最近检测 戊唑醇在小麦 土壤中的残留,查见许多方法提到醋酸铅,检测方法大体如下“称取10g左右样品于 250 ml具塞三角瓶中,加85 ml丙酮加20 ml甲醇浸泡过夜,振荡提取1 h,合并滤液于500 ml抽滤瓶,加100 ml 水3 ml [color=#fe2419]醋酸铅饱和溶[/color]液,10 ml氯化钠饱和溶液,摇匀静止5 min后抽滤,滤渣用30 ml二氯甲烷洗2次,合并滤液于500 ml分液漏斗中,充分振摇,收集二氯甲烷,浓缩至1~2 m.,用氮气吹至近干,用丙酮定容5~10 ml待[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]测定。“别的方法也大同小异 我看也有用761的方法 直接用乙腈提取的,不知道以前的方法 用到醋酸铅 是什么意思 他在这起到什么作用,能推广到其他实验吗?着急期待中。
进样空白里面有残留峰,不进样就没有残留峰,残留物质 萘甲醇,我考虑是六通阀被污染,拆下来用水超声还是有残留,请问还有哪里可能被污染了。也反冲了针座。
进样空白里面有残留峰,不进样就没有残留峰,残留物质 萘甲醇,我考虑是六通阀被污染,拆下来用水超声还是有残留,请问还有哪里可能被污染了。也反冲了针座。
如题,最近做实验常用无水乙醇(分析纯)擦洗器皿,但是感觉会残留一些味道,又不像是酒精的味道,再说酒精挥发也很快,不知这样正常吗?据说酒精提纯会加苯系物,会不会是那些东西?大家用酒精时是什么样的?如果换优级纯的会好吗?谢谢~~~
谁有柑桔中烯唑醇农药的残留检测方法?
动物组织中检测喹乙醇标示残留物:3-甲基-喹恶啉-2-羧酸,不知哪位大虾有这方面经验,请赐教。
求助NY 1500.69.1-2009 农药最大残留限量 已唑醇 葡萄 有这个标准的达人请共享一下,谢谢!!
版友问题,做乙醇甲苯残留溶剂顶空进样不出峰,做其他的直接进样正常,会有什么原因呢?
甲醇残留溶剂直接进样法可以做吗?水当溶剂,为什么直接进样峰型超级难看,而且出了2个峰,仪器用的是岛津2014,FID检测器[img=,690,517]http://ng1.17img.cn/bbsfiles/images/2018/01/201801081507_4859_3352426_3.png[/img]
顶空仪器做甲醇,乙醇,二氯甲烷,用DMSO做溶剂后有残留,后来还做过甲醇,乙醇,二氯甲烷,醋酸用水做溶剂。现在是做空白水样,和空顶空瓶都有很大的残留 最高峰面积三千多,最小也有一千多。拆了顶空里的气路都用丙酮和清水超声过,还有就是做了空哇哈哈水样高温清洗顶空系统。现在的结果要好一些,但是在进醋酸水做溶剂时又出现残留。各位有没有遇到这样的问题?是DMSO残留比较严重,还是在顶空里做醋酸有影响呢?请各位老师有这方面经验的解答一下
根据欧盟委员会(EC)No 396/2005法规第6节的规定,英国收到一份来自Syngenta Crop Protection公司要求修改油菜籽(rape seed)中的环唑醇(cyproconazole)最大残留限量(MRL)的申请。为了使环唑醇在英国的使用能达到预期效果,欧盟拟建议将油菜籽中环唑醇的最大残留限量由0.1mg/kg提高至0.3mg/kg。EMS依据欧盟委员会(EC)No 396/2005法规第8节的规定起草了评估报告草案,并提交至欧盟委员会,并于2010年9月13日转至欧洲食品安全局(EFSA)。此前在对申请进行评估的过程中,EFSA发现申请的环唑醇MRL值并不符合法律规定。因此,MRL申请被搁置,直至2011年4月26日英国再次提出有效申请。欧洲食品安全局对提交的评估材料进行评估后,得出的结论为:代码商品现行MRL值(毫克/千克)建议MRL值(毫克/千克)建议理由执行的残留物质:环唑醇0401060油菜籽0.10.3该建议MRL值有充足的数据支持,在使用时被鉴定为不会对消费者产生风险。
在做食品添加剂分析时,检测甲醇乙醇残留,最近进空样都有 甲醇残留,导致检测出来甲醇偏高,有什么好的清理办法吗。
不知道有没有哪位做过乙二醇的残留分析,你们是用何种方法测定的。目前我使用的方法是用正戊醇做内标,DB-624的柱子,目前可以勉强把它们分离开,但是乙二醇拖尾严重,尤其走样品时,不知道是不是因为样品的问题,乙二醇出峰就像山包一样,无法准确积分。色谱条件:进样器:250度 检测器:280度 柱温:60度保持3分钟,10度/分升至150度
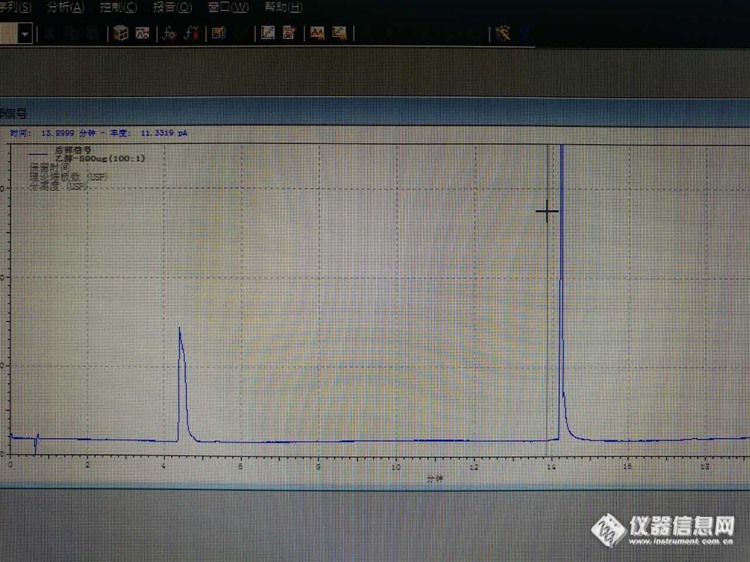
安捷伦7820A+7697测乙醇残留,空白有干扰,柱子DB-624,30m*0.53mm,程序升温:40度,恒温2分钟,以每分钟10度的速率升至160度,保持2分钟,开始怀疑有残留,工程师对进样针,定量环都清洗了,还是有。最后结论是空气中的。用氮气吹过的顶空瓶也有干扰。求助各位大神帮忙指导一下。照片是空白(水),4.3分钟是乙醇出峰时间。[img]https://ng1.17img.cn/bbsfiles/images/2018/11/201811172235170476_4006_3102197_3.png[/img]
最近做一个原料药的 溶剂残留检测 DMF 和乙醇单位的气相比较简陋 没有顶空进样器。搜索了下论坛发现直接进样检测,用DMSO 做溶剂。 我就参照两个方法。设定了检测条件 用二氯甲烷做内标物 程序升温检测结果:供试溶液的内标物峰面积要比对照溶液的小一半 !PS:产品能溶在溶剂里 但是产品的沸点很高 300多 请问各位是不是溶剂选择不对 还是内标物选择不对?
[size=16px] 金标仪能检测哪些药物残留 金标仪(或金标读卡仪)能检测的药物残留主要包括以下几类: 真菌毒素残留类:如食用油、粮食及饲料中的黄曲霉毒素B1、液态奶中的黄曲霉毒素M1、食品中的呕吐毒素、玉米赤霉烯酮、赭曲霉毒素A等。 激素残留类:如莱克多巴胺、克伦特罗、沙丁胺醇、己烯雌酚等。 水产品安全类:包括呋喃妥因代谢、呋喃西林代谢、呋喃它酮代谢、呋喃唑酮代谢、孔雀石绿、氯霉素等。 抗生素残留类:如磺胺、喹诺酮、喹乙醇等。 此外,金标仪还可以检测干式法试纸条食品中的有毒有害物质、非法添加剂类、残留类等。 需要注意的是,具体的检测项目可能会因仪器型号、生产厂家、使用场景等因素而有所差异。因此,在选择和使用金标仪时,应仔细了解其检测范围和使用方法,并根据实际需求进行选择。[img=,690,690]https://ng1.17img.cn/bbsfiles/images/2024/05/202405091054340133_2087_6098850_3.jpg!w690x690.jpg[/img][/size]
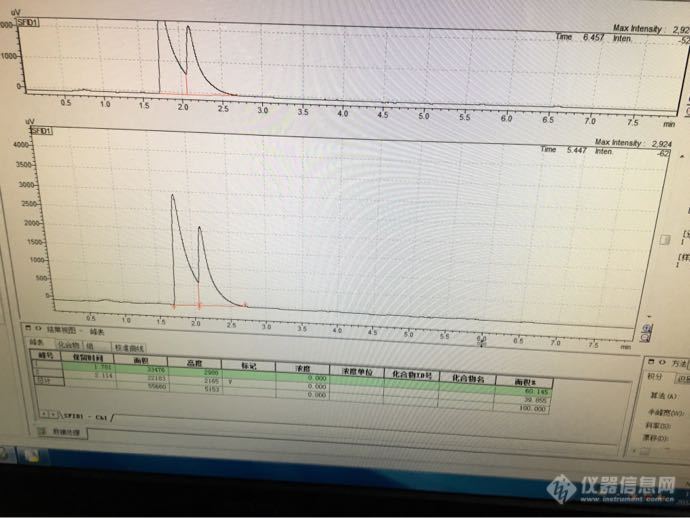
做乙醇残留实验,正在前期摸索阶段,但是峰型一直不好~~~我们用的是安捷伦气相色谱7820A,色谱柱是DB-624(30*0.32mm,1.4μm);FID检测器,柱温40℃,恒温2min,10℃/min升至220℃,恒温2min,检测器温度250℃;进样口温度240℃,流速1.5ml/min,分流比50:1;顶空条件:样品温度80℃,加热箱100℃,传输线105℃,平衡时间30min。我们前期摸索是用乙醇,加DMSO稀释的,跑出来的峰型是这样的,怎么解决,是什么原因造成的呢?前边的峰是乙醇峰,后边的是DMSO峰http://ng1.17img.cn/bbsfiles/images/2016/11/201611291119_01_2822825_3.jpg
1.分析目标化合物 恶唑菌酮 2、仪器设备 带紫外分光光度检测器的高效液相色谱仪(HPLC(UV)) 液相色谱--质谱仪(LC/MS) 3、试剂 丙酮 氯化钠溶液 正己烷 无水硫酸钠 乙腈:高效液相色谱用 甲醇:高效液相色谱用 恶唑菌酮标准品:含恶唑菌酮98%以上,熔点为140℃~143℃。 4.试验溶液的制备 1) 提取方法 豆类:称取10.0g样品,加入20mL水,放置2小时。 水果和蔬菜:称取20.0g样品。 加入100mL丙酮,均质后,抽滤。滤纸上的残留物中加入50mL丙酮,均质后,按上述同样操作,合并所得的滤液。40℃以下浓缩至约30mL。浓缩液中加入100mL 10%氯化钠溶液,分别用100mL和50mL正己烷振荡提取两次。提取液中加入无水硫酸钠脱水,滤去无水硫酸钠后,滤液在40℃以下浓缩,除去溶剂。残留物中加入5mL乙醚:正己烷(1:19)混合溶液溶解。 2)净化方法 ①硅胶柱色谱法 在硅胶小柱(690mg)中注入5mL正已烷,舍弃流出液,注入1)所得到的溶液,舍弃流出液。注入10mL乙醚:正己烷(1:19)混合溶液,舍弃流出液。再注入20mL乙醚:正己烷(3:7)混合溶液,溶出液40℃以下浓缩,除去溶剂。残留物中加入2mL丙酮:正己烷(1:19)混合溶液溶解。 ②酰胺丙基甲硅烷基化硅胶柱色谱法 在酰胺丙基甲硅烷基化硅胶小柱(500 mg) 中依次注入5mL丙酮:正已烷(1:19)混合溶液和5mL正已烷,舍弃各流出液。注入①所得的溶液,舍弃流出液。再注入8mL丙酮:正已烷(1:19)混合溶液,舍弃流出液。再注入20mL丙酮:正已烷(1:9)混合溶液,流出液40℃以下浓缩,除去溶剂。残留物中2.5mL甲醇溶解后,再加入2.5mL水。 ③ 十八烷基甲硅烷基化硅胶柱色谱法 在十八烷基甲硅烷基化硅胶小柱((500 mg ) 中依次注入5mL甲醇和5mL水,舍弃各流出液。注入②所得的溶液,舍弃流出液。再注入15 mL水:甲醇(1:1)混合溶液,舍弃流出液。再注入8mL乙腈:水(7:3)混合溶液,溶出液在45℃以下浓缩,除去溶剂。残留物溶解在乙腈:水(1:1)混合溶液中,准确至2mL(豆类为1 mL)作为试验溶液。
喹乙醇(N-羟乙基-3-甲基-2-喹啉酰胺-1,4-二氧化物)是一种化学合成抗菌促生长剂。1965年由德国拜尔公司等首先发现它对动物具有促生长作用。由于喹乙醇有中度至明显的蓄积毒性,对大多数动物有明显的致畸作用,对人也有潜在的三致性,即致畸形,致突变,致癌。因此喹乙醇在美国和欧盟都被禁止用作饲料添加剂。《中国兽药典》(2010版)也有明确规定,喹乙醇被禁止用于家禽及水产养殖。农业部在2001年第168号公告中就作了严格规定:只能用于体重低于35千克的猪。由于喹乙醇曾经的广泛使用和较大危害性,对其进行残留监控十分必要。喹乙醇本身不稳定,在动物体内能够在短时间内代谢,其在动物体内有十多种代谢产物,其中3-甲基喹噁啉-2-羧酸(MQCA)是主要代谢物,在体内相对稳定。因此,在检测饲料时,可检测喹乙醇原形物,但在检测食品及动物产品(肉、肝脏、水产品等)时应检测喹乙醇代谢产物。目前喹乙醇及其代谢产物的液相色谱及液相色谱-质谱检测标准主要有:1.饲料类GB/T8381.7-2009 饲料中喹乙醇的测定 高效液相色谱法DB43/T 891-2014 饲料中喹乙醇、氰乙基-(2-亚甲基肼喹噁啉基)-N,N-二氧化物(喹赛多)、卡巴氧的测定 液相色谱-串联质谱法(暂无文本)农业部2086号公告-5-2014 饲料中卡巴氧、乙酰甲喹、喹烯酮和喹乙醇的测定 液相色谱-串联质谱法2.食品及动物产品GB/T 20746-2006 牛、猪的肝脏和肌肉中卡巴氧和喹乙醇及代谢物残留量的测定 液相色谱-串联质谱法GB/T 20797-2006 肉与肉制品中喹乙醇残留量的测定GB/T 22984-2008 牛奶和奶粉中卡巴氧和喹乙醇代谢物残留量的测定 液相色谱-串联质谱法SC/T 3019-2004 水产品中喹乙醇残留量的测定 液相色谱法SN/T 0197-2014 出口动物源性食品中喹乙醇代谢物残留量的测定 液相色谱-质谱/质谱法(暂无文本)农业部1077号公告-5-2008 水产品中喹乙醇代谢物残留量的测定 高效液相色谱法从上述标准可以看出,大部分食品及动物产品标准检测喹乙醇代谢物(MQCA)。但少数标准如GB/T 20797-2006、SC/T 3019-2004在动物产品及水产品中检测喹乙醇原形物,存在瑕疵,显得不是非常严谨。
求助乙醇残留测定的方法
本人在测定乙醇残留时用的是水做溶剂,在只进溶剂测定时色谱图上与乙醇位置很接近处总有一小峰,我知道水是没有峰的,请问那是什么东西呢?我改变色谱条件都无法将其除去,也不能与乙醇位置分离,请问该怎么办?谢谢了
我们的一个产品用到乙二醇甲醚,需对产品中的乙二醇甲醚的残留进行控制。方法如下:色谱柱:DB-624进样口温度:200,检测器温度:300柱温 40保持4min,10/min升温至200保持7min流速:4.0ml/min分流比:1:1顶空条件顶空瓶平衡温度:140,定量环温度:150,传输线温度:160我们遇到的问题是:乙二醇甲醚在这种条件下的响应很低,500ppm的限度下峰面积为41.2,50ppm限度下无响应。而乙二醇甲醚在药典里规定的限度为50ppm请问,我们应该怎么改进条件来检测乙二醇甲醚残留?
有网友问:测定溶剂残留(甲醇、乙醇、苯、甲苯、三氯甲烷、二氯甲烷,醋酸),混合对照品、单个对照品、样品均有两个巨型矩形平头峰、残留物的锋型只有一个,保留时间一致,并且很小,1000多点。看不出对照品和样品之间的差异。 按照药典标准 第二法 ECD检测 顶空进样(加热箱温度100 定量环110 传输线115度) 检测残留物:甲醇、乙醇、苯、甲苯、三氯甲烷、二氯甲烷,醋酸 如果可以的话 做以上几种残留物 按照什么方法和条件做 比较可行 谢谢
这几天LC/MS/MS(thermo)在做沙星类药物,基质效应和回收率都不错,稳定性什么也挺好, 但没怎么考查残留。昨天才发现走,走完UOQ(2500NG/ML)后,在走空白针,有残留,信号和LLOQ一样大,内标也有残留。要冲好多针,才能冲干净,我的方法是100UL血样加1ML异丙醇 提取,100UL流动相复溶。为了让信号弱点,改成50血样,加1ML异丙醇 ,200体积流动相复溶。信号是下来了,还是有残留。 卸了柱子,用二通,进样针吸取UOQ后进样,在走空白样,还会有点残留,(那应该是进样针的问题了??)。 我的流动相是(0.1%甲酸)水:甲醇(0.1%甲酸)。 走个梯度, 离子源清洗过了,洗液也换成纯乙腈了,走LLOQ在走空白样,还是有百分之几的残留。不知道有什么好办法。会不会是进样针有污染,冲不干净?请大家给点意见,谢谢,
要做个燃烧残留物看有没有汽油等,看文献都要正己烷萃取,请问正己烷萃取具体怎么操作,然后怎么做GC/MS
今天遇到一个样品的残留了,而且很关键,所以一定要重新干净。 经过更换色谱柱确认,不是柱子残留;经过进空针和空白对比,确认进样才有残留,如果进空针的话无残留,确认残留是在进样针部分。 由于残留样品易溶于甲醇,故使用甲醇认真冲洗进样针,但是仍然有残留,貌似没有多大的变化,就差把针座拆下来超声清洗了。 求助,可能存在于哪里?如何清洗?实在不想拆针座,老是让我装的漏液,郁闷了!!!
求助各位老师,依然是残留问题,换了长、短柱,也换了苯基柱和C18柱,进标准样品后的空白总是有不同程度的残留,这个问题困扰我很久了,洗针液也加了异丙醇仍然不行,今天连接两通发现不会有残留,请问各位老师这样该如何判断残留出在哪里?进样量从5微升改为2微升还是有残留。现在怀疑是有机相甲醇无法将分析物全洗脱下来?但梯度里99%甲醇的时间是2.5分钟,换乙腈试试?还怀疑是柱前柱后的管路连接有问题?但的确诸多分析物中,含氯的分析物残留更严重,两三针空白才能冲洗干净,其他不含氯的分析物的残留一针空白后就可以冲洗干净了。谢谢各位老师指点,残留问题困扰我快一个月了、、、、
我们有一个酯类API,合成过程中加入正己醇生成的酯,该API需要测定正己醇残留,正己醇沸点约157℃,因此采用150℃的顶空条件测定样品,发现样品中的正己醇残留色谱峰面积其大,可能是该API在顶空条件下降解生成了正己醇,那么这样我如何实验证明该正己醇是降解的,样品中的正己醇残留应该怎么测定呢?谁有过类似的经历,分享一下。
[font=&]各位大神,我遇到顶空残留的问题,顶空进空气和水样都会有残留峰出现,而且测水峰面积比测空气大3-4倍,出峰时间在3.9和6.5分钟左右,但是进氮气就没有峰出现,顶空针,六通阀,切换阀,GC进样口,检测器口都洗过了,柱子也截了老化了,测试水和空气还是一样的峰[/font][font=&]空白溶剂是水 测乙醇残留 [/font][font=&]条件:恒温炉 80 样品流路 90 传输线105 [/font][font=&] 进样口 200 柱子程序升温50-220 [/font]检测器250 进样口压力42KPa 总流量8.1ml 柱流量 0.46ml 分流比10[font=&]GC是顶空和液体混用 液体进样测试的样品出峰面积有1亿多面积,也有1千多万的面积 [/font][font=&]最有疑问的是进了氮气就没有峰出现 [/font][font=&]现在怀疑分流流路和吹扫流路有残留[/font]且实验条件的流量偏小 但是测氮气也是一样的条件就没有峰就又矛盾了[font=&][/font][font=&] [/font][font=&] [/font]
[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相[/color][/url]做溶剂残留甲醇,用什么柱子最好,最低检出限能做到多少?







 我要推广仪器
我要推广仪器
 下载APP
下载APP