液质联用,由于测定药物溶解度的关系,在药物母液中加盐酸调了PH。对柱子、质谱有伤害嘛?知道是流动相肯定是不能用盐酸调PH的,但是,有其他前辈有的说母液加盐酸可用,有的说不可以。也问了维修工程师,说应该影响不大,但是他说具体找应用方面的工程师,今天休息。拜托各位了~
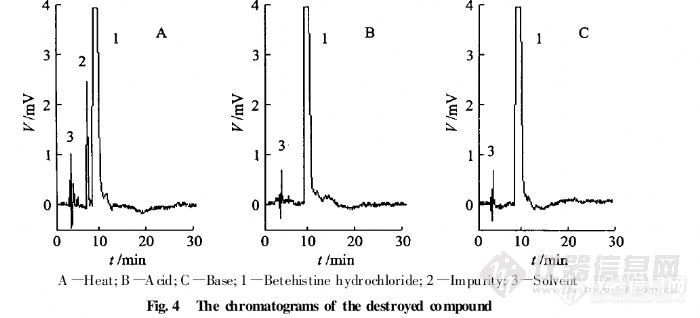
【作者】 陆榕; 孙进; 方金玲; 孙英华; 刘晓红; 何仲贵;【机构】 沈阳药科大学药学院; 沈阳药科大学药学院 辽宁沈阳110016; 辽宁沈阳110016;【摘要】 目的用离子对HPLC法测定盐酸倍他司汀注射液中药物及有关物质含量。方法色谱柱:Diamonsil C18柱(200 mm×4.6 mm,5μm);流动相:甲醇10 mmol.L-1醋酸钠溶液(含5 mmol.L-1庚烷磺酸钠,体积分数为0.2%的三乙胺,用冰醋酸调pH至3.3)(25∶75),检测波长:261 nm,流速:1.0 mL.min-1,柱温:室温。结果盐酸倍他司汀质量浓度在1~250 mg.L-1内线性关系良好(r=0.999 9),平均回收率为99.8%,RSD=1.0%。盐酸倍他司汀的理论塔板数大于2 000,盐酸倍他司汀与其主要杂质分离度不低于1.5。结论适用于盐酸倍他司汀注射液中药物与有关物质的含量测定。http://ng1.17img.cn/bbsfiles/images/2012/07/201207251621_379666_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207251623_379668_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207251623_379670_2379123_3.jpg

99.999%) 流速:0.9 mL/min 质谱条件 接口温度:280 ℃ 溶剂延迟:8 min EI 温度:230 ℃ 四极杆温度:160 ℃离子监测:定性离子:86、243、262、277 ;定量离子:86文章出处:P097关键字:克伦特罗;沙丁胺醇;西马特罗;莱克多巴胺,β -激动剂药物,动物组织,SPE,ProElut PXC,瘦肉精摘要:适用于动物肌肉和肝脏中盐酸克仑特罗、沙丁胺醇等4 种β -激动剂药物的测定谱图:http://ng1.17img.cn/bbsfiles/images/2016/05/201605130959_593204_1610895_3.jpg图例:1. 克伦特罗;2. 沙丁胺醇;3. 西马特罗;4. 莱克多巴胺
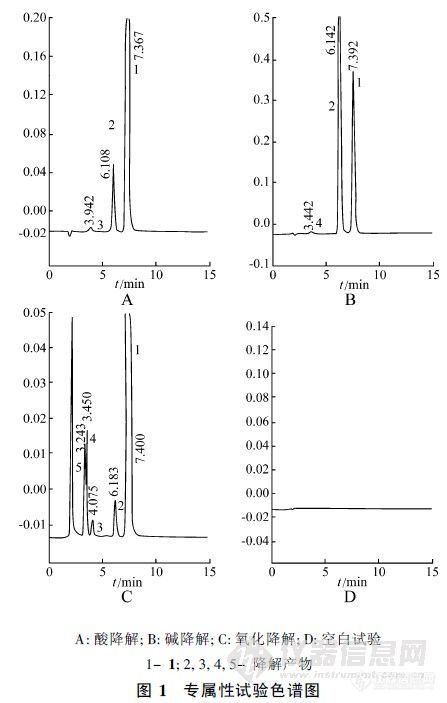
【作者】 王璐璐; 郑稳生; 谭小川; 张宇佳;【Author】 WANG Lu-Lu, ZHENG Wen-Sheng, TAN Xiao-Chuan, ZHANG Yu-Jia (Chinese Academy of Medical Sciences, Institute of Materia Medica, Beijing 100050)【机构】 中国医学科学院协和医科大学药物研究所; 中国医学科学院协和医科大学药物研究所 北京100050; 北京100050; 北京100050;【摘要】 采用 HPL C法测定注射用盐酸地尔硫的含量及有关物质。色谱柱为 Diamonsil C1 8( 15 0 mm× 4.6 m m,5μm ) ,0 .1m ol/ L醋酸钠缓冲液 ( p H6 .2 ) -甲醇 -乙腈 ( 5∶ 6∶ 6 )为流动相 ,检测波长为 2 40 nm。检测限为 0 .2 ng,线性范围为 6~ 14μg/ ml( r=0 .9998) ,RSD为 0 .11% ,平均回收率为 10 0 .2 % ,RSD为 0 .5 2 %。 更多还原【Abstract】 A HPLC method for the determination of diltiazem hydrochloride for injection and its related substances was presented. Diamonsil C18 column was used, with mobile phase of 0.1 mol/L sodium acetate buffer solution (pH6.2)-methanol-acetonitrile (5∶6∶6) and at detection wavelength of 236 nm. The detection limit was 0.2 ng. The linear range was 6~14 μg/ml, and average recovery was 100.2% with RSD of 0.52%. 更多还原【关键词】 注射用盐酸地尔硫; 有关物质; HPLC; 测定; 【Key words】 diltiazem hydrochloride for injection; related substance; HPLC; determination; http://ng1.17img.cn/bbsfiles/images/2012/08/201208201140_384595_2352694_3.jpg

盐酸***(涉密)原料药GC分析方法1、所在地区 山东淄博 ,从事行业 制药,分析的物质名称或大致样品组成:分析原料药中有机残留,甲醇、二氯甲烷、四氢呋喃、正己烷、正溴丁烷的残留。2、分析方法: 自主研发 企业标准一、样品前处理:盐酸***中有机残留,外标法计算,残留有机溶剂不得高于药典相关规定。第一部分、甲醇 二氯甲烷 正己烷 四氢呋喃 第二部分、 正溴丁烷对照溶液的配制:[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906142043_155418_1612824_3.jpg[/img]5种残留对照[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906142045_155419_1612824_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906142045_155420_1612824_3.jpg[/img]配制对照所用到得量取工具,在小于10ul时用微量注射液量取,(一种溶剂一只针)[img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906142048_155421_1612824_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2009/06/200906142049_155422_1612824_3.jpg[/img]精密量取甲醇38ul(相当于30mg)二氯甲烷4.6ul(6.0mg)四氢呋喃8.1ul(7.2mg)正己烷4.4ul(2.9mg)置100ml 加水适量充分振摇,并稀释到刻度: 精密量取0.2ml于顶空瓶中,密封 作为对照5个对照

盐酸可乐定的测定和盐酸丁咯地尔片含量的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021833_180179_1896702_3.jpg
国家食药总局:警惕部分食品中违法添加“盐酸西布曲明”和“酚酞”一、背景信息 近期,食品药品监管部门发现有减肥类产品添加“盐酸西布曲明”和“酚酞”的违法行为,并对该违法行为进行了查处。“盐酸西布曲明”和“酚酞”究竟是什么?是否可添加到食品中?有何相关标准和法规?本期为您解读。 二、专家观点 (一)盐酸西布曲明曾为处方药,但目前已在全球大多数国家停止使用。 盐酸西布曲明(Sibutramine Hydrochloride)是西布曲明(Sibutramine)的氯化物,是一种中枢神经抑制药物,曾用于肥胖症的治疗。 盐酸西布曲明曾于1997年经美国食品药品监督管理局(FDA)批准上市,随后在欧盟、澳洲、加拿大、日本等多个国家获得批准上市,2000年在我国上市。 2009年12月,欧洲药品管理局(EMEA)发布研究报告显示,与安慰剂对照组相比,服用盐酸西布曲明的患者发生严重、非致死性心血管事件的风险增加。欧盟、英国等国家和地区先后停止使用盐酸西布曲明类药品。原国家食品药品监督管理局也组织相关专家对西布曲明在我国使用的安全性进行了评估,认为其减肥治疗的风险大于效益,并于2010年10月通知要求停止生产销售使用西布曲明制剂及原料药。目前盐酸西布曲明作为减肥药已在全球大多数国家停止使用。
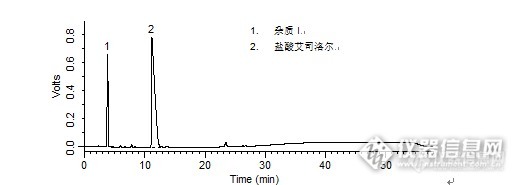
1. 杂质I2. 盐酸艾司洛尔 盐酸艾司洛尔样品制备 制备方法有关物质衍生溶液:取盐酸艾司洛尔对照品约10 mg,置10 mL量瓶中,加入1 mol/L盐酸溶液1 mL,放置30分钟,加1 mol/L的氢氧化钠溶液1 mL使中和,用流动相A 稀释至刻度,摇匀。分析条件 色谱柱Diamonsil C18(2) 250 x 4.6 mm,5 μm (Cat#:99603)流动相流动相A:乙腈:甲醇:磷酸盐缓冲液(取磷酸二氢钾3.0 g,加水至650 mL)=15:20:65流动相B:甲醇梯度流速1 mL/min柱温30 ℃检测器UV 222 nm进样量20 μL 色谱图有关物质衍生溶液http://ng1.17img.cn/bbsfiles/images/2016/04/201604211737_591070_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 分离度 1 3.842 6280189 655879 2747.059 0.670 -- 2 11.157 29271705 784686 1512.532 5.026 10.154 本品种同时使用了SpursilC18色谱柱,在药典规定条件下进行检测,满足药典要求。

今天将为大家介绍两个使用资生堂色谱柱对盐酸小檗碱进行分析的文献数据,请参考。盐酸小檗碱(Berberine Hydrochloride)分子式:C20H18ClNO4·2H2O 分子量:407.85黄色结晶性粉末;无臭,味极苦。在热水中溶解,在水或乙醇中微溶,在三氯甲烷中极微溶解,在乙醚中不溶。对痢疾杆菌、大肠杆菌、肺炎双球菌、金葡菌、链球菌、伤寒杆菌及阿米巴原虫有抑制作用。临床主要用于肠道感染及菌痢等。还发现本品有抗心律失常的作用。 小檗碱有较强的体内外抗肿瘤活性并能诱导B16细胞分化;同盐酸阿糖胞苷在体外具有协同作用。(一)加味左金丸 http://ng1.17img.cn/bbsfiles/images/2017/03/201703021026_01_2222981_3.png【色谱条件】色谱柱:CAPCELL PAK C18 S5; 4.6mm i.d.×250 mm流动相:乙腈/0.05mol/L磷酸二氢钠溶液(含0.2%的三乙胺,用磷酸调pH值为3.0)=27/75流 速:1.0mL/min温 度:40℃检 测:UV263nm进样量:10μL*摘自:中国医药指南,2013年5月,第11卷,第14期,401-402 (二)三黄片 http://ng1.17img.cn/bbsfiles/images/2017/03/201703021026_02_2222981_3.png【色谱条件】色谱柱:CAPCELL PAK C18 MGII S5; 4.6 mm i.d.×150 mm流动相:乙腈/水(每1000ml流动相中含磷酸二氢钾3.4g,十二烷基硫酸钠1.7g)=1/1流 速:1.0mL/min检 测:UV265nm注:文献中所用液相方法与2015年版《中国药典》中三黄片检测方法一致。*摘自:药物研究,2013年,第7期,26-28
1. 可用作医药、农药、染料及其他有机合成中间体。可用来合成2-氨基嘧啶、2-氨基-6-甲基嘧啶、2-氨基-4,6-二甲基嘧啶,是制造磺胺嘧啶、磺胺甲基嘧啶、磺胺二甲基嘧啶等磺胺药物的中间体。 2. 盐酸胍(或硝酸胍)与氰乙酸乙酯反应,环合为2,4-二氨基-6-羟基嘧啶,用于合成抗贫血药叶酸。还可用作合成纤维的防静电剂。 3. 也可用于蛋白质变性剂。化学性质如下:1.性状:白色或微黄色块状物2.熔点(℃):181-1833.相对密度(g/mL,20/4℃):1.3544.溶解性:在20℃时在100g水中可以溶解228g,在100g甲醇中可以溶解76g,在100g乙醇中可以溶解24g。几乎不溶于丙酮、苯和乙醚。5.pH 值(4%水溶液,25℃):6.4
药典里盐酸阿米洛利 含量测定方法如下:取本品约0. 2g,精密称定,加0.01mol/L盐 酸溶液5ml与乙醇50ml使溶解,照电位滴定法(附录VE A),用 氢氧化钠滴定液(0. lmol/L)滴定,两个突跃点体积的差作为 滴定体积。每lml的氢氧化钠滴定液(0. lmol/L)相当于 26. 61mg的C6 H8 C1N7 0 • HC1。请问加盐酸和乙醇的作用分别是什么?急急急,谢谢了
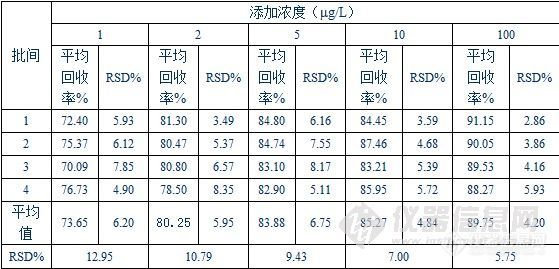
1.实验材料1.1 固相萃取小柱:PCX(150mg/6mL)1.2 四种β-激动剂药物:盐酸克仑特罗、沙丁胺醇、西马特罗、莱克多巴胺等4种β-激动剂药物。 2. 试料的制备取空白样品,经过液液萃取初步处理后,添加适宜浓度的标准溶液作为空白添加试料。 3. 净化 依次用甲醇5mL、水5mL和30mmol/L盐酸5mL润洗固相萃取小柱,将上述备用液过柱,依次用水5mL、甲醇5mL淋洗,真空抽干,用4%氨化甲醇5mL洗脱PCX小柱,收集洗脱液于具塞玻璃试管中,50℃下氮气吹干。在样液过柱和洗脱过程中流速控制在1mL/min左右。 4. 衍生化及检测将上述盛有残渣的具塞玻璃试管放入50℃烘箱中加热片刻,除去水分后,加入甲苯100mL和双三甲基硅基三氟乙酰胺(BSTFA)100mL,涡旋振荡20s,密封玻璃塞,置于80℃恒温烘箱中加热1小时,冷却后加入300mL甲苯,作为试样溶液,供气相色谱-质谱分析(色谱柱:DA-5MS, 30m×0.25mm×0.25μm,P/N:1525-3002)。 5. 结果5.1 回收率实验(精密度和准确度)将猪肝空白样品经过液液萃取初步处理后,分别添加一定量的标准溶液,配制1μg/L、2μg/L、5μg/L、10μg/L和100μg/L五个浓度的试样溶液,每批次内同一浓度做5次平行实验,共4个批次(样品典型回收率色谱图见附图)。 猪肝中实验结果列表如下:http://ng1.17img.cn/bbsfiles/images/2010/11/201011221740_261251_801_3.jpg猪肝中实验结果列表如下5.2重复性实验(批间误差实验): 猪肝中实验结果列表如下:http://ng1.17img.cn/bbsfiles/images/2010/11/201011221741_261252_801_3.jpg附图:猪肝中0.5μg/L、1μg/L、2μg/L、5μg/L、10μg/L和100μg/L六个浓度检测结果总离子流图(TIC)代表图谱:http://www.agela.com.cn/UploadFile/2009622153255641.jpg
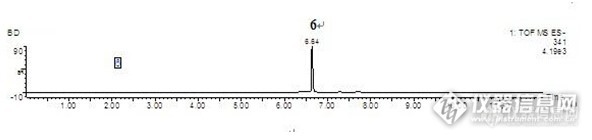
盐酸芬戈莫德在大鼠体内代谢的尿液及胆汁样品分析 芬戈莫德最初是由冬虫夏草(子囊菌亚门赤僵菌)培养液中提取的抗生素成分经化学修饰后合成的免疫抑制剂。芬戈莫德是鞘氨醇的结构类似物,研究显示,该药具有与其他药物完全不同的免疫抑制机制,在体内磷酸化后与位于淋巴细胞上的鞘氨醇-1-磷酸受体(S1PR)结合,通过改变淋巴细胞的趋化,促使淋巴细胞在淋巴组织内滞留,从而减少自身反应性淋巴细胞再次进入循环的几率,进而防止这些细胞浸润中枢神经系统(CNS)。进而达到免疫抑制效果。而且该过程是可逆的,停药后淋巴细胞水平即可以恢复正常。临床研究表明,口服制剂芬戈莫德针对复发-缓解型多发性硬化症疗效确切,优于目前的常用MS治疗药物干扰素β-1a注射剂(Avonex,已用于多发性硬化症的临床治疗药物)。芬戈莫德可靶向作用于对中枢神经系统(CNS)有潜在自身攻击性的淋巴细胞,促进神经保护与修复过程,降低MS的复发率,延缓损伤的进展过程,减少颅内核磁共振成像(MRI)病灶的数量,减轻病灶的严重程度。 药物及实验动物:盐酸芬戈莫德为本所研制,实验用大鼠为Wistar雄性大鼠,6-8周龄,体重范围约200-250g/只,本所实验中心提供;大鼠代谢笼为苏州动物实验仪器厂产品。色谱条件色谱柱:Acquity BEH C18 (100mm×2.1mm, 1.7μm)流动相:A:水(0.05%TFA)B:乙腈(0.05%TFA)http://ng1.17img.cn/bbsfiles/images/2014/12/201412302201_530374_2217446_3.jpg质谱条件Waters LCT Premier XETM型飞行时间质谱仪,W-负离子模式;毛细管电压2200 V;锥孔电压35 V;离子源温度120℃;脱溶剂气温度350℃;脱溶剂气流量10L /h;锥孔气流量700 L /h;质量扫描范围m /z 50 ~ 1200[
这个问题涉及到很多方面,故放在综合区,望版主谅解。近日在考察注射用盐酸阿糖胞苷是否可用USP37的标准。发现一个问题,USP37收载的该剂型的活性物质是阿糖胞苷,而不是盐酸阿糖胞苷;而在查阅国外的几个制药公司的该产品的说明书,注明的活性物质也是阿糖胞苷原型,而非盐酸盐。故在此想请教对药物化学和药物分析很了解的童鞋们几个问题:1、国外的该产品说明书上注明的是阿糖胞苷原型药,USP37收载的也是原型药,是否说明在国外该制剂采用的原料药是阿糖胞苷,而非其盐酸盐?2、国内的制药厂家的该产品说明书上注明的活性物质是盐酸阿糖胞苷,且中国药典收载的也是盐酸阿糖胞苷,毫无疑问说明原料药采用的是盐酸阿糖胞苷,为什么要用盐酸盐,而不跟国外一样采用原型药?是为了避开专利还是盐酸盐的形式更有利于人体吸收?3、USP37收载原型药,被检测的药物是盐酸盐,是否说明该药物不能用USP37的标准来检?4、在哪里可以查到国外药物的专利,及其详细信息,如药物的化学结构、化学式、其专利到期的时间等?5、如何可以准确地知道国外一些药品制剂所使用的原料的详细信息?尤其是理化方面的信息,这些信息在说明书上体现的很少。望高手不吝赐教!谢谢!
第十四章 生物碱类药物的分析(上)掌握盐酸麻黄碱、硫酸阿托品、硫酸奎宁、盐酸吗啡和硝酸士的宁的鉴别、杂质检查和含量测定方法。第一节 盐酸麻黄碱的分析一、鉴别1.双缩脲反应 氨基醇特征反应与硫酸铜和氢氧化钠试液反应生成紫色配位化合物,加入乙醚后醚层呈紫红色,水层呈蓝色。2.红外光谱3.Cl-反应二、含量测定:非水滴定法。本品为盐酸盐,需预先加入醋酸汞。第二节 硫酸阿托品的分析一、鉴别1.Vitali反应 托烷生物碱特征反应:与发烟硝酸共热,冷后加醇制氢氧化钾,显深紫色。2.红外光谱3.硫酸盐鉴别反应(1)加氯化钡生成白色沉淀,沉淀在盐酸或硝酸中不溶解(2)加醋酸铅生成白色沉淀,沉淀在醋酸铵或氢氧化钠试液中溶解(3)加盐酸不生成白色沉淀,与硫代硫酸盐区别二、特殊杂质检查1.莨菪碱检查:阿托品为外消旋体,消旋不完全时引入的莨菪碱具有旋光性。测定供试品溶液旋光度来控制莨菪碱限量。2.其他生物碱检查:制备过程中引入的莨菪碱、颠茄碱等杂质碱性弱于阿托品。取供试品盐酸水溶液,加入氨试液,其他生物碱立即游离发生浑浊。规定加氨试液不得立即发生浑浊。三、含量测定1.原料药 非水滴定法1mol硫酸阿托品消耗1mol的高氯酸2.片剂、注射剂 酸性染料比色法在一定pH值条件下,生物碱盐的阳离子和酸性染料的阴离子定量结合生成配位化合物,即离子对,此离子对易溶于有机溶剂,经有机溶剂(氯仿等)提取后,比色测定含量。(BH+)W +(In-)W → (BH+?In-)W →(BH+?Iln-)O对照品比较法测定含量。第三节 硫酸奎宁的分析一、鉴别1.荧光反应 加水溶解后,加稀硫酸成酸性,显蓝色荧光2.绿奎宁反应 含氧喹啉衍生物特征反应在药物微酸性水溶液中滴加微过量溴水,再加入过量氨试液,显翠绿色。3.硫酸盐鉴别4.红外光谱二、特殊杂质检查1.氯仿-乙醇中不溶物检查检查制备过程中引入的无机盐与其他生物碱。取供试品2g,不溶物滤过干燥至恒重,遗留残渣不得过2mg。2.其他金鸡纳碱的检查:薄层色谱高低浓度对比法三、含量测定1.原料药 非水滴定法 1mol硫酸奎宁消耗3mol的高氯酸2.片剂 提取中和法 将适
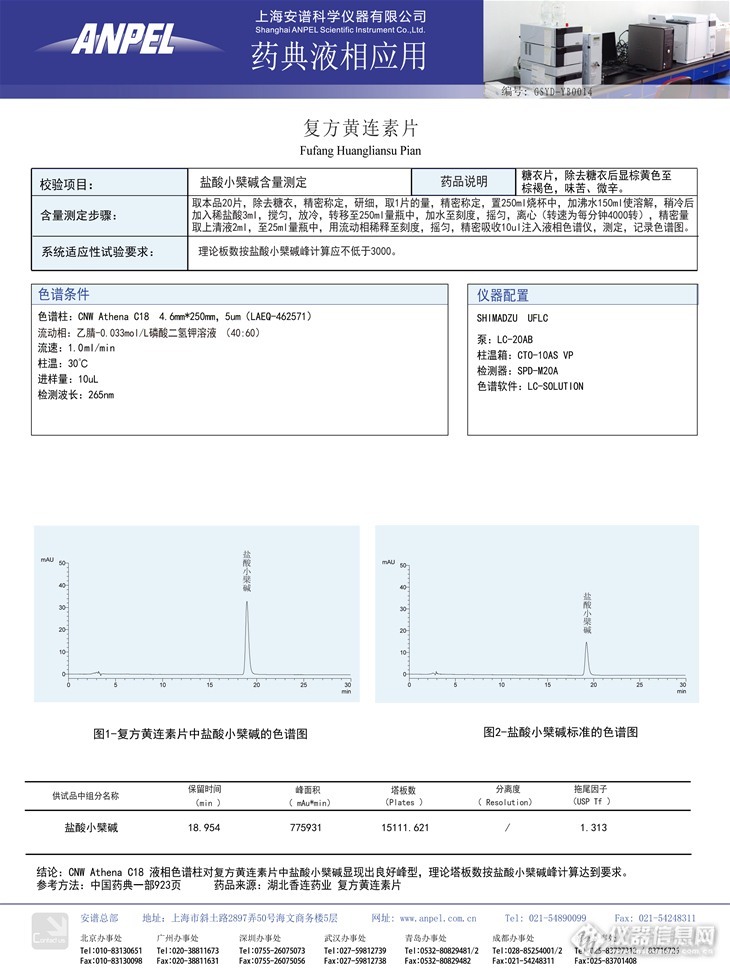
CNW Athena C18液相色谱柱复方黄连素片中盐酸小檗碱一 样品结构信息一种异喹啉生物碱。分子式+。又称黄连素 。存在于小檗科等4科10属的许多植物中。小檗碱从乙醚 中可析出黄色针状晶体。熔点145℃。溶于水,难溶于苯、乙醚和氯仿。小檗碱为一种季铵生物碱,其盐类在水中的溶解度都比较小,例如盐酸盐为1∶500,硫酸盐为1∶30。小檗碱从水或稀乙醇中析出的晶体带有5.5分子结晶水;若从氯仿、丙酮或苯中结晶,也带有相应的结晶溶剂分子。小檗碱用不同的碱处理,可得到季铵式、醛式和醇式等三种不同形式的小檗碱,其中以季铵式最稳定。小檗碱对溶血性链球菌、金黄色葡萄球菌、淋球菌和弗氏、志贺氏痢疾杆菌均有抗菌作用,并有增强白血球吞噬作用。小檗碱的盐酸盐(俗称盐酸黄连素)已广泛用于治疗胃肠炎、细菌性痢疾等,对肺结核、猩红热、急性扁桃腺炎和呼吸道感染也有一定疗效http://ng1.17img.cn/bbsfiles/images/2017/01/201701191657_651678_2442872_3.jpg二、样品来源记录药品名:复方黄连素片生产厂家:湖北香连药业http://ng1.17img.cn/bbsfiles/images/2014/01/201401031101_486507_2442872_3.jpg
第十八章 抗生素类药物掌握青霉素钠、氨节西林和头孢羟氨苄的鉴别、杂质检查和含量测定方法;青霉素V钾及其片剂的鉴别、杂质检查和含量测定方法。 掌握硫酸链霉素、硫酸庆大霉素的鉴别、检查和含量测定方法。 熟悉罗红霉素的鉴别、检查和含量测定方法。 熟悉盐酸美他环素的鉴别、检查和含量测定方法。第一节 青霉素钠的分析青霉素族中的母核为6-氨基青霉烷酸(简称6-APA),游离羧基酸性,能与无机碱或某些有机碱成盐。青霉素母核无紫外吸收,而苄基取代基有紫外吸收。β-内酰胺环不稳定,遇酸、碱、青霉素酶及某些金属离子等作用,易发生水解和分子重排,导致β-内酰胺环的破坏而失去抗菌活性。一、鉴别 1.抑菌实验 通过对金黄色葡萄球菌的抑制作用进行鉴别。加入青霉素酶培养后无抑菌作用,同法检查未经青霉素酶灭活的有抑菌作用。2.沉淀反应 本品为钠盐,加稀盐酸使成酸性,生成分子型,难溶于水即白色沉淀。此沉淀能在乙醇、醋酸戊酯、氯仿、乙醚或过量的盐酸(与酰胺基成盐)中溶解。2.红外光谱法4.钠盐焰色反应 火焰鲜黄色。二、检查1.吸收度 侧链苯环在264nm有最大吸收,而降解产物在280nm有最大吸收。规定二波长处吸收度值范围。测定264nm吸收度为控制青霉素钠含量。规定280nm吸收度为控制杂质限量。2.水分 本品遇水易水解,费休法测定水分不得超过0.5%。2.细菌内毒素 细菌内毒素检查法。利用鲎试剂与细菌内毒素发生凝集反应,来判断内毒素是否符合规定。4.无菌 灭活后,无菌检查法检查。三、含量测定:汞量法。青霉素水解后,其碱性水解产物青霉噻唑酸及青霉胺都能与汞盐定量反应,根据消耗汞盐量可计算青霉素含量。注意事项1.滴定前加1mol/L氢氧化钠5ml,使药物水解为青霉噻唑酸并继续水解为青霉胺,才能与Hg2+反应。2.水解后加等量硝酸中和,再加pH 4.6醋酸盐缓冲液,方能滴定。2.采用电位滴定法。滴定曲线出现两个突跃,计算时以第二个突跃为准,此时反应摩尔比为1:1。4.空白试验也要称取样品,但不经水解直接滴定,以消除样品中降解产物对测定影响。5.青霉素百分含量为总青霉素百分含量与降解产物百分含量之差。第二节 青霉素V钾及其片剂的分析一、鉴别1.水解反应 在青霉素酶作用下,β-内酰胺环水解开环,生成羧酸,使溶液转为酸性。本法专属性强。2.紫外分光光度法 核对药物的λ[f
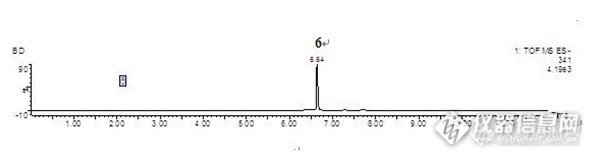
盐酸芬戈莫德在大鼠体内代谢的尿液及胆汁样品分析芬戈莫德最初是由冬虫夏草(子囊菌亚门赤僵菌)培养液中提取的抗生素成分经化学修饰后合成的免疫抑制剂。药物及实验动物:盐酸芬戈莫德为本所研制,实验用大鼠为Wistar雄性大鼠,6-8周龄,体重范围约200-250g/只,本所实验中心提供;大鼠代谢笼为苏州动物实验仪器厂产品。色谱条件色谱柱:Acquity BEH C18 (100mm×2.1mm,1.7μm)流动相:A:水(0.05%TFA)B:乙腈(0.05%TFA)质谱条件结果分析:通过比较大鼠灌胃盐酸芬戈莫德溶液后收集的尿液样品、空白尿液样品及分到的代谢产物的高分辨质谱和多级质谱数据,在给药后的尿液中共鉴定出了8个代谢产物(如下图)所有代谢产物的高分辨质谱数据的准确度均小于1PPm。通过比较大鼠灌胃盐酸芬戈莫德溶液后收集的胆汁样品、空白胆汁样品及分到的代谢产物的高分辨质谱和多级质谱数据,在给药后的胆汁中共推测出了4个代谢产物(如下图)。所有代谢产物的高分辨质谱数据的准确度均小于1PPm。结果与讨论:经过对于给药后大鼠尿液及胆汁样品分析,初步推测盐酸芬戈莫德在大鼠体内的代谢产物有8种。
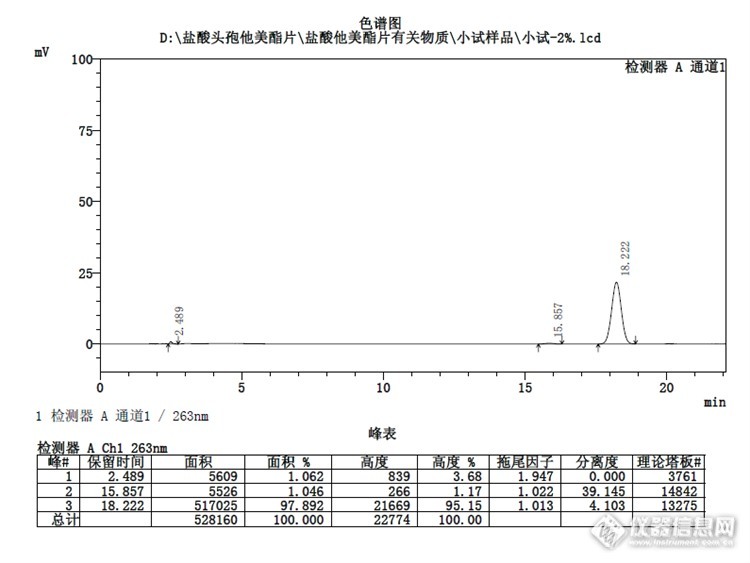
盐酸头孢他美酯是种广谱抗生素,可用于对它敏感细菌感染所引起的炎症。该产品为口服用。化学名:(6R,7R)-3-甲基-7-结构式:http://ng1.17img.cn/bbsfiles/images/2014/03/201403302127_494735_1621890_3.gif 英文名:Cefetamet Pivoxil Hydrochloride Tablets 药物别名:威锐片 成份:本品主要成分为盐酸头孢他美酯 性状:本品为薄膜衣片,除去包衣后呈白色、类白色,有引湿性。 药代动力学:本品单一剂量和多剂量的药代动力学参数基本一致。本品口服后,经过肠黏膜或首次经过肝脏时盐酸头孢他美酯被迅速代谢,在体内转变为头孢他美而发挥作用。本品随食物口服后,平均约55%的剂量转变为头孢他美。口服本品500mg后3~4小时,血药浓度达峰值4.1±0.7mg/L,分布容积为0.29L/kg,与细胞外水平一致。约22%头孢他美与清蛋白结合。年龄、肾脏及肝脏疾病对盐酸头孢他美酯的生物利用度无影响。抗酸剂(镁、铝、氢氧化物等)或雷尼替丁不改变本品生物利用度。本品90%以头孢他美形式随尿液排出,清除半衰期为2~3小时。肾衰竭患者,头孢他美的清除情况同肾功能成正比。 适应症:本品适用于敏感菌引起的下列感染:1.耳、鼻、喉部感染,如中耳炎、鼻窦炎、咽炎、扁桃体炎等。2.下呼吸道感染,如慢性支气管炎急性发作、急性气管炎、急性支气管炎等。3.泌尿系统感染,如非复杂性尿路感染、复杂性尿路感染(包括肾盂肾炎)、男性急性淋球菌性尿道炎等。注意事项 1.对青霉素类药物过敏者慎用。 2.若发生严重过敏反应,应立即停药,并紧急治疗。 3.在使用本品期间,由于肠道微生物的改变,可能导致伪膜性肠炎。若发生假膜性肠炎,应积极治疗(推荐使用万古毒素)。 4.本品应放到儿童触及不到的地方。 孕妇及哺乳期妇女用药:1.由于缺乏有关人类胎儿的临床数据,妇女妊娠期间,不推荐使用本品。若有对该药敏感的微生物严重感染时,必须充分权衡利弊。2.在乳汁中尚未发现本品的代谢物。 儿童用药:本品对新生儿的有效性和安全性尚无可靠的临床数据。 药物相互作用: 抗酸剂,H2受体拮抗剂对本品的药代动力学无影响。目前尚未见到本品对实验室检测值和/或方法有影响的报道,也未观察到伴随利尿药治疗的患者在使用本品时对肾功能的损伤。 药物过量: 若过量服用,发生严重反应,应洗胃,并采取对应治疗。 贮藏:遮光、密封、在干燥处保存。详见:http://baike.so.com/doc/6048874.html该品种国内批准文号有40个,见国家药监局网。截图如下:http://ng1.17img.cn/bbsfiles/images/2017/01/201701191656_646694_1621890_3.png试验条件:主要参照中国药典2010年版二部。用十八烷基硅烷键合硅胶为填充剂;以乙腈-甲醇-水-磷酸盐缓冲液(取无水磷酸氢二钠5.8g与磷酸二氢钾3.5g,加水溶解并稀释成1000ml)(360:95:500:45)为流动相;检测波长为263nm。取头孢他美酸和头孢他美酯对照品适量,用乙腈溶液(9→20)溶解并稀释制成每1ml中约含头孢他美酸0.05mg和含头孢他美酯1.4mg的混合溶液,取10μl注入液相色谱仪,头孢他美酯峰与头孢他美酸峰的分离度应不小于28.0,头孢他美酯峰与其相对保留时间约为0.9和1.1处杂质峰的分离度均应大于于2.0,理论板数按头孢他美酯峰计算不低于3000。取本品的细粉适量,加乙腈溶液(9→20)溶解并定量稀释制成每1ml中约含头孢他美1.0mg的溶液,滤过,取续滤液作为供试品溶液;精密量取供试品溶液适量,用乙腈溶液(9→20)定量稀释制成每1ml中约含头孢他美20μg的溶液,作为对照溶液。照含量项下的色谱条件,取对照溶液10μl注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的25%;再精密量取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的4.5倍。供试品溶液色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积(2.0%),各杂质峰面积的和不得大于对照溶液主峰面积的2.5倍(5.0%)。色谱柱信息:月旭Welchrom C18, 5μm, 4.6×250mm(货号:00310-02043;序列号:w13211564)试验图谱:1.系统适用性溶液:http://ng1.17img.cn/bbsfiles/images/2014/03/201403302057_494730_1621890_3.png2.供试品溶液:http://ng1.17img.cn/bbsfiles/images/2014/03/201403302112_494732_1621890_3.png3.对照溶液:http://ng1.17img.cn/bbsfiles/images/2014/03/201403302121_494734_1621890_3.png
二、样品储存和稳定性考察:取样后最好立即进行分析,冷藏(4℃)、冰冻(-20℃)有时也不能完全保证样品不起变化。尿液是很好的细菌生长液,若需收集24小时或更长时间的样品或不能立即测定的,应置冰箱冷藏或加防腐剂(1%甲苯、过饱和氯仿)保存。分析样品贮存时应考虑:储存条件;样品在贮存中会对分析结果产生什么影响;评述样品稳定性时会发生什么问题;如何预防或校正不稳定样品的分析结果。 测定前样品的制备除少数体液经简单处理后直接测定外,通常在最后一步测定前要采取适当的样品制备,即进行分离、净化、浓集、必要时尚需对待测组分进行化学改性,为测定创造良好条件。一、样品的制备要考虑:药物的理化性质、待测物的浓度范围、药物测定的目的、选用的生物体液和组织的类型、样品制备与分析技术的关系。二、蛋白质的处理:是测定血浆、血清、全血及组织匀浆等样品中药物时的最先处理步骤。(一)加入沉淀剂和变性试剂:硫酸铵是经典的蛋白质沉淀剂,它与蛋白质分子竞争系统中水分子,而使蛋白质析出。阴离子型沉淀剂(三氯醋酸、高氯酸、钨酸、焦磷酸)与带电荷的蛋白质在氏于等电点的pH时形成不溶性盐;反之,阳离子型沉淀剂(含锌盐、铜盐)与蛋白质分子中带阴电荷的羧基,在高于蛋白质等电点时,形成不溶性盐。有关机制不十分清楚。(二)加入可与水混合的有机溶剂:乙醇过量存在时,能使与蛋白结合状态的药物释放可将混合物离心,取上清液(含药),但这不能解决样品的净化问题。蛋白沉淀法对于与蛋白结合力强的药物的回收率较差。也有采用酸消化法(Acid digestion)使药物自蛋白结合处释出,但常导致药物的分解。(三)组织的酶消化法:蛋白水解酶(Proteolytic enzyme)中的枯草菌溶素(Subtilisin Carlsberg)不仅可使组织酶解,且可使药物析出。优点:1、因是在平稳条件下进行的,可避免某些药物在酸中水解及较高温度时降解;2、可显著改善对蛋白结合率强的药物的回收率;3、可用有机溶剂直接提取消化液而无乳化生成的危险;4、在用HPLC时,无需再进行过多的净化操作。缺点是不适用于一些在高pH时易水解的药物。三、提取:(一)溶液的pH调节:最佳pH选择主要与药物的pKa值有关。pH与pKa相当时,50%的药物以非电离形式存在。碱性药物最佳pH值要高于pKa值1~2个pH单位;反之,则低1~2个单位。可使90%药物以非电离开形式存在,易为溶剂提取。而对于碱性很强的药物往往采用“离子对”技术进行提取和定量。体内酸性物质较多,在碱性条件下不会被萃取出来,故在pH值偏高的情况下进行提取较好。(二)提取溶剂的极性:选好第一个提取溶剂可减少以后的净化操作,在液-液提取中多采用极性小的溶剂。加入少量醇类可克服极性小溶剂提取能力弱和减小药物在容器表面的吸附损失的不足。也有利用不同极性的混合溶剂来提取药物和净化脂肪酸类。(三)提取技术:由于体液样品量少且药物含量低,一次分析的样品数量较多。与常量和微量分析相比,提取时通常不采用反复提取的方法,多半进行一次(至多二次)提取,在改变pH后,从有机相回提至水相也只进行一次。一般并不考虑“提尽药物”,测定含量时则应精确加入提取溶剂,提取液也要定量分出。为避免进样时带来的误差,多采用提取前加入等量内标,以待测组分峰高(或峰面积)与内标峰高(或峰面积)之比对浓度作标准曲线。这样,即使在一系列操作过程中有微量损失,对比值影响也较小。混合时可在密塞情况下将试管平置于振荡器内振荡,振荡时间与强度视情况而定。可采用以“药物转入溶剂中量”与“混合时间”作图法,选取理想和符合实际的提取方式和时间。(四)提取溶剂的蒸发:提取液常为数ml,往往不能直接供GC或HPLC测定,需采取浓集办法,常用真空蒸发(注意暴沸)或吹氮气流使溶剂挥散。
随着食品安全曝光出的问题 药品的安全问题国家也开始重视了 其中手性药物最容易钻空子 下面的资料有助于大家对手性药物分离有所了解 由于药物对映体之间在药理、毒理及吸收等方面存在较大差异,因此,建立分离和测定对映体化合物的方法十分重要。HPLC法在分离和测定药物对映体的常用方法,包括手性衍生化试剂、手性流动相和手性固定相在药物对映体分离测定中的应用。对对映体化合物的分析鉴定有指导意义。 手性化合物的拆分是当前分析化学中最为活跃的领域之一,自然界中的许多化合物都是有旋光性的,而合成手性药物中大多(88%)是外消旋体,许多手性药物的对映体在生理过程中显示了不同生理活性。据研究反应停的致畸作用主要是由于其(S)-(-)异构体所致。因此,建立高专属性、高灵敏度、高分离度的对映体拆分和测定方法,对提高药物的活性、减小副作用,深入研究药物的作用机理等具有重要的理论和实际意义。 对映体化合物之间除对偏振光的偏转方向不同外,具有完全相同的理化性质,因而其分离比较困难。传统的拆分方法有分步结晶、微生物和酶消化法等,或者用手性衍生化试剂将其转化成非对映体,然后根据其物理性质不同进行分离,但这些方法难于进行微量的分离和测定。80年代以来,随着快速、准确、微量的光学异构体的HPLC拆分及测定方法的建立和发展,使HPLC迅速成为药物对映体分离和测定最为广泛应用的方法。 手性HPLC拆分法是以现代HPLC技术为基础,引入手性环境使对映异构体间呈现物理特征的差异而进行分离。通常分间接法和直接法,前者是对映体混合物以手性试剂作柱前衍生,形成一非对映体,然后以常规(偶也见手性)固定相分离。后者是直接以手性流动相(CMP)或手性固定相(CSP)直接进行分离。 1、手性衍生化试剂法 手性衍生化试剂(CDR)法是在分子间引入手性中心,其产物为非对映异构体(diastereomer,DSTM),从而进行分离。 下列情况通常选用CDR法进行拆分:(1)不宜直接拆分。添加某些基团,以增加色谱系统的选择性。如游离胺类在CSP上往往是颇弱的色谱性质,生成中性化合物后则获显著改善。(2)提高紫外或荧光检测的效果。刘雁鸣等用NBD-(L)-APY荧光试剂柱前衍生化测定布洛芬对映体,提高了检测灵敏度。对CDR的要求通常为:溶质分子至少有一个(多个时其性质各不相同)功能团供衍生(多为-NH2,-OH或-COOH)。光学活性试剂必需是手性高纯度;反应条件必须温和、简便;宜附有发色或荧光基团。 目前,已有许多商品化的CDR可供选用,常见的CDR可分为以下几类:(1)异硫氰酸酯和异氰酸酯类此类试剂易与大多数醇类及胺类化合物反应进而被分离,如麻黄素类,肾上腺素类,肾上腺素拮抗剂,儿茶酚胺类等。王亚芹等采用S( )-1-(1-苯基)乙基异氰酸酯为衍生化试剂分析了血浆中普罗帕酮的对应体,并研究了其在健康人体内的药代动力学。邱宗荫等用乙酰葡萄糖异硫氰酸酯(GITC)为柱前CDR,以反相HPLC法测定血浆中地佐西平对映体的血药浓度,线性范围为5~200μg.L-1。陈冰等用GITC为柱前CDR,用反相HPLC法测定血浆中普罗帕酮对映体的血药浓度,适合用于临床药动药效学研究。(2)萘衍生物类由于此类化合物有利于提高立体选择性和检测灵敏度,因此萘的各种衍生物用作手性试剂十分普遍。Wainer等选用萘甲醛(NDH)为手性试剂,与其缩合成恶唑烷衍生物,成功地分离了麻黄碱、4-甲氧基麻黄碱、伪麻黄碱。Bhatti等用S-( )-1-(1-萘基)-乙基异氰酸酯为CDR,用HPLC法测定了人血浆中美托洛尔对映体浓度。(3)酰氯与磺酰氯类此类试剂可与化合物直接缩合,或与样品反应后,再引入其它基团,合成更有利于拆分与检测的衍生物。Sallustio等以SOCl2与芳丙酸类消炎镇痛药如2-苯丙酸、酮洛芬及非诺洛芬的血浆样品提取物反应,然后再与R-2-苯乙胺成酰胺衍生物,产物以NP(Sil,乙腈∶二氯甲烷,5∶95)分离,异构体均可完全拆分。(4)光学活性氨基酸类为最早采用的色谱手性试剂,为提高反应活性和定量回收率,常将羧基转化成酰氯、酸酐等。此类试剂广泛用于胺、羧酸及醇类药物,尤其是氨基酸类,其衍生化法多基于肽合成原理。 本类方法要求手性药物具有活泼反应基团,同时两个对映体的衍生化速度应相同,否则会引起非对映体与原对映体的组成产生差异,另外要求手性衍生化试剂光学纯度高,反应要迅速、彻底,因此应用受到一定限制。 2、直接方法 直接方法是在分子间引入手性环境,即采用手性流动相或手性固定相不经柱前衍生化直接分离药物对映体的方法,该法近年发展迅速。 2.1 手性流动相拆分法向流动相中加入一手性试剂,它与溶质常以氢键、离子键或金属离子的配位健生成非对映体缔合物,从而以常规HPLC固定相分离。分离机理为:(1)在流动相中形成立体选择性复合物;(2)手性流动相添加剂(CMPA)与固定相之间发生作用,形成动态的CSP,该法可通过改变CMPA的种类、浓度及流动相的组成而优化分离条件。 常用的CMPA主要有:(1)环糊精类主要是α-、β-和γ-环糊精及其衍生物。Eto等用β-环糊精测定了数种巴比妥类和乙内酰脲类药物在生物体液中的对映体浓度。谢剑炜等用β-环糊精手性流动相添加剂,用反相HPLC法首次抗胆碱能药物盐酸戊乙奎醚、盐酸苯环壬酯和盐酸卡马特灵,3个手性药物4对对映体完全达到基线分离。(2)手性离子对试剂,karlsso等以N-苯甲酰甘氨酰脯氨酸作为CMPA,分离测定了血浆中(R)-和(S)-普萘洛尔。与HPLC中的离子对法的差别主要在于前者是手性离子对试剂,由于CMPA价格昂贵,其体系稳定性差等原因,应用受到一定的限制。范柏等用L-苯丙氨酸为配合剂,Cu2 为配合离子,用简便的手性配合交换反相HPLC法成功拆分了氧氟沙星对映体,手性流动相为6mmol.L-1L(D)-苯丙氨酸,3mmol.L-1硫酸铜-甲醇(83∶17)。 2.2 手性固定相拆分法由于CSP技术的飞速发展,采用CSP分离对映体化合物的方法应用越来越广泛。目前,商品化的手性柱已有数十种,却无一具有类似ODS柱那样普遍的适应性,且价格昂贵。随着手性识别机理的深入研究,新方法、新理论不断提出,预计将会有价廉、适应性广的CSP面世。(1)环糊精键合相α-、β-和γ-环糊精(CD)是分别由6~8个葡萄糖单位通过α-(1、4)连接构成的环状低聚糖,CD-CSP通过共价键将CD分子键合到硅胶上,形成对水稳定的键合相。β-CD键合相的立体选择性较好,应用最多。β-CD柱上分离较好的化合物通常其手性中心为分子中环状结构的一部分,或至少与两个SP2杂化碳原子相连。Berthod等采用商品的β-CD柱(CydobondⅠ)和γ-CD柱(CydobondⅡ)拆分了25种不同类型的手性药物,其中对映体之间达基线分离的有11种。(2)吸附络合物形成相要想实现手性识别,手性化合物与CSP之间至少应存在三种相互作用,称为三点识别模式。这些作用可以是氢键、静电作用、疏水作用、π-π作用、偶极-偶极作用或空间作用,一般通过将某些氨基酸,如(R)-或(S)-苯基甘氨酸等分子中的α-氨基与3,5-二硝基苯甲酰氯反应后,离子或共价键合到氨丙基硅胶上而制得。该类固定相通常按正相方式操作,其在药物分析中应用较少,后来,RUSTUM等发现也可使用反相分离系统,从而扩展了其应用范围。(3)手性聚合物相用不同方法将纤维素衍生物涂复于大孔硅胶上而制得,在此类固定相上得到成功分离的化合物大都含有苯基、羰基、腈基、磺酰基或羟基等。目前,纤维素—三(3.5一二甲基苯基氨基甲酸酯)手性固定相应用较多。例如,用三(3,5-二甲苯基氨基甲酸酯)纤维素衍生物为CSP对血浆中普萘洛尔对映体的测定。Shibukawa等人采用3,5-二甲苯基氨基甲酸酯衍生化的直链淀粉手性固定相(AD-CSP)分离了维拉帕米及其代谢产物去甲维拉帕米的对映体,方法的线性范围为2.5~100μg.L-1。(4)蛋白质键合相以离子键(或共价键)和蛋白交联作用将蛋白质固定于硅胶上,利用蛋白质分子与手性化合物分子间的立体选择性作用,进行药物对映体分离,其机理一般有氢键、静电作用、疏水作用、离子对和离子交换作用。将α1-酸性糖蛋白(α1-AGP)固定到硅胶上而制得AGP柱可直接分离许多碱性、酸性及中性药物对映体。钟大放等用CHIRAL-AGP柱,选择不同流动相分别拆分了SFZ-47、KMBZ-009和地丙苯酮3种药物的4对对映体,并研究了SFZ-47在家犬体内的药代动力学。Schmidt等人以α1-酸性糖蛋白为CSP测定人体血浆中美沙酮对映体的含量。Orn等以α1-酸性糖蛋白为CS
第十章 胺类药物的分析掌握盐酸普鲁卡因、盐酸利多卡因、盐酸丁卡因和对乙酰氨基酚的鉴别、杂质检查和含量测定方法。 掌握肾上腺素、盐酸去氧肾上腺素及其制剂的鉴别、杂质检查和含量测定方法。 第一节 盐酸普鲁卡因的分析有芳伯氨基特性,显重氮化-偶合反应。含酯键易水解,产物主要为对氨基苯甲酸(PABA)。脂烃胺侧链为叔胺氮原子,具有弱碱性。盐酸盐系白色结晶性粉末,具有一定的熔点,易溶于水和乙醇,难溶于有机溶剂。一、鉴别1.重氮化-偶合反应 分子结构中具有芳伯氨基或潜在芳伯氨基的药物,均可发生重氮化反应,生成的重氮盐可与碱性β-萘酚偶合生成有色的偶氮染料。 2.水解反应 取本品约0.1g,加水2ml溶解后,加10%氢氧化钠溶液1ml,即生成白色沉淀,加热变为油状物(普鲁卡因);继续加热,产生的蒸汽(二乙氨基乙醇)能使湿润的红色石蕊试纸变为蓝色;热至油状物消失后(生成可溶于水的对氨基苯甲酸钠),放冷,加酸酸化,即析出白色沉淀。此沉淀能溶于过量的盐酸。3.氯化物反应沉淀反应 在硝酸酸性条件下与硝酸银生成白色沉淀,沉淀加氨试液即溶解。氧化还原反应 加二氧化锰混匀,硫酸润湿,加热产生氯气,能使湿润淀粉碘化钾试纸显蓝色。4.红外光谱法二、特殊杂质检查普鲁卡因分子结构中有酯键,易发生水解反应。其注射液制备过程中受灭菌温度、时间、溶液pH值、贮藏时间以及光线和金属离子等因素的影响,可发生水解反应生成对氨基苯甲酸和二乙氨基醇。其中对氨基苯甲酸随贮藏时间的延长或高温加热,可进一步脱羧转化为苯胺,而苯胺又可被氧化为有色物,使注射液变黄,疗效下降,毒性增加。故中国药典90年版规定,本品注射液应检查水解产物对氨基苯甲酸,其限度不得超过1.2%。检查方法 薄层色谱法。供试品溶液2.5mg/ml与对照品溶液30μg/ml分别点于硅胶H薄层板上,展开后用对二甲氨基苯甲醛溶液喷雾显色。供试品溶液如显与对照品溶液相应的杂质斑点,其颜色与对照品溶液主斑点比较,不得更深。三、含量测定 分子结构中具有芳伯氨基,可用亚硝酸钠滴定法测定含量。
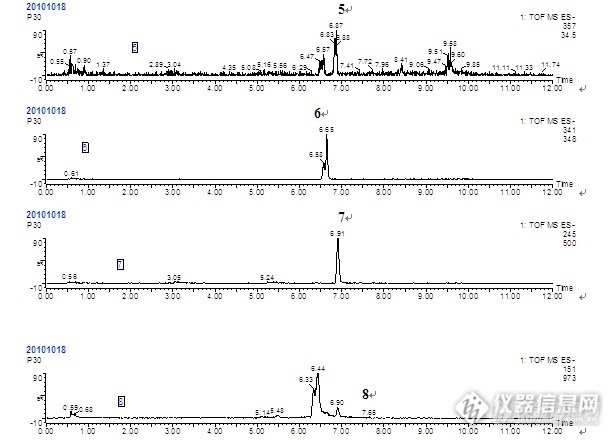
LC-MS对盐酸芬戈莫德在大鼠体内的代谢分析 芬戈莫德最初是由冬虫夏草(子囊菌亚门赤僵菌)培养液中提取的抗生素成分经化学修饰后合成的免疫抑制剂。芬戈莫德是鞘氨醇的结构类似物,研究显示,该药具有与其他药物完全不同的免疫抑制机制,在体内磷酸化后与位于淋巴细胞上的鞘氨醇-1-磷酸受体(S1PR)结合,通过改变淋巴细胞的趋化,促使淋巴细胞在淋巴组织内滞留,从而减少自身反应性淋巴细胞再次进入循环的几率,进而防止这些细胞浸润中枢神经系统(CNS)。进而达到免疫抑制效果。而且该过程是可逆的,停药后淋巴细胞水平即可以恢复正常。临床研究表明,口服制剂芬戈莫德针对复发-缓解型多发性硬化症疗效确切,优于目前的常用MS治疗药物干扰素β-1a注射剂(Avonex,已用于多发性硬化症的临床治疗药物)。芬戈莫德可靶向作用于对中枢神经系统(CNS)有潜在自身攻击性的淋巴细胞,促进神经保护与修复过程,降低MS的复发率,延缓损伤的进展过程,减少颅内核磁共振成像(MRI)病灶的数量,减轻病灶的严重程度。 药物及实验动物: 盐酸芬戈莫德为本所研制,实验用大鼠为Wistar雄性大鼠,6-8周龄,体重范围约200-250g/只,本所实验中心提供;大鼠代谢笼为苏州动物实验仪器厂产品。 色谱条件色谱柱:Acquity BEH C18 (100mm×2.1mm,1.7μm)流动相:A:水(0.05%TFA)B:乙腈(0.05%TFA)http://ng1.17img.cn/bbsfiles/images/2014/11/201411281531_525077_2217446_3.jpg 质谱条件 Waters LCT Premier XETM型飞行时间质谱仪,W-负离子模式;毛细管电压2200 V;锥孔电压35 V;离子源温度120℃;脱溶剂气温度350℃;脱溶剂气流量10L /h;锥孔气流量700 L /h;质量扫描范围m /z 50 ~ 1200;扫描时间0.2s。 给药方案与样品的收集: 血浆样品的收集健康雄性wistar大鼠3只,体重180-220g,1只为空白对照组,2只为给药组(取血时间30min和120min),给药前禁食12h,期间自由饮水。灌胃给药剂量为35 mg/kg,给药体积为1.5mL/只,给药30min和120min后,分别于颈动脉取全血,置于涂有肝素的离心试管中,3500prm离心10min,分离血浆,于-20℃冰箱中保存,直至分析。 血浆样品的预处理 取0.5ml血浆,置于离心管中,加入5倍的乙腈,3500prm离心10min,除去蛋白,取上清液,在40℃,旋转蒸干,用50%甲醇溶解,涡旋,11000prm离心10min,取2μL进行分析。 结果分析 对大鼠灌胃盐酸芬戈莫德溶液后收集的血浆样品用乙腈沉淀蛋白前处理方法处理之后,进行TOF-MS/MS分析,将所得HR-MS,MS2等数据与空白血浆和对照品比较后,在血浆样品中共推测出7个代谢产物。http://ng1.17img.cn/bbsfiles/images/2014/11/201411281532_525078_2217446_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/11/201411281532_525079_2217446_3.jpg结果与讨论:1、经过对于给药后大鼠血浆样品分析,初步推测盐酸芬戈莫德在大鼠体内的代谢产物有7种,其结构进一步鉴定中。2、流动相的选择方面进行了优化。流动相的选择主要从溶剂种类和梯度洗脱设置两方面进行优化。分析方法中采用了乙腈作为有机相,原因是乙腈比甲醇具有更大的洗脱强度,从而可以减少色谱峰的展宽,得到较好的峰型,此外,使用乙腈洗脱,其粘度较低,可以减小系统压力。在水相中加入TFA,可以进一步改善化合物的峰型,减少拖尾,此外,TFA的存在还可以提高样品在离子源中的离子化效率,因此,使用乙腈-0.05%TFA水溶液为流动相梯度洗脱,可以使样品分析在 9min之内完成。3、 生物样品中含有许多内源性物质,血浆中含量较高的内源性物质主要是蛋白类成分。蛋白质在测定过程中会形成泡沫,浑浊或沉淀,有时还会与加入的试剂发生反应,从而干扰测定。蛋白还会污染仪器。如果直接进样用液相色谱分析含蛋白的体液样品,蛋白质会逐渐变性沉结在色谱柱上,导致柱效降低,柱压上升,甚至堵塞色谱柱;含有蛋白的样品如果进入离子源,会造成离子源的严重污染和损坏,降低检测的灵敏度,所以血浆样品需进行合理的前处理。常用的生物样品前处理方法有蛋白沉淀法、固相萃取法和液液萃取法。由于待测的代谢产物的极性都比较大,采用液液萃取法(溶剂用乙酸乙酯)对化合物的提取效率差,因此不宜使用。主要比较了蛋白沉淀法和固相萃取法,两种方法均能有效提取待测化合物,经过实验发现,蛋白沉淀法比较好,并且考虑到血浆样品量较少,因此选择蛋白沉淀法。
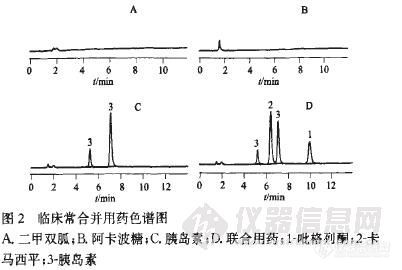
【作者】 方昱; 张虹; 李英;【机构】 同济大学附属同济医院;【摘要】 目的:建立高效液相色谱法测定人血浆中盐酸吡格列酮浓度的方法。方法:以卡马西平为内标,血浆样品用醋酸乙酯萃取,采用Diamonsil(TM)C18柱(250mm×4.6mm,5μm)分析。流动相为乙腈-0.05mol.L-1磷酸二氢钾(pH6.5)(42∶58),流速1.2mL.min-1,检测波长229nm,柱温35℃。结果:本法在0.025~4.0mg.L-1间线性关系良好,r=0.999 9,RSD为2.08%,(n=6),最低检测质量浓度为0.02mg.L-1。日内、日间精密度RSD均小于10%,低、中、高浓度的提取回收率均大于99%(n=5)。结论:此方法灵敏度高、重复性好,尤其适用于临床治疗合并用药时的药物浓度监测。【谱图】
各位老师,求帮忙,,,我用高效液相色谱分析盐酸地尔硫卓,发现峰型拖尾严重(拖尾因子都有4.9)流动相:醋酸盐缓冲液(用氢氧化钠调PH6.2):乙腈:甲醇=50::25:25用无水乙醇溶解的样品,浓度为0.1㎎/ml柱子是新的C18,250X4.6的
名 称 盐酸滴定液的配制及标定操作规程 一、 目 的:建立盐酸滴定液(1mol/L、0.5mol/L、0.2mol/L、 0.1mol/L)的配制及标定的标准操作规程,确保检验的准确性。二、 适用范围:适用于盐酸滴定液(1mol/L、0.5mol/L、0.2mol/L、0.1mol/L)的配制及标定。三、 责 任 者:QC化验员、QA。四、 引用标准:中华人民共和国药典(2000年版)二部附录。五、 规 程:1、误差要求: 法定标准标定 标定份数≥3份,相对偏差≤0.1%复标 复标份数≥3份,相对偏差≤0.1%标定、复标 二者的相对偏差≤0.15%滴定液浓度 (F)应在1.000-1.050之间2、反应原理:2HCl+Na2CO3 2 NaCl+H2O+CO23、指示剂:甲基红-溴甲酚绿混合指示液4、基准试剂:基准无水碳酸钠5、仪器与用具:三角烧瓶、滴定管6、操作步骤配制:盐酸滴定液(1 mol/L) 取盐酸90 ml,加水适量使成1000 ml,摇匀。 盐酸滴定液(0.5 mol/L、0.2 mol/L或0.1 mol/L) 照上法配制,但盐酸的取用量分别为45 ml、18 ml或9.0 ml。标定:盐酸滴定液(1 mol/L) 取在270~300 ℃干燥至恒重的基准无水碳酸钠约1.5g,精密称定,加水50ml使溶解,加甲基红-溴甲酚绿混合指示液10滴,用本液滴定至溶液由绿色转变为紫红色时,煮沸2分种,冷却至室温,继续滴定至溶液由绿色变为暗紫色。每1ml盐酸滴定液(1mol/L)相当于53.00mg的无水碳酸钠。根据本液的消耗量与无水碳酸钠的取用量,算出本液的浓度,即得。盐酸滴定液(0.5mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.8g。每1ml盐酸滴定液(0.5mol/L)相当于26.50mg的无水碳酸钠。盐酸滴定液(0.2mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.3g。每1ml盐酸滴定液(0.2mol/L)相当于10.60mg的无水碳酸钠。盐酸滴定液(0.1mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.15g。每1ml盐酸滴定液(0.1mol/L)相当于5.30mg的无水碳酸钠。如需用盐酸滴定液(0.05 mol/L、0.02 mol/L或0.01 mol/L)时,可取盐酸滴定液(1mol/L或0.1mol/L)加水稀释制成。必要时标定浓度。7、计算公式: 盐酸滴定液(1 mol/L) F=M/(V×0.053)盐酸滴定液(0.5 mol/L) F=M/(V×0.0265)盐酸滴定液(0.02mol/L) F=M/(V×0.0106)盐酸滴定液(0.1 mol/L)F=M/(V×0.0053)式中:M无水碳酸钠的质量(g) V滴定所耗盐酸滴定液的体积(ml) 8、注意事项反应致近终点时,应加热煮沸2分钟,以赶走溶解于水中的CO2,排除CO2对反应的干扰。
林可霉素,盐酸克林霉素,大观霉素,3种药物的ASE和手动2种方式提取大观霉素回收率低,药物极性强,易溶于水,溶于甲醇。乙腈,甲醇,水,甲酸水都已试过,求各位大神指教。
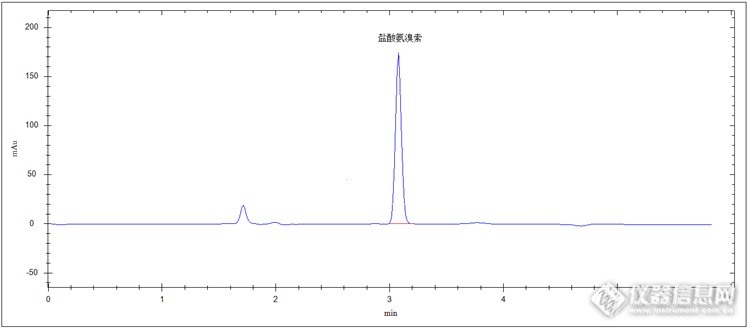
盐酸氨溴索片超快速检测 盐酸氨溴索片是一种良好的祛痰药,具有促进粘液排除作用及溶解分泌物的特性,有良好的祛痰及润滑呼吸道功效,并可促进肺表面物质的活性等药效。对于咳嗽、痰多、急慢性呼吸道疾病、支气管分泌异常等症状的治疗效果显著。 盐酸氨溴索片中主要药物成分为盐酸氨溴索,药物的品质和该物质在该药物中的含量有着直接的关系,所以该药品中该药物成分的检测不可小觑。 下面我们就来介绍高效液相色谱法检测盐酸氨溴索片中盐酸氨溴索含量。 对照品溶液的制备: 精密称取盐酸氨溴索对照品7.5mg于具塞离心管中,加甲醇0.2ml溶解,再加甲醛溶液40μl,摇匀,置于60℃恒温水域中加热5分钟,氮吹仪吹干。残渣用5ml水充分溶解,后置于25ml容量瓶中,加流动相稀释至刻度,制成300μg/ml对照品溶液,备用。 准确量取盐酸氨溴索对照品溶液2.5ml于25ml容量瓶中,加流动相溶解至刻度,制成30μg/1ml对照品溶液(命名为工作液),备用。 供试品溶液的制备: 取该样品适量,充分研碎,精密称取1.5mg于50ml容量瓶中,加流动相溶解并定容至刻度,0.45μm微膜滤过,待测。色谱条件:检测器:紫外检测器色谱柱:pGrandsil-STC C18,4.6 X 150mm,5μm检测波长:248nm流动相:0.01mol/L磷酸氢二铵溶液(用磷酸调节pH值至7.0)-乙腈(40:60)流速:1.0 mLmin
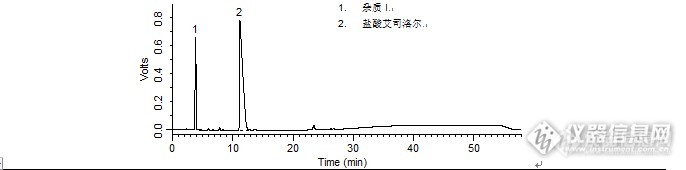
问题:注射用盐酸艾司洛尔:有关物质衍生溶液中杂质和盐酸艾司洛尔的分离度是多少?答案:10.154获奖名单:zengzhengce163(ID:zengzhengce163)WUYUWUQIU(ID:wulin321)dyd3183621(ID:dyd3183621)http://ng1.17img.cn/bbsfiles/images/2016/01/201601081731_581399_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601081732_581400_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601081732_581401_708_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/01/201601081732_581402_708_3.jpg活动奖励】幸运奖(2钻石币):抽奖软件,当天随机抽取3个回答正确的版友ID号(最后一个ID号,截止至下午3:00),每人奖励2个钻石币积分奖励:所有回答正确的版友奖励10个积分(幸运奖获得者除外)。【注意事项】同样的答案,每人只能发一次PS:该贴浏览权限为“回贴仅作者和自己可见”,回复的版友仅能看到版主的题目及自己的回答内容,无法看到其他版友的回复内容。下午3点之后解除,即可看到正确答案、获奖情况及所有版友的回复内容。注射用盐酸艾司洛尔样品制备 制备方法含量测定对照品溶液:取盐酸艾司洛尔对照品,加水溶解并定量稀释制成每1 mL 约含50 μg 的溶液。有关物质衍生溶液:取盐酸艾司洛尔对照品约10 mg,置10 mL量瓶中,加入1 mol/L盐酸溶液1 mL,放置30分钟,加1 mol/L的氢氧化钠溶液1 mL使中和,用流动相A 稀释至刻度,摇匀。分析条件(含量测定) 色谱柱Diamonsil C18(2) 250 x 4.6 mm,5 μm (Cat#:99603)流动相乙腈:甲醇:磷酸盐缓冲液(取磷酸二氢钾3.0g,加水溶解并稀释至650 mL)=15:20:65 流速1 mL/min柱温30 ℃检测器UV 222 nm进样量20 μL 分析条件(有关物质) 色谱柱Diamonsil C18(2) 250 x 4.6 mm,5 μm (Cat#:99603)流动相流动相A :乙腈:甲醇:磷酸盐缓冲液(取磷酸二氢钾3.0 g,加水至650 mL)=15:20:65流动相B:甲醇梯度流速1 mL/min柱温30 ℃检测器UV 222 nm进样量20 μL 色谱图含量测定对照品溶液http://ng1.17img.cn/bbsfiles/images/2016/01/201601081023_581265_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数* N USP拖尾因子 分离度 1 9.345 677855 56730 13763.887 1.061 -- *药典要求理论板数按盐酸艾司洛尔峰计算不低于2000有关物质衍生溶液http://ng1.17img.cn/bbsfiles/images/2016/01/201601081025_581266_708_3.png 峰号 保留时间 min 峰面积 μV*s 峰高 μV 理论塔板数 N USP拖尾因子 分离度 1 3.842 6280189 655879 2747.059 0.670 -- 2 11.157 29271705
第十三章 杂环类药物的分析 (上)掌握异烟肼、尼可刹米的鉴别、杂质检查和含量测定方法。 掌握盐酸氯丙嗪、奋乃静的鉴别、杂质检查和含量测定方法。 掌握地西泮、氯氮卓及其制剂的鉴别、杂质检查和含量测定方法。 本章讨论化学合成的杂环类药物,选择应用比较广泛的三类杂环药物中的几个典型药物予以重点介绍:即吡啶类中的尼可刹米、异烟肼;吩噻嗪类中的氯丙嗪、奋乃静;苯骈二氮杂卓类中的地西泮和氯氮卓。 第一节 异烟肼的分析一、结构与性质 本类药物母核吡啶环上的氮原子为碱性氮原子,吡啶环γ位上被酰肼取代,酰肼基具有较强的还原性,并可与某些含羰基的试剂发生缩合反应。 二、鉴别试验 1.制备衍生物测定熔点酰肼基与芳醛缩合形成腙,析出结晶,可测定其熔点。最常用的芳醛为香草醛。2.银镜反应 取异烟肼约10mg,置试管中,加水2ml溶解后,加氨制硝酸银试液1ml,即发生气泡与黑色浑浊,并在试管壁上生成银镜。 3.红外光谱三、异烟肼中游离肼的检查 异烟肼是一种不稳定的药物,其中的游离肼是由制备时原料引入,或在贮存过程中降解而产生。而肼又是一种诱变剂和致癌物质,因此国内外药典多数规定了异烟肼原料药及其制剂中游离肼的限量检查。中国药典对异烟肼原料和注射用异烟肼中游离肼的检查均采用薄层色谱法。 检查方法 取本品,加水制成每1ml中含50mg的溶液,作为供试品溶液。另取硫酸肼加水制成每1ml中含0.20mg(相当于游离肼50μg)的溶液,作为对照溶液。吸取供试液10μl 与对照溶液2μl,分别点于同一硅胶薄层板(用羧甲基纤维素钠溶液制备)上,以异丙醇-丙酮(3:2)为展开剂,展开后,晾干,喷以乙醇制对二甲氨基苯甲醛试液,15min后检视,在供试品主斑点前方与硫酸肼斑点相应的位置上,不得显黄色斑点。 异烟肼斑点呈棕橙色的清晰斑点,Rf值约为0.21。游离肼斑点呈鲜黄色,Rf值约为0.3。本法检出肼的灵敏度为0.1μg,检出限量约为0.02%。四、含量测定:异烟肼具有还原性,可用氧化还原滴定法测定含量。中国药典采用溴酸钾法,用甲基橙指示剂指示终点;同时采用本法测定异烟肼片和注射剂的含量。 测定方法 取本品约0.2g,精密称定,置100ml量瓶中,加水使溶解并稀释至刻度,摇匀;精密量取25ml,加水50ml、盐酸20ml与甲基橙指示剂1滴,用溴酸钾滴定液(0.01667mol/L)缓缓滴定(温度保持在18~25℃)至粉红色消失。 还可采用碘量法、溴量法、非水滴定法测定含量。







 我要推广仪器
我要推广仪器
 下载APP
下载APP