溶液(用三乙胺调pH3.0)-乙腈(75:25);流速:1mL/min;检测波长:240nm;柱温:室温。 程成等用HPLC法测定头孢氨苄胶囊的含量。Waters色谱系统,色谱柱采用Nova Pak ODs柱(4μm,4.6mm×250mm);流动相为0.01mol/L醋酸铵溶液(冰醋酸调pH4.0)-甲醇(70:30);流速:0.8ml/min;检测波长262nm;柱温25℃。 周静安等用HPLC法测定头孢氨苄胶囊的含量。仪器:岛津LC-10AD 液相色谱仪。色谱柱为Shim-pack CLC-ODS柱(150mm×6mm);流动相为甲醇-水(70:30);流速为1.0ml/min;检测波长为262nm。 邓永辉等HPLC法测定头孢氨苄的含量。仪器:岛津LC-10AD 液相色谱仪。色谱柱:250mm×4.6mm 不锈钢柱,填料为Hypersil ODS;磷酸二氰钾溶液(0.01mol/L)-乙氰-甲醇(90:8:2),并用磷酸溶液调节PH 至4.5±0.1;检测波长:254mm。 崔慈等用HPLC法测定头孢氨苄的含量。仪器:上海伍丰LC-100液相色谱仪。色谱柱:150mm的C18柱子,以0.025mol/L磷酸溶液(用20%NaOH调节PH至3左右)-乙腈88:12为流动相:检测波长为235nm。如果各位还有不同方法,欢迎发布交流,谢谢!
GB/T 22942-2008 蜂蜜中头孢唑啉、头孢匹林、头孢氨苄、头孢洛宁、头孢喹肟残留量的测定 液相色谱-串联质谱法
【第十一届原创】HPLC法测定注射用头孢他啶聚合物含量1样品简介注射用头孢他啶,主要成份为头孢他啶,加适量碳酸钠做助溶剂。2.仪器设备、试剂与对照品2.1仪器设备:waters e2695高效液相色谱仪赛托利斯 CPA225D分析天平色谱柱:天津开发区色谱分析仪器有限公司葡聚糖G-10 凝胶色谱柱 400mm×14mm2.2试剂:硫酸铵(AR) 广州化学试剂厂磷酸二氢钠(AR) 广州化学试剂厂磷酸氢二钠(AR) 广州化学试剂厂碳酸钠(AR) 广州化学试剂厂2.3 对照品:头孢他啶(来源是齐鲁安替制药有限公司,批号为WST-C-8034EJ82JC,含量85.81%)蓝色葡聚糖2000(来源是Bei Jing Biodee Biotechnology Co.Ltd,批号为9004-54-01,含量100%)3.色谱条件流动相:以含3.5%硫酸铵的pH7.0的0.1mol/L磷酸盐缓冲液为流动相A,以水为流动相B,波长:254nm 流速:0.8ml/min 温度:室温洗脱方式:等度 进样体积:100ul4.样品制备4.1系统适用性溶液制备4.1.1 1.5mg/ml蓝色葡聚糖2000溶液制备:称取37.5mg蓝色葡聚糖2000至25ml容量瓶中,加水溶解并稀释至刻度,摇匀。4.1.2 系统适用性溶液制备:称取头孢他啶约0.2g 与碳酸钠20mg,置10ml量瓶中,用1.5mg/ml的蓝色葡聚糖2000溶液溶解并稀释至刻度,摇匀。4.2 对照溶液制备取头孢他啶对照品约12mg,精密称定,加水溶解并定量制成每1ml中约含0.1mg的溶液。4.3供试品溶液制备取本品,按标示量加水溶解并定量稀释制成每1ml中含20mg的溶液,照头孢他啶项下的方法测定,含头孢他啶聚合物的量不得过标示量的1.0%。4.4测定法4.4.1 量取100μl系统适用性溶液注入液相色谱仪,用流动相A进行测定,记录色谱图。高聚体的峰高与单体和高聚体之间的谷高比应大于1.5。4.4.2 量取1.5mg/ml蓝色葡聚糖2000溶液100μl注入液相色谱仪,分别以流动相A,B进行测定,记录色谱图。按蓝色葡聚糖2000峰计算理论板数均不低于500,拖尾因子均应小于2.0。在两种流动相系统中蓝色葡聚糖2000峰的保留时间比值应在0.93~1.07之间,对照溶液主峰与供试品溶液中聚合物峰与相应色谱系统中蓝色葡聚糖2000峰的保留时间的比值均应在0.93~1.07之间。4.4.3 另以流动相B为流动相,精密量取对照溶液100μl,连续进样5次,峰面积的相对标准偏差应不大于5.0%。5. 结果讨论系统适用性结果报告:流动相A中蓝色葡聚糖2000理论塔板数为1875,拖尾因子为0.93。流动相B中蓝色葡聚糖2000理论塔板数为1356,拖尾因子为0.79。在两种流动相中蓝色葡聚糖2000峰保留时间比值0.98,对照溶液主峰中聚合物峰与相应色谱系统中蓝色葡聚糖峰的保留时间比值为1.00,供试品溶液主峰中聚合物峰与相应色谱系统中蓝色葡聚糖峰的保留时间比值为1.00。系统适用性溶液以流动相A测定,记录图谱,高聚体的峰高与单体和高聚体之间的谷高比值为3.9。结果表明:葡聚糖G-10凝胶色谱柱能满足分析要求。[img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221931500102_136_3170710_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221931501152_5601_3170710_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221931503923_2007_3170710_3.jpeg[/img][img]https://ng1.17img.cn/bbsfiles/images/2018/08/201808221931506933_7008_3170710_3.jpeg[/img]
美国药典标准测定头孢地尼含量色谱条件溶液A:14.2mg/ml无水磷酸氢二钠溶液B:13.6mg/ml磷酸二氢钾缓冲溶液:将溶液A与溶液B按2:1混合(pH7.0)溶液C:0.1%四甲基氢氧化铵水溶液,用1/10稀磷酸调节pH至5.5溶液D:37.2mg/mlEDTA二钠流动相:乙腈、甲醇、溶液C、溶液D (350:200:4500:2)系统适应性溶液:用缓冲溶液配制每0.2mg/ml头孢地尼RS和0.5mg/ml头孢地尼有关物质A RS标准溶液:用缓冲溶液配置0.2mg/ml头孢地尼RS标准品样品溶液:用缓冲溶液配制0.2mg/ml头孢地尼样品色谱柱:C18柱(5um,4.6*150mm)推荐:YMC-Pack ODS-AM(P/N:AM12S05-1546WT)检测波长:254nm柱温:40摄氏度流速:1.0ml/min进样量:5ul系统适应性样品:系统适应性溶液和标准溶液(注:头孢地尼有关物质A RS有4个色谱峰)拖尾因子:以头孢地尼峰计不得超过1.5(系统适应性溶液)分离度:头孢地尼有关物质A RS的第二个峰与头孢地尼峰的分离度不得小于1.2

Venusil XBP C18(L)分析头孢噻肟钠的分析报告摘要:本实验按照头孢噻肟钠2010版中国药典方法进行测定,以含量测定和有关物质检测方法共同进行色谱柱的筛选。在选定的色谱柱上,进一步考察了该色谱柱用于头孢噻肟钠系统适用性、含量、有关物质等项测试的结果,并初步考察了色谱柱的使用寿命,结果表明VenusilXBP C18(L)适用于头孢噻肟钠的分析:(1)系统适用性溶液中共检出7个杂质,分离度均符合标准要求;(2)对照品平行进样6针RSD为0.13%,保留时间17.039分钟,柱效5068,拖尾因子1.116,均符合标准要求;(3)含量测定中,头孢噻肟钠保留时间16.936分钟,柱效4779,拖尾因子1.118,符合标准要求;(4)有关物质测定中共检出6个杂质,分离度均符合标准要求;(5)用于头孢噻肟钠含量测试时,连续进样300针,保留时间、柱效和拖尾因子均无显著变化,表明VenusilXBP C18(L)对该样品和流动相有较好耐受性。关键词:头孢噻肟钠;VenusilXBP C18(L);2010版药典;液相色谱法前言头孢噻肟钠为中国药典2010版二部收录品种,本实验按照该标准进行测试,通过测试得出VenusilXBP C18(L)适用于头孢噻肟钠分析,其测试结果令人满意。实验部分试剂材料超纯水、甲醇、无水磷酸氢二钠、磷酸高效液相色谱柱:Venusil XBP C18(L);5 μm,150 Å,4.6 × 150mm样品制备系统适用性溶液制备:取头孢噻肟对照品适量,加流动相溶解并稀释制成每1 ml约含1 mg的溶液,作为系统适用性试验溶液。实验结果系统适用性测定结果表1. Venusil XBP C18(L)用于头孢噻肟钠系统适用性溶液测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410220596_01_2864683_3.png表2.Venusil XBP C18(L)用于头孢噻肟钠对照品溶液测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410251562_01_2864683_3.png含量测定结果表3. Venusil XBP C18(L)用于头孢噻肟钠含量测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410281917_01_2864683_3.png有关物质测定结果表4.Venusil XBP C18(L)用于头孢噻肟钠有关物质测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410305808_01_2864683_3.png色谱柱批次验证结果表5. Venusil XBP C18(L)批次验证结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410355084_01_2864683_3.png色谱柱寿命测试结果表6.Venusil XBP C18(L)用于头孢噻肟钠寿命测试结果http://ng1.17img.cn/bbsfiles/images/2015/08/201508241038_562414_2864683_3.png
[color=black]头孢哌酮钠(Cefoperazone Sodium)为第三代头孢菌素,属β-内酰胺类抗生素。[/color][align=left][img=,297,163]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120919066287_2141_2222981_3.jpg!w297x163.jpg[/img][/align][align=left][color=black]头孢哌酮钠 (M.W.667.65)Cefoperazone Sodium[/color][/align][color=black]CAPCELLPAK C18 MG[/color][color=black]和C18 UG120使用聚合物包被技术,抑制残留硅醇基的二次效应,同时采用超高纯硅胶,金属杂质更低,峰形更尖锐,非常适合具有配位结构、易与金属离子鳌合进而导致峰形拖尾的配位性化合物的分析。[/color][color=black]CAPCELLPAK C18 UG120[/color][color=black]的表面极性非常低,因此极性化合物可以以尖锐峰形快速溶出,同时也能取得良好的分离效果,具有独特的分离特性。而CAPCELL PAK C18 MG的整体保留能力较UG120更强,从极性化合物到疏水性化合物,对应各种极性的化合物都具有高保留能力和高柱效,适合大多数的一般分析。[/color][img=,400,231]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120924348057_9069_2222981_3.jpg!w900x522.jpg[/img][img=,400,289]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120924401677_8334_2222981_3.jpg!w900x651.jpg[/img][align=left][b][color=#0070c0][/color][/b][/align][align=left][b][color=#0070c0][/color][/b][/align][align=left][b][color=#0070c0]实验方法[/color][/b][/align][color=black]按照2015年版《中国药典》头孢哌酮钠含量测定项下方法对头孢哌酮钠原料药进行分析。[/color][color=black][/color][img=,400,236]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120930296387_4566_2222981_3.jpg!w735x435.jpg[/img][img=,400,251]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120930391737_1789_2222981_3.jpg!w755x474.jpg[/img][align=left][color=black] 图1 CAPCELL PAK C18 UG120分析结果 [/color][color=black][color=black]图2 CAPCELL PAK C18 MG分析结果[/color][/color][/align][color=black][/color][align=center][color=black] [/color][/align][align=left][color=black] [img=,400,117]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120933467027_8088_2222981_3.jpg!w900x264.jpg[/img][img=,400,134]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120933567447_2275_2222981_3.jpg!w900x303.jpg[/img][/color][/align][align=left][color=black] 表1 CAPCELL PAK C18 UG120结果详表 [/color][color=black]表2 CAPCELLPAK C18 MG 结果详表[/color][/align][align=left][color=black][img=,400,185]https://ng1.17img.cn/bbsfiles/images/2019/03/201903120934501237_9662_2222981_3.jpg!w900x418.jpg[/img][/color] [/align][color=black]综上所述,CAPCELL PAK C18 MG和C18 UG120两款色谱柱均能实现头孢哌酮钠的良好分离,头孢哌酮主峰不对称因子分别为1.23和1.38;其中,MG色谱柱的保留能力相对较强,而UG120可使极性物质快速溶出,用户可结合实际情况进行选择。[/color]
老师您好,做头孢拉定的含量,4.6*5um,150mm,C18柱,色谱柱只用了100针就不行了,开始使用时没有用甲醇洗,请教原因。谢谢
头孢丙烯片检测报告一.样品分子结构http://ng1.17img.cn/bbsfiles/images/2013/12/201312031058_480423_1621890_3.gif二. 样品来源记录样品化学名:头孢丙烯样品商品名:施复捷样品测定描述:主成分溶出度生产厂家:中美上海施贵宝制药有限公司三. 液相方法条件方法来源:(根据中国药典2010年版二部)具体方法:色谱柱:月旭Welchrom C18, 5μm, 4.6×150 mm(货号:WEL518415;序列号:W11212195;批号:W1811.02)波长:280nm流动相:磷酸二氢铵溶液(取磷酸二氢铵20 .7g,加水1800ml使溶解,用磷酸调节pH值至4. 4)-乙腈(90:10)梯度:等度柱温:室温流速:1.0ml/min进样量:10μl流动相的配制:色谱纯乙腈和磷酸二氢铵溶液均抽滤,过0.45μm滤膜;溶出度测定对照品溶液的配制:精密称取头孢丙烯对照品(来源:中检所,批号:130567-200902,含量:94.9%,规格:100mg)适量,置100ml量瓶中,加水溶解并定量稀释制成每lml中约含 0.28mg的溶液,作为对照品溶液;溶出度测定样品溶液的配制:取本品,照溶出度测定法(附录X C第一法),以水900ml为溶出介质,转速为每分钟100转,依法操作,45分钟时取溶液适量,滤过,取续滤液作为供试品溶液;系统适应性要求:头孢丙烯(Z)异构体峰与(E)异构体峰的分离度应大于2.5。以对照品溶液作为系统适用性溶液,头孢丙烯主峰由两峰构成,两峰面积之和以外标法计算每片的溶出量(%),出峰保留时间较前为头孢丙烯(Z)异构体峰,较后为头孢丙烯(E)异构体峰。备注: 1. 该项目对溶出介质的体积应控制在±1%; 2. 该项目对溶出介质的温度应控制在±0.5℃; 3. 流动相最好临用临配。四.
例如:头孢呋肟酯对照品,HPLC含量测定用时,含量96.9%,而用于UV检测溶出度时,含量为98.8%,为什么一个对照品有两个含量?检测方式不同么?
请问一下头孢氨苄有关物质取样量是怎么计算的上液相用的
进口兽药质量标准硫酸头孢喹肟注射液Liusuan Toubaokuiwo ZhusheyeCefquinome sulfate Injection本品为硫酸头孢喹肟与油酸乙酯等配制而成的混悬注射液。含头孢喹肟(C23H24N6O5S2)应为标示量的90.0%~105.0%。【性状】 本品为类白色至浅褐色混悬液体;久置分层。【鉴别】(1)含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品峰的保留时间一致。(2)取摇匀后的供试品2 ml,加水5 ml,稀盐酸1 ml,混匀,置超声浴中超声10分钟,弃去有机层,溶液显硫酸盐的鉴别反应(附录15页)。【检查】有关物质 照含量测定项下的方法。取摇匀后的供试品1.0 ml,加入流动相25.0 ml,置超声浴中超声5分钟,弃去有机层,取水层滤过,取续滤液10µ l,注入液相色谱仪,记录色谱图,2,3-环己基吡啶与头孢喹肟相对保留时间为0.20。按峰面积归一化法计算,2,3-环己基吡啶应不得过3.0%,其他单一杂质应不得过0.50%,杂质总量应不得过4.0%。水分 取本品,照水分测定法(附录58页,第一法)检查,含水分不得过0.2%。细菌内毒素 取摇匀后的供试品2 ml与细菌内毒素检查用水3 ml混匀,分成2等份,振摇30秒,离心15分钟(2000g),吸取水层1 ml,加1 mol/L氢氧化钠溶液0.06 ml调节pH值至6.5~7.5。用细菌内毒素检查用水按1:10稀释后,照细菌内毒素检查法(附录73页)检查,每1 mg头孢喹肟中含细菌内毒素的量应小于0.1 EU。无菌 取供试品8瓶,混合均匀,加入含6%吐温-80的蛋白胨缓冲液(1g/L)400ml,混匀,加入800×106单位青霉素酶(每1ml供试品溶液,加2×106单位青霉素酶),充分振摇,将供试品倒置,在37℃放置4小时;取供试品溶液,依法检查(附录79页,直接接种法),应符合规定。分散性 取本品1瓶,振摇30秒,将供试品转移置玻璃容器中,不得观察到结块或沉淀物。沉降 取本品1瓶,振摇30秒,取供试品10 ml置刻度试管中(内径1.0~1.5 cm),10分钟内不得沉淀。粒度 取摇匀后的供试品,置显微镜下检查,颗粒直径在5µ m以下应不得少于80%,10µ m以下不得少于90%,20µ m以下不得少于95%,50µ m以下不得少于100%。装量 按最低装量检查法(附录67页)检查,应符合规定。【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;取一水合高氯酸钠3.45g溶于1000 ml水中,加磷酸12 ml和乙腈90 ml,用三乙胺调节pH至3.6为流动相;检测波长为270 nm。取头孢噻肟约25 mg,溶于100.0 ml流动相中,另取头孢喹肟约25 mg,置25 ml量瓶中,精密加入上述头孢噻肟溶液1 ml,用流动相稀释至刻度。精密量取10µ l注入液相色谱仪,记录色谱图;计算头孢喹肟与头孢噻肟的分离度,应大于1.0。
头孢类抗生素在食用动物中的有效分析一直是热点。分享一种分子印迹固相萃取联合高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱仪[/color][/url]紫外检测器(MISPE-HPLC-UV)检测食品样品中头孢噻呋钠(CTFS)的方法。在该方法中,首先合成了一种环保的分子印迹聚合物(MIP)并用作吸附剂,它对水中的CTFS表现出优异的选择性,并且可以在1小时内达到吸附平衡。在优化的条件下,CTFS 在 0.005–1.0 mg L -1范围内获得了良好的线性,定量限为 0.0015 mg L -1,在牛奶、鸡肉、猪肉和牛肉样品的三个加标水平下,平均回收率高于 91.9%(RSD 小于 8.5%)。20个循环后,用于CTFS的MISPE小柱的回收率仍高于95%,证明了MISPE-HPLC-UV方法对食品样品中CTFS的分析具有较高的灵敏度和选择性。相关研究详见https://doi.org/10.1016/j.foodchem.2021.129013
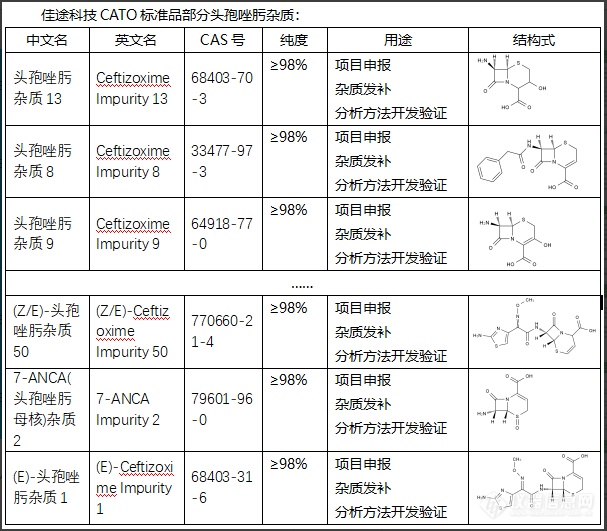
有关头孢唑肟杂质的作用,以下是要注意的一些可能性:1.负面作用:过多的杂质可能导致药物效力下降,并可能引发不良反应或副作用。例如,有些杂质可能导致过敏反应。2.毒性:某些杂质可能具有毒性。例如,某些杂质可能具有致癌性。3.影响药效:杂质可能会影响药物的生物利用度,即药物进入体内后能达到预期药效的能力。CATO标准品药品生产中的质量控制步骤非常重要,目的就是要尽可能减少杂质的存在。任何药品都必须经过严格的质量检测,确保其安全有效。[img=,607,531]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041449269442_4660_6381668_3.png!w607x531.jpg[/img]
[size=3][b]石墨炉测定植物中Pb,Cr的含量[/b][/size]请问专家老师,我是一个新手,想测定植物中 Pb,Cr的含量,我需要怎么做?

[size=4]简述HPLC测定药品含量的步骤[/size][U]目 的:[/U]对于刚接触HPLC(液相色谱仪)的工作人员来说,在实验前感觉有很多事情都要同时去做,且思路不是很清晰。现在,我就用十个步骤来说明用HPLC测定药品含量的全过程,供大家参考。[U]仪 器:[/U]安捷伦1100型HPLC(A、B、C、D四元泵,配200mm的C18柱)[U]药 品:[/U]头孢噻肟钠(头孢类无菌原料)[U]关键词:[/U]HPLC、色谱柱、步骤、流动相、基线、平衡、天平、溶解、校正因子、含量测定安捷伦1100型HPLC图1:[img]http://ng1.17img.cn/bbsfiles/images/2009/08/200908192122_166853_1622024_3.jpg[/img]

近日接触到一个药品——头孢拉定胶囊。其中检查项目有项是检查头孢氨苄的含量。见下图。http://ng1.17img.cn/bbsfiles/images/2014/09/201409051634_512996_0_3.jpg下图是头孢拉定项下的做法http://ng1.17img.cn/bbsfiles/images/2014/09/201409051637_512998_0_3.jpg 对此的疑问如下: 1、头孢拉定胶囊中为何要检查头孢氨苄,是否说明在生产头孢拉定中不可避免的会有头孢氨苄呢? 2、在检查中测定出头孢氨苄含量,还得测定出头孢拉定含量,这样才能计算出结果是吗? 3、标准规定的好奇怪,直接在胶囊剂的检查项下规定“含头孢氨苄不得过5.0%(举例说个数字啊,也可以是6.0%)”这样不就可以了吗,为何要规定成“含头孢氨苄不得过头孢拉定和头孢氨苄总量的6.0%”
请教各位老师:我现在遇到一个问题,购买了批号为HOJ296的头孢曲松钠对照品,测得水分是10.5%,标签给出的无水含量是92.4%,折算成这批对照品的含量是82.7%。之前我们常用的EP对照品的含量是83.7%。我们现在有个怀疑,测出的水分是不是太高了,不知道是真实的水分就是这么高还是我们测水分的仪器的问题。而且USP对照品也比较贵,我们也不能多次测试。想请问下还有没有人用这批对照品,或者用过USP头孢曲松对照品,它的水分经验值是多少?谢谢
我这可以做有含量的质量控制塑料样品(含重金属的和邻苯的),大家可以来做比对的。
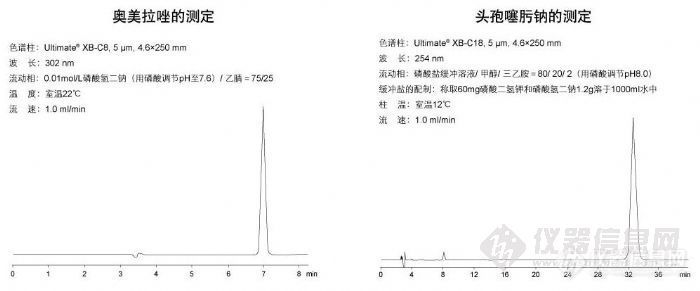
奥美拉唑的测定和头孢噻肟钠的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021837_180180_1896702_3.jpg
以头孢曲松钠聚合物举例:按外标法以峰面积计算,含头孢曲松聚合物以头孢曲松计,不得过0.5%。 A供*M对*对照纯度聚合物%=----------------------------- A对*供试量*?举例来说,供试量为0.2g,百分含量为76.0%,?这个地方应不应该乘以76.0%,有何依据,不乘又有何依据?期盼得到各位老师的指导。个人意见:应该乘以头孢曲松的纯度76.0%,“含头孢曲松聚合物以头孢曲松计”,这里的头孢曲松我理解为供试品中的纯头孢曲松,因为是它产生的聚合物。如果那位老师有兴趣可以用QQ305115490和邮箱zongrui0911@163.com和我联系。寇宗睿
我按照药典规定的方法测定头孢他啶聚合物含量,可做了好多次也检测不出头孢他啶聚合物峰,这是怎么回事?望各位大虾指教!
求头孢c类中间体7aca的液相分析条件下内标法测7aca的含量(请注明所用流动相以及所用内标物质,谢谢)
应符合中国药典2000年版附录V D高效液相色谱法及附录XIX A药品质量标准分析方法验证的有关规定。若另有规定者应于正文中写清楚,如拖尾因子,附录中规定应在0.95~1.05,某品种若不能达到,可另制订合适指标。 一. “含量测定”可按下述格式表述 多数抗生素类药品的含量测定采用的是HPLC-紫外检测器法,其书写格式包括以下内容: (一)照高效液相色谱法(附录V D)测定。 (二)色谱条件与系统适用性试验:包括以下内容:色谱柱填料种类,流动相组成及配制方法,流速,检测波长,柱理论板数,拖尾因子,分离度测定溶液配制方法及分离度要求,进样量,数次进样对照品溶液应达到的精密度等。 注:1. 如果理论板数对分离度及定量测定结果影响不显著时可省略。 2 如果色谱峰的对称性较好,可不规定拖尾因子,但对称性差的品种应规定拖尾因子。 3. 考核分离度的方法有: (1)用已知杂质对照品,测定其与主组分峰的分离度,如头孢克洛含量测定时规定头孢克洛与d-3异构体对照品混合溶液的分离度应符合规定。 (2)杂质对照不能获得的情况下,可通过样品的适当处理使降解,测定降解物与主组分峰的分离度,如头孢呋辛钠含量测定中,取对照品溶液在60℃水浴中加热10分钟,冷却,使部分头孢呋辛转变为去氨基甲酰头孢呋辛后,测定头孢呋辛峰与去氨基甲酰头孢呋辛峰的分离度。 (三)对照品溶液的制备:应写清楚对照品溶液制备所用溶剂及溶液浓度。若储备液与测定液溶剂(稀释液)有差异时,应分别写清楚配制方法。 (四)测定法:应写清供试品溶液的配制方法。对于制剂若由于辅料会影响取样均一性者,还应规定合适的取样量。对于溶液稳定性差的品种,应写清“临用前配制”或“供试液应冷处保存”等。含量测定溶液浓度的确定,除了应在线性范围内外,应以规定的进样量测定的峰面积积分值在6位数以上为宜。 另外,也有一些无特征紫外吸收物质,例氨基糖苷类抗生素,可以采用HPLC-蒸发光散射检测器(Evaporative Light-Scattering Detector, ELSD)法测定含量。当采用该法测定含量时,书写格式可参考上述HPLC-紫外检测器法,但应注意以下几点: (1)“色谱条件与系统适用性试验”项:液相色谱条件中应注明流速,因为流速大小直接影响ELSD的检测器条件;检测器条件中应注明漂移管温度,载气流速。系统适用性试验一般要包括数次进样对照品溶液应达到的精密度(RSD),以确保检测器的稳定性(一般来说,该法要求RSD不得过2%)。 (2)“对照品溶液的制备”项:由于ELSD的响应值(A)与进样浓度(C)间呈指数关系(A=aCb,a,b均为常数),在采用该法测定含量时,计算方法宜采用随行校正曲线法(lgA=algC+b,a,b分别为校正曲线的斜率、截距),故对照品溶液的浓度至少应包括高、中、低3个不同浓度,浓度设计以能覆盖供试品溶液浓度为宜。 (3)“供试品溶液的制备与测定”项:同HPLC-紫外检测器法,但计算方法采用的是随行校正曲线法,即供试品浓度是根据供试品溶液中主峰峰面积和随行校正曲线(lgA=algC+b)来确定的,具体计算方法可参见“3 项撰写细则”一章中的“1.7 组分检查或纯度检查”部分。 二. 有关物质测定* (一) 有关物质测定的色谱条件与含量测定相同者,可参照中国药典2000版中头孢呋辛钠有关物质检查法表述。但其中有如下几点不完美: 1. 供试品溶液浓度低于含量测定时供试品浓度,有可能影响杂质的检测,应适 当提高。 2. 结果评价应以加校正因子或不加校正因子的主组分自身对照法计算出单个最大杂质或表观含量大于0.1%含量以上的杂质总和的含量或表观含量,而不以单个最大杂质或杂质峰面积总和不得大于对照品溶液主峰峰面积的多少倍表示。 (二) 有关物质测定的色谱条件与含量测定不同者,可参照中国药典2000年版中头孢克洛有关物质检查法表述,但其中有如下表述方法需作修改。 1. 流动相梯度洗脱方式以列表方式表述如下: 流动相A与流动相B按下表变换方式进行洗脱: 时间(分) 流动相A(%,V/V) 流动相B(%,V/V) 0~30 95%®75% 5%®25% 30~45 75%®0% 25%®100% 45~55 0 100% 55~56 0®95% 100%®5% 56~71 95% 5% 2. 结果评价方式同上,应计算出单个最大杂质含量或表观含量大于0.1%以上的杂质总和的含量或表观含量。 (三)有关物质测定中明确规定对照品溶液数次进样后相对标准偏差的限度,并以数次进样的平均值计算出各有关物质的量。 (*注:最低检出限或最低定量限的确定应按照中国药典的要求进行验证,以保证结果的可*性。)
请教各位,如果我要分析纯三大酸,双氧水中的金属元素含量,我该怎么稀释做? 我用的是ICP-OES
请问,我要检测土壤中的氨氮含量怎么做?是先用蒸馏水稀释为液体的在检测吗?我有水质检测仪检测氨氮的仪器是连华的,但是现在要做土壤的,请问能做吗?







 我要推广仪器
我要推广仪器
 下载APP
下载APP