采用YMC-Pack TMS (C1)色谱柱按照EP6.0和USP 32nd规定色谱条件分离盐酸帕罗西汀及有关物质(杂质)。
【成果类别】 应用技术【成果水平】 国内领先【研究起止时间】 【评价形式】 行业准入【成果入库时间】 2004
试验目的:为该品种后期研究如有关物质和含量测定摸索流动相。试验条件:【含量测定】照高效液相色谱法(中国药典2010年版二部附录V D)测定。色谱条件与系统适用性试验用十八烷基硅烷键合硅胶 为填充剂;取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相;检测波长为295nm。分别取盐酸帕罗西汀、去氟帕罗两汀与N-甲基帕罗西汀对照品各5mg,置同一10ml量瓶中,加流动相 溶解并稀释至刻度,摇匀,作为系统适用性试验溶液,取20μl注人液相色谱仪,记录色谱图,出峰顺序为去氟帕罗西汀峰、盐酸帕罗西汀峰、N-甲基帕罗西汀峰,理论板数按盐酸帕罗西汀峰计算不低于3000,盐酸帕罗西汀峰、去氟帕罗西汀峰与 N-甲基帕罗西汀峰之间的分离度均应符合要求。测定法 取本品10片,精密称定,研细,精密称取适量(约相当于帕罗西汀10mg),用流动相溶解并稀释制成每 lml中约含0.lmg的溶液,摇匀,滤过,精密量取续滤液20μl注人液相色谱仪,记录色谱图;另取盐酸帕罗西汀对照品,同法测定,按外标法以峰面积计算,即得。色谱柱信息:序列号(SN):W11212195。典型色谱图:http://ng1.17img.cn/bbsfiles/images/2013/02/201302051437_424683_1621890_3.gif对照品和样品色谱峰比较:http://ng1.17img.cn/bbsfiles/images/2013/02/201302051501_424684_1621890_3.gif试验总结:采用中国药典2010年版二部收载的方法,进行原料药的含量标定,其系统适用性结果符合要求,拖尾因子和分离度不是很理想,在后期研究准备调节流动相成分比例,以期得到改善。
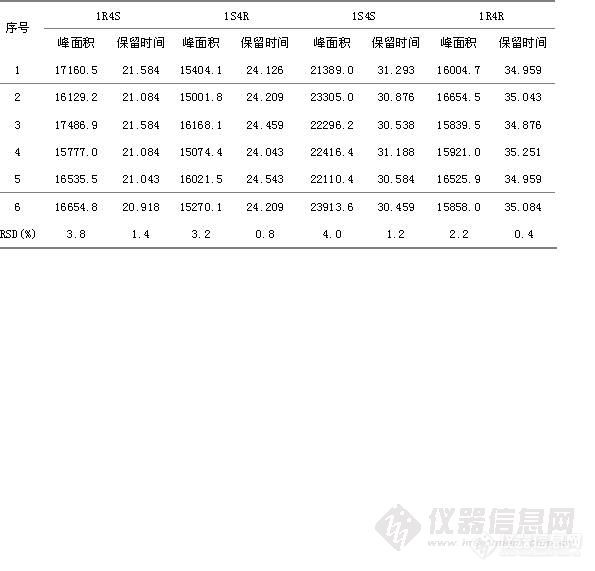
继“投石问路----记一次流动相的摸索”后,这篇是后续的研究部分。项目:含量测定(3.2.P.5.2.7)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:色谱柱(柱长:150mm,内径:4.6mm,填料:C18,填料粒径:5μm ,SN:W11212195)UV检测器(检测波长:295nm)柱温:室温流动相:取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相流速:1.0ml/min运行时间:约20分钟系统适用性:去氟帕罗西汀峰、盐酸帕罗西汀峰、N-甲基帕罗西汀峰,理论板数按盐酸帕罗西汀峰计算不低于3000,盐酸帕罗西汀峰、去氟帕罗西汀峰与 N-甲基帕罗西汀峰之间的分离度均应符合要求。具体试验操作:取本品10片,精密称定,研细,精密称取适量(约相当于帕罗西汀10mg),用流动相溶解并稀释制成每 lml中约含0.lmg的溶液,摇匀,滤过,精密量取续滤液20μl注人液相色谱仪,记录色谱图;另取盐酸帕罗西汀对照品,同法测定,按外标法以峰面积计算,即得。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。http://ng1.17img.cn/bbsfiles/images/2013/06/201306211717_446850_1621890_3.gif3.2.P.5.3.5.2流动相选择(色谱图见附件941~955)参照中国药典2010年版二部收载的盐酸帕罗西汀片质量标准中含量测定项, 取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相。(1)、试验方法与具体步骤:精密称取盐酸帕罗西汀对照品23.0mg、去氟帕罗西汀对照品5.25mg、N-甲基帕罗西汀对照品5.41mg,同置10ml量瓶中,加流动相适量超声使溶解并稀释至刻度,摇匀,滤过,作为系统适用性试验供试液1;精密称取盐酸帕罗西汀对照品10.2mg,置100ml量瓶中,加流动相适量超声使溶解并稀释至刻度,摇匀,滤过,作为系统适用性试验供试液2。分别精密量取以上供试液20μl注入液相色谱仪记录色谱图。(2)、试验结果见下表图。http://ng1.17img.cn/bbsfiles/images/2013/06/201306211718_446851_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306211718_446852_1621890_3.gif以上试验结果表明,以取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相,盐酸帕罗西汀与去氟帕罗西汀和N-甲基帕罗西汀均能有效分离,各峰理论板数均大于3000,适合含量测定。3.2.P.5.3.5.3进样精密度试验(色谱图见附件956~961)精密称取盐酸帕罗西汀对照品10.4mg,置100ml量瓶中,加流动相适量超声使溶解并稀释至刻度,摇匀,滤过,取续滤液作为供试液,精密量取供试液20μl注入液相色谱仪,记录色谱图,重复操作6次,试验结果见下表。http://ng1.17img.cn/bbsfiles/images/2013/06/201306211719_446854_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306211720_446855_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306211721_446857_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306211722_446858_1621890_3.g
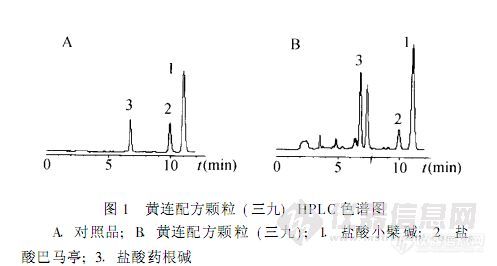
【作者中文名】雷鹏; 刘韶; 李新中; 徐平声;【作者英文名】Lei Peng; Liu Shao; Li Xin-zhong; Xu Ping-sheng(Department of Pharmacy of Xiangya Hospital; Central South University; Changsha 410008);【作者单位】中南大学湘雅医院药剂科; 中南大学湘雅医院药剂科 长沙;【摘要】目的 考察不同厂家黄连配方颗粒中盐酸小檗碱、盐酸巴马亭和盐酸药根碱含量。方法 用高效液相色谱法测定盐酸小檗碱、盐酸巴马亭和盐酸药根碱含量。色谱柱为Diamonsil C18 (4 6 mm×250 mm, 5μm); 流动相为0 2 mol·L-1磷酸二氢钠(用磷酸调节pH至3 0) 乙腈(70∶30); 流速为1 mL·min-1; 柱温为30 ℃; 检测波长为345 nm。结果 不同厂家黄连配方颗粒中盐酸小檗碱含量为3 .67~72. 53 mg·包-1, 盐酸巴马亭含量为0 .60~28. 70 mg·包-1, 盐酸药根碱含量为5. 40~26 .54 mg·包-1。结论 不同厂家产品盐酸小檗碱、盐酸巴马亭和盐酸药根碱含量差异显著。http://ng1.17img.cn/bbsfiles/images/2012/08/201208061421_381879_2379123_3.jpg

盐酸可乐定的测定和盐酸丁咯地尔片含量的测定http://ng1.17img.cn/bbsfiles/images/2009/11/200911021833_180179_1896702_3.jpg
继““色”路蹒跚,证得正果,某品种含量测定方法学部分”后整理的耐用性试验部分。藤下葡萄,一颗知涩、一颗知酸、一颗知甜、三颗知百味。检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:色谱柱(柱长:150mm,内径:4.6mm,填料:C18,填料粒径:5μm ,SN:W11212195)UV检测器(检测波长:295nm)柱温:室温流动相:取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相流速:1.0ml/min运行时间:约20分钟系统适用性:去氟帕罗西汀峰、盐酸帕罗西汀峰、N-甲基帕罗西汀峰,理论板数按盐酸帕罗西汀峰计算不低于3000,盐酸帕罗西汀峰、去氟帕罗西汀峰与 N-甲基帕罗西汀峰之间的分离度均应符合要求。具体试验操作:取本品10片,精密称定,研细,精密称取适量(约相当于帕罗西汀10mg),用流动相溶解并稀释制成每 lml中约含0.lmg的溶液,摇匀,滤过,精密量取续滤液20μl注人液相色谱仪,记录色谱图;另取盐酸帕罗西汀对照品,同法测定,按外标法以峰面积计算,即得。计算公式:标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。http://ng1.17img.cn/bbsfiles/images/2013/06/201306281008_448170_1621890_3.gif统计结果表明,各试验条件变化对含量测定结果无明显影响,各试验项目及结果见下。申报质量标准条件。取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相,流速为每分钟1.0ml,检测波长为295nm。http://ng1.17img.cn/bbsfiles/images/2013/06/201306281014_448171_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306281014_448172_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306281014_448173_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306281014_448174_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306281015_448175_1621890_3.gifhttp://ng1.17img.cn/bbsfiles/images/2013/06/201306281015_448176_1621890_3.gif
建立毛细管气相色谱法测定盐酸氮卓斯汀中残留溶剂。采用DB-624毛细管色谱柱,FID检测器,二甲基甲酰胺为溶剂,程序升温,外标法同时检测盐酸氮卓斯汀中乙醇、丙酮、二氯甲烷、正己烷4种有机溶剂残留量。各待测组分完全分离,线性关系良好,检测限分别为3.51μg/mL、0.35μg/mL、2.12μg/mL、0.11μg/mL,精密度RSD5%,平均回收率99.1%~102.4%。本法操作简单,结果准确可靠,可用于盐酸氮卓斯汀中残留溶剂的检测。
领导希望能探索出一个流动相,可以对辛伐他汀,辛伐他汀铵盐,盐酸环丙沙星,环丙羧酸用一种流动相进行预检,以确定其有关物质情况,有没有人做过这方面的研究,求帮助
请问谁做过这个药品的含量,谁知道为什么要用氮气吹干呀,没有氮气自然干燥给以吗???[font=SimSun][size=4][size=2][font=宋体] 色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;以0.01mol/L磷酸氢二铵溶液(用磷酸调节pH值至7.O)-乙腈(50:50)为流动[/font][/size][/size][font=SimSun][size=3]相;检测波长为248nm。取盐酸氨溴索对照品约5mg,加甲醇0.2ml溶解,再加甲醛溶液(1→100)401,摇匀,置60℃加热5分钟,氮气吹干。残渣用水5ml溶解,再加流动相稀释至20ml,取20靗注入液相色谱仪,盐酸氨溴索与杂质B(相对保留时间约为0.8)的分离度应大于4.0。[/size][/font][/font][size=3][font=SimSun][size=2][font=宋体] 测定法 取本品20片,精密称定,研细,精密称取适量,加流动相溶解并定量稀释制成每lml中约含盐酸氨溴索30[/font][/size][size=2][font=宋体]靏[/font][/size][size=2][font=宋体]的溶液,滤过,精密量取续滤液[/font][/size][size=2][font=宋体]20[/font][/size][size=2][font=宋体]靗[/font][/size][size=2][font=宋体]注入液相色谱仪,记录色谱图;另取盐酸氨溴索对照品适量,精密称定,用流动相溶解并定量稀释制成每lml中约含30[/font][/size][size=2][font=宋体]靏[/font][/size][size=2][font=宋体]的溶液,同法测定。按外标法以峰[/font][/size][/font][size=2][font=宋体][font=SimSun]面积计算,即得。[/font][/font][/size][/size]
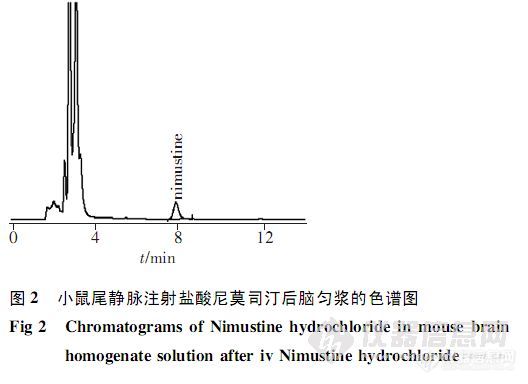
【作者】 王雪; 巫孟君; 贾钰铭; 张文彬; 陈华黎; 覃瑶; 何勤;【Author】 WANG Xue1,WU Meng-jun1,JIA Yu-ming2,ZHANG Wen-bin3,CHEN Hua-li1,QIN Yao1,HE Qin1 (1. West China School of Pharmacy,Sichuan University,Chengdu,Sichuan,610041 P. R. China;2. The No. 2 People’s Hospital in Yib- in,Yinbin,Sichuan,644000 P. R. China;3. The Tumor Hospital in Sichuan,Chengdu,Sichuan,610041 P. R. China )【机构】 四川大学华西药学院; 宜宾市第二人民医院; 四川省肿瘤医院;【摘要】 目的采用HPLC法测定小鼠脑匀浆中的盐酸尼莫司汀。方法使用Diamonsil C18柱,流动相为甲醇-水-三乙胺(20∶79.5∶0.5,磷酸调pH3),流速1.0mL·min-1,检测波长241nm,柱温35℃。结果盐酸尼莫司汀0.20~5.00mg·L-1与峰面积的线性关系良好(r=0.9999);平均方法回收率95.7%,平均萃取回收率101.2%,日内、日间RSD均5%。结论该方法简便、灵敏、准确,可用于测定脑匀浆中的盐酸尼莫司汀。http://ng1.17img.cn/bbsfiles/images/2012/07/201207301543_380591_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207301544_380592_2379123_3.jpg
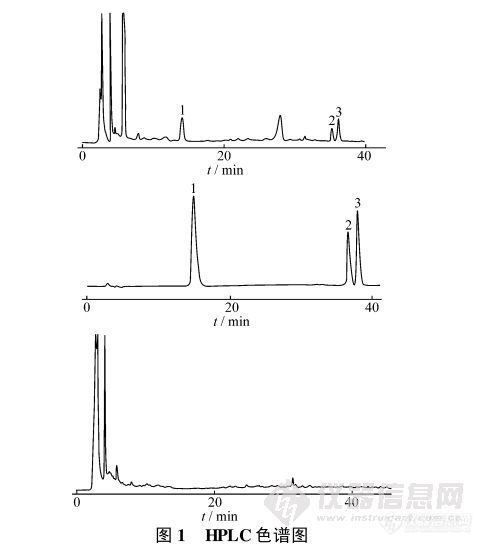
【作者】:申兰慧, 石 涛, 邹爱峰, 任金山, 张胜强【摘要】: 目的: 建立儿童Ò 号口服液( 黄柏、龙胆草、知母等) 中龙胆苦苷, 盐酸巴马汀及盐酸小檗碱高效液相色谱测定方法。方法: 以Diamonsil TM C18柱( 250mm @ 4. 6mm, 5Lm) 为色谱柱, 以乙腈- 磷酸- 三乙胺- 水为流动相, 检测波长为270nm, 流速为1mL#min- 1。结果: 龙胆苦苷, 盐酸巴马汀及盐酸小檗碱的线性范围分别为72. 8~ 728Lg#mL- 1( r = 0. 9998) , 7. 55~75. 5Lg#mL- 1( r = 0. 9998) , 12. 45~ 124. 5Lg#mL- 1( r = 0. 9999) , 平均加样回收率( n = 6) 分别为98. 17% , 98. 78% ,98. 60% ; RSD 分别为1. 60% , 1. 35% , 1. 25% 。结论: 该方法准确, 重复性好, 专属性好, 适合于儿童Ò 号的质量控制。【作者单位】: 中国药科大学药物分析教研室, 东南大学药剂科, 南京威尔曼药物研究所【关键词】: RP- HPL C; 梯度洗脱; 龙胆苦苷; 盐酸巴马汀; 盐酸小檗碱http://ng1.17img.cn/bbsfiles/images/2012/07/201207311305_380827_1838299_3.jpg
NYT 438-2001中,偏磷酸+甲醇提取后,氢氧化钠调pH 11-12,然后用乙醚萃取,取乙醚层吹干定容过柱后测定。 同时搜到两篇文献,1是《中国卫生检验杂志》2005年第9期摘录,地址http://www.gotoread.com/vo/2620/page274153.html这篇完全是按照NYT 438来做的,用乙醚来萃取2是饲料工业 FEED INDUSTRY 2004 Vol.25 No.5 P.58-60http://www.wanfangdata.com.cn/qikan/periodical.Articles/silgy/silg2004/0405/040519.htm摘 要:探索了饲料中盐酸克伦特罗的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法测定方法.饲料试样中盐酸克伦特罗经0.01mol/l盐酸溶液提取,乙醚脱脂净化请问:1、氢氧化钠调pH的作用。2、乙醚。一个用乙醚萃取,一个用乙醚净化,标准说的是用乙醚萃取,但是用百度搜索“盐酸克伦特罗 乙醚”,所有网页里面都有很显目一句“不溶于乙醚”,何解?本来在前处理版发了同样帖子,但是好几天都没人理,只好在这再发一个了,请高手指点!谢谢!
加热浓盐酸,HCL挥发大于水,当HCL的质量分数下降到20.2%时,HCL与水的挥发相同,液体会保持20.2%的质量分数不变。这是在网上看到的结论。不知是不是这样的?加热稀盐酸(质量分数远远小于20.2%的),盐酸的质量分数会不会也返回到并保持20.2%不变呢?网上找到的:因为浓度为20.24%的HCl溶液恰好是恒沸溶液,即达到平衡状态时,蒸气的组成与溶液的组成相同且HCl的质量分数都是20.24%。易于理解的说法就是:当浓度大于20.24%时,HCl的蒸发速率大于H2O的蒸发速率;当浓度小于20.24%时,HCl的蒸发速率小于H2O的蒸发速率;当浓度为20.24%时,HCl的蒸发速率与H2O的蒸发速率相同,达到了动态平衡。因此无论盐酸的初始浓度是多少,只要外压是101.3kPa,蒸馏最后都得到20.24%的盐酸,其沸点最高,是383K。
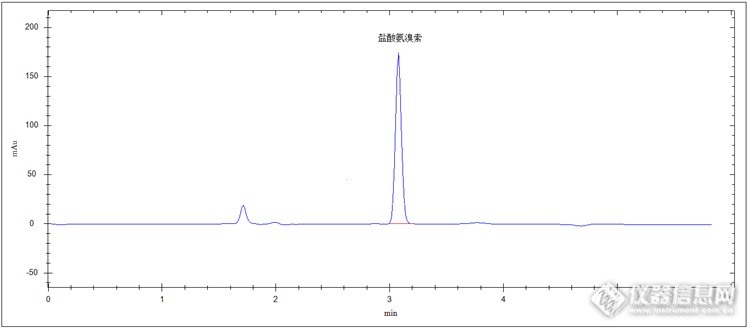
盐酸氨溴索片超快速检测 盐酸氨溴索片是一种良好的祛痰药,具有促进粘液排除作用及溶解分泌物的特性,有良好的祛痰及润滑呼吸道功效,并可促进肺表面物质的活性等药效。对于咳嗽、痰多、急慢性呼吸道疾病、支气管分泌异常等症状的治疗效果显著。 盐酸氨溴索片中主要药物成分为盐酸氨溴索,药物的品质和该物质在该药物中的含量有着直接的关系,所以该药品中该药物成分的检测不可小觑。 下面我们就来介绍高效液相色谱法检测盐酸氨溴索片中盐酸氨溴索含量。 对照品溶液的制备: 精密称取盐酸氨溴索对照品7.5mg于具塞离心管中,加甲醇0.2ml溶解,再加甲醛溶液40μl,摇匀,置于60℃恒温水域中加热5分钟,氮吹仪吹干。残渣用5ml水充分溶解,后置于25ml容量瓶中,加流动相稀释至刻度,制成300μg/ml对照品溶液,备用。 准确量取盐酸氨溴索对照品溶液2.5ml于25ml容量瓶中,加流动相溶解至刻度,制成30μg/1ml对照品溶液(命名为工作液),备用。 供试品溶液的制备: 取该样品适量,充分研碎,精密称取1.5mg于50ml容量瓶中,加流动相溶解并定容至刻度,0.45μm微膜滤过,待测。色谱条件:检测器:紫外检测器色谱柱:pGrandsil-STC C18,4.6 X 150mm,5μm检测波长:248nm流动相:0.01mol/L磷酸氢二铵溶液(用磷酸调节pH值至7.0)-乙腈(40:60)流速:1.0 mLmin
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=108875]NYT 438-2001 饲料中盐酸克伦特罗的测定[/url]NYT 438-2001中,偏磷酸+甲醇提取后,氢氧化钠调pH 11-12,然后用乙醚萃取,取乙醚层吹干定容过柱后测定。 同时搜到两篇文献,1是《中国卫生检验杂志》2005年第9期摘录,地址http://www.gotoread.com/vo/2620/page274153.html这篇完全是按照NYT 438来做的,用乙醚来萃取2是饲料工业 FEED INDUSTRY 2004 Vol.25 No.5 P.58-60http://www.wanfangdata.com.cn/qikan/periodical.Articles/silgy/silg2004/0405/040519.htm摘 要:探索了饲料中盐酸克伦特罗的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱法测定方法.饲料试样中盐酸克伦特罗经0.01mol/l盐酸溶液提取,乙醚脱脂净化请问:1、氢氧化钠调pH的作用。2、乙醚。一个用乙醚萃取,一个用乙醚净化,标准说的是用乙醚萃取,但是用百度搜索“盐酸克伦特罗 乙醚”,所有网页里面都有很显目一句“不溶于乙醚”,何解?请高手指点!谢谢!
【作者】 陆榕; 孙进; 方金玲; 孙英华; 刘晓红; 何仲贵;【机构】 沈阳药科大学药学院; 沈阳药科大学药学院 辽宁沈阳110016; 辽宁沈阳110016;【摘要】 目的用离子对HPLC法测定盐酸倍他司汀注射液中药物及有关物质含量。方法色谱柱:Diamonsil C18柱(200 mm×4.6 mm,5μm);流动相:甲醇10 mmol.L-1醋酸钠溶液(含5 mmol.L-1庚烷磺酸钠,体积分数为0.2%的三乙胺,用冰醋酸调pH至3.3)(25∶75),检测波长:261 nm,流速:1.0 mL.min-1,柱温:室温。结果盐酸倍他司汀质量浓度在1~250 mg.L-1内线性关系良好(r=0.999 9),平均回收率为99.8%,RSD=1.0%。盐酸倍他司汀的理论塔板数大于2 000,盐酸倍他司汀与其主要杂质分离度不低于1.5。结论适用于盐酸倍他司汀注射液中药物与有关物质的含量测定。http://ng1.17img.cn/bbsfiles/images/2012/07/201207251621_379666_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207251623_379668_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207251623_379670_2379123_3.jpg
话说广东加强了对危险化学品的控制,经销购买盐酸硫酸的都需要取得公安局的安全证明,就是拿N多万去买个证。而且现在都办不到证了。(貌似硝酸却不在管制范围???)这两天盐酸用完了,请购盐酸时,采购才说没证买不到。偏偏供货商也没证。于是。。。。。。。。。。。。杯具啊!请问各位有遇到这样的情况吗?怎么解决的?我们一个小工厂,为了化验室半个月才用掉的一瓶盐酸去花钱办证似乎是很那个的行为。。。。。
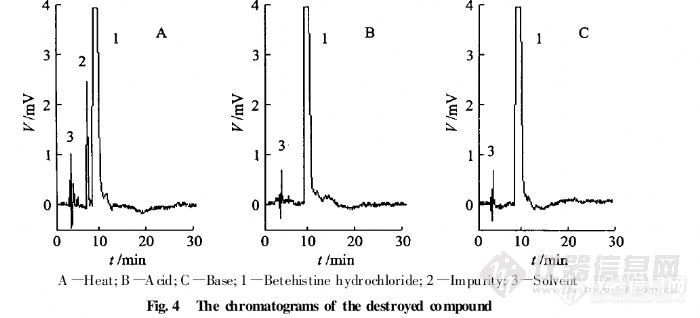
HPLC法测定盐酸舍曲林片的有关物质 摘要:目的:建立盐酸舍曲林片有关物质的测定方法,其外标法测定其四种光学异构体,自身对照法测定其它有关杂质。方法:采用依利特Hypersil ODS2色谱柱(4.6mm×150mm,5μm);流动相为磷酸二氢钠缓冲液 β-环糊精12.8g,HP-β-环糊精17.3g,加水至1000ml]-乙腈[font=Times New Roman]-[font=宋体]三乙胺([font=Times New Roman]800[font=宋体]:[font=Times New Roman]200[font=宋体]:[font=Times New Roman]10[font=宋体])[font=Times New Roman],[font=宋体]用磷酸调节[font=Times New Roman]p H [font=宋体]为[font=Times New Roman]2.5±0.5为流动相,流速为每分钟1ml/min,检测波长为214nm。结果:建立了β-环糊精、HP-β-环糊精手性流动相拆分盐酸舍曲林光学异构体,的方法,结论:该方法专属性好,无干扰,盐酸舍曲林异构体与主峰分离良好。可作为控制盐酸舍曲林片质量的方法。关键词:HPLC外标法;HPLC自身对照法;盐酸舍曲林片;光学异构体; β-环糊精; HP-β-环糊精。盐酸舍曲林是一种选择性抑制[font=Times New Roman]5-[font=宋体]羟色胺再摄取的抗抑郁药([font=Times New Roman]SSRI[font=宋体]),为新一代抗抑郁药的代表药物,目前已跃成为抗抑郁治疗的一线药和首选药。盐酸舍曲林由美国辉瑞公司开发,并于[font=]1990[font=宋体]年[font=]12[font=宋体]月最初在英国上市,目前为止已经在[font=]30[font=宋体]个国家陆续上市。盐酸舍曲林(商品名:郁尔复片剂)于[font=Times New Roman]1996[font=宋体]年[font=Times New Roman]12[font=宋体]月获得中国药品行政保护,并于当年在中国以片剂形式(不含原料药)上市,用于治疗精神抑郁症。现在国内各大医院、药店均有销售。盐酸舍曲林在中国无专利保护。[font=Times New Roman]2004[font=宋体]年[font=Times New Roman]6[font=宋体]月该行政保护终止。由于盐酸舍曲林存在4种光学异构体,在合成工艺过程中可能会引入中间杂质盐酸舍曲林顺式左旋异构体,盐酸舍曲林反式左旋异构体,反式右旋异构体,因其均为手性异构体,眼前其异构体检测方法为毛细管电泳法,且只对其异构体顺式左旋异构体进行检测,其它杂质检测采用反相HPLC,且其盐酸舍曲林主峰不能很好的与其相邻异构体分开,参照相关文献,并经方法学研究,建立了盐酸舍曲林有关物质的检测方法,采用HPLC外标法测定[
我最近看AAS得贴子配标液添加的酸都是硝酸 而且浓度都很低我这标准 加的(1+1)的盐酸这浓度挺高的在安装仪器的时候 工程师说可以不加酸 直接用去离子水不知道这浓度高低有啥影响?盐酸和硝酸种类有啥不同
最近做了火焰两个Fe盲样,1.直接稀释,以纯水做空白,不加酸。2.用5%盐酸稀释,以5%盐酸作为空白。 结果:1. 7.546 和 47.5 回收率103% 2. 7.934 和 50.5 回收率103.4%标曲基体5%盐酸。按照道理减去相应的空白结果应该一样的,为什么加酸后偏高?(操作程序不会有什么问题。)应该相信哪个?为什么?

【作者】 李云兰; 林志华; 杨帆; 李菲菲; 李青山;【机构】 山西医科大学药学院; 山西医科大学药学院 山西太原030001; 山西太原030001;【摘要】 目的:建立高效液相色谱法测定注射用盐酸帕洛诺司琼的含量。方法:采用Shimadzu LC-10 ATvp液相色谱系统,色谱条件:Diamonsil ODS(4.6mm×200mm,5μm)色谱柱,以甲醇-磷酸盐缓冲液(45∶55)为流动相,流速为1.0mL.min-1,紫外检测波长241nm,柱温25℃。结果:在16.8~151.2mg.L-1范围内峰面积与浓度呈良好的线性关系,回归方程为A=6 026.82C-10 143,r=0.999 9(n=5),方法精密度的RSD为0.95%(n=6),稳定性的RSD为0.56%(n=9),重复性的RSD为0.90%(n=9),平均回收率为100.7%(n=9,RSD为0.43%)。结论:该方法简便、灵敏、专属、准确,可用来测定注射用盐酸帕洛诺司琼制剂中盐酸帕洛诺司琼的含量。http://ng1.17img.cn/bbsfiles/images/2012/08/201208142255_383928_1609970_3.jpg
听同事说,纯王水配置好后,放置一段时间,或者低温加热,如果颜色发红,说明盐酸硝酸没有问题。如果还是颜色发黄,且很浅,说明盐酸硝酸有问题,不知道大家有这方面的经验没有,是否可以靠这个来判断呢?
仪器及检测元素:AFS3000,Se溶样过程:样品消解完后,我加10ml浓盐酸(GR),样品呈墨绿色,加水定容到50ml,溶液颜色褪去,可测得的结果和直接用20%盐酸定容测得的结果相比,前者严重偏低。还有一个问题,先加浓盐酸,加标和不加标的样测得到结果基本一致,严重偏低。疑问:会不会是先加浓盐酸,使得Se挥发?
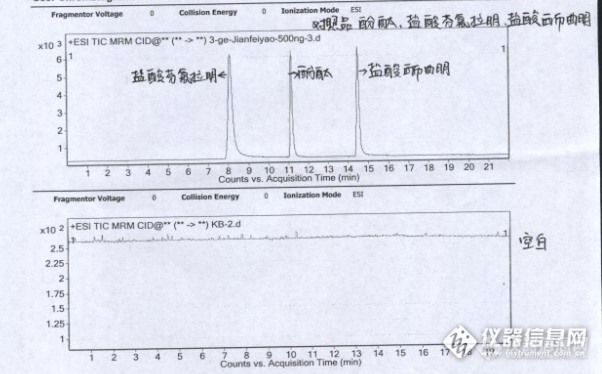
减肥产品中酚酞、盐酸西布曲明及盐酸芬氟拉明的检测盐酸西布曲明,盐酸芬氟拉明,酚酞已被国家食品药品监督管理总局明令禁止用于食品(含保健品)了,目前还有部分违规企业在减肥产品中违法添加。盐酸西布曲明曾为处方药,但目前已在全球大多数国家停止使用。盐酸西布曲明(Sibutramine Hydrochloride)是西布曲明(Sibutramine)的氯化物,是一种中枢神经抑制药物,曾用于肥胖症的治疗。酚酞是化学品和临床处方药,有严格的适应症,需在医生指导下应用,若长期过量服用可能引发严重的副作用。在制药上作为医药原料,其药品名称为酚酞片(Phenolphthalein Tablets),主要用于治疗习惯性、顽固性便秘。过量或长期滥用,可造成人体电解质代谢紊乱,严重时甚至可诱发心律失常。婴儿和哺乳期妇女禁用,幼儿和孕妇慎用。市场上抽检的三批次的减肥产品违法添加盐酸西布曲明,酚酞的检测:仪器型号及编号 Agilent 1200 LC/MS 6410B 天平型号及编号 BP211D,LD310-2 色谱条件:色谱柱:phenomenex C18柱(100x3.0 mm,2.6 μm) 预柱 Agilent 预柱流速(mL/min) 0.2 进样量(uL) 5 柱温(℃) 25℃ http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_668681_2166779_3.png质谱条件:电喷雾离子化源(ESI) 碰撞气压力(Mpa) 0.15 Nebulizerpressure(Psi) 15 drying GasFlow(L/min) 6 Dry Temp(℃) 350 电离源 ESI ,正离子模式 http://ng1.17img.cn/bbsfiles/images/2017/10/2016071708440193_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071708440482_01_2166779_3.png将标准品分别配制成1mg/mL的酚酞,盐酸芬氟拉明,盐酸西布曲明标准储备液,分别吸取标准储备溶液进行稀释,得到100ng/mL,80ng/mL,50ng/mL,20ng/mL,10ng/mL,5ng/mL的标准工作溶液。2.标准曲线的制作取各标准工作溶液5 uL注入液质仪,采集数据。以峰面积为纵坐标(Y),以标准工作溶液浓度(X)为横坐标绘制标准曲线。酚酞,盐酸芬氟拉明,盐酸西布曲明标准品及空白的色谱图、质谱图及工作曲线:http://ng1.17img.cn/bbsfiles/images/2017/10/2016071717420205_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717421213_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717422190_01_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717433884_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717434225_01_2166779_3.png3.试样提取取各试样适量(约相当于一次用量),置50mL离心管中,精密加入甲醇20mL,超声处理15min,放冷至室温,10000r/min离心5min,取上清液用50%甲醇稀释。稀释过程:①0.2→2.0ml (稀释10倍);②0.1→2.0ml(共稀释200倍); ③0.1→2.0ml(共稀释4000倍)③0.1→2.0ml(共稀释80000倍)样品1、2、3号的酚酞,盐酸西布曲明,盐酸芬氟拉明色谱图http://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600844_2166779_3.png样品1号酚酞,盐酸西布曲明,盐酸芬氟拉明的质谱图http://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600845_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600846_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607171746_600847_2166779_3.png从图中可见1号未检出盐酸芬氟拉明;2、3号样品的质谱图略。定量分析 酚酞的含量(mg/粒)= C样×V样 ×样品稀释倍数×W平 /W样×10-6 供试品编号 10粒内容物装量(g) 平均装量(g) 取样量 (g) 检测结果(ng/mL) 含量 (mg/粒) 平均含量 (mg/粒) 1号 2.6327 0.2633 0.2692 16.5391 25.88 27.8 0.2623 18.5693 29.82 2号 2.4988 0.2499 0.2691 15.0804 22.41 [align=c
EN71-3:2013+A1:2014方法规定工作溶液使用0.07M盐酸作为基体问题就来了通过比较使用硝酸作为基体的工作溶液与0.07M盐酸作为基体的工作溶液测试一个相同的未知浓度含有锡的溶液两种基体的工作溶液测试结果差别非常大:盐酸基体工作溶液测试结果比硝酸基体工作溶液测试结果大2倍左右到底哪个结果才是准确的呢?到底是什么原因导致的呢?我了解到的是:锡的母液(1000mg/L)是5%盐酸作为基体的,难道使用0.07M的盐酸(约0.6%)配成工作溶液之后,因为盐酸浓度降低了,而造成水解,导致测试结果偏高了?
项目:含量测定(3.2.P.5.2.7)检查方法:照高效液相色谱法(中国药典2010年版二部附录Ⅴ D)测定试验条件:色谱柱(柱长:150mm,内径:4.6mm,填料:C18,填料粒径:5μm)http://ng1.17img.cn/bbsfiles/images/2013/01/201301081630_419121_1621890_3.gifUV检测器(检测波长:225nm)柱温:30℃流动相:0.2mol/L磷酸二氢钾-0.2mol/L磷酸-乙腈(7:7:5)流速:1.0ml/min运行时间:约25分钟系统适用性:理论板数按盐酸坦洛新峰计算不低于2000。具体试验操作:取本品20片,精密称定,研细,精密称取适量(约相当于盐酸坦洛新0.2mg),置50ml量瓶中,加流动相适量振摇后超声使溶解,放冷至室温,加流动相稀释至刻度,摇匀,滤过,精密量取续滤液20μl注入液相色谱仪中,记录色谱图;另精密称取盐酸坦洛新对照品2mg置50ml量瓶中,加流动相振摇后超声使溶解并稀释至刻度,摇匀,滤过,精密量取续滤液1ml置10ml量瓶中,加流动相稀释至刻度,作为对照品溶液,精密量取对照品溶液20μl注入液相色谱仪中,记录色谱图。按外标法以峰面积计算,即得。计算公式:http://ng1.17img.cn/bbsfiles/images/2013/01/201301081634_419123_1621890_3.gif标示量百分含量(%)=××100%式中:Cs为对照品的浓度(mg/ml);At为供试液的主峰面积;Nt为供试液的稀释倍数;AS为对照品溶液的主峰面积;W为供试品取样量(mg)。3.2.P.5.3.5含量测定含量测定方法学验证结果概要项目验证结果波长选择225nm。流动相选择0.2mol/L磷酸二氢钾-0.2mol/L磷酸-乙腈(7:7:5)。进样精密度试验连续测量6次,主峰保留时间和峰面积RSD均小于2.0%。线性关系试验浓度在2460ng/ml~5740ng/ml,直线方程为y=107.8x﹢5880.1,R2=0.9992,相关系数r=0.9996,截距为5880,小于100%浓度溶液主峰面积的2.0%(8841)。溶液稳定性试验室温条件下置12个小时,测得峰面积的RSD为1.3%回收率试验平均回收率为100.3%,在98.0%~102.0%范围内,RSD为1.0%(n=9),小于2.0%。重复性试验试验结果在95.3%~100.5,RSD值为2.0%。耐用性同有关物质项含量测定结果上市品和自制品均符合规定3.2.P.5.3.5.1波长选择参照盐酸坦洛新缓释片药品质量标准YBH19552005含量测定项、盐酸坦洛新原料药质量标准YBH03262005有关物质项和本品有关物质研究试验结果,检测波长选定为225nm。3.2.P.5.3.5.2[
如题所述,最近在测定汞时(以硝酸作为介质),线性总是感觉不太好,以前用硝酸做介质时一直都挺好,这是最近的事,但也不是偏差很大,能达到995左右,但是换盐酸做介质时线性就没问题,不知道大家有没有遇到过这样的问题,一起讨论下这是什么原因。
名 称 盐酸滴定液的配制及标定操作规程 一、 目 的:建立盐酸滴定液(1mol/L、0.5mol/L、0.2mol/L、 0.1mol/L)的配制及标定的标准操作规程,确保检验的准确性。二、 适用范围:适用于盐酸滴定液(1mol/L、0.5mol/L、0.2mol/L、0.1mol/L)的配制及标定。三、 责 任 者:QC化验员、QA。四、 引用标准:中华人民共和国药典(2000年版)二部附录。五、 规 程:1、误差要求: 法定标准标定 标定份数≥3份,相对偏差≤0.1%复标 复标份数≥3份,相对偏差≤0.1%标定、复标 二者的相对偏差≤0.15%滴定液浓度 (F)应在1.000-1.050之间2、反应原理:2HCl+Na2CO3 2 NaCl+H2O+CO23、指示剂:甲基红-溴甲酚绿混合指示液4、基准试剂:基准无水碳酸钠5、仪器与用具:三角烧瓶、滴定管6、操作步骤配制:盐酸滴定液(1 mol/L) 取盐酸90 ml,加水适量使成1000 ml,摇匀。 盐酸滴定液(0.5 mol/L、0.2 mol/L或0.1 mol/L) 照上法配制,但盐酸的取用量分别为45 ml、18 ml或9.0 ml。标定:盐酸滴定液(1 mol/L) 取在270~300 ℃干燥至恒重的基准无水碳酸钠约1.5g,精密称定,加水50ml使溶解,加甲基红-溴甲酚绿混合指示液10滴,用本液滴定至溶液由绿色转变为紫红色时,煮沸2分种,冷却至室温,继续滴定至溶液由绿色变为暗紫色。每1ml盐酸滴定液(1mol/L)相当于53.00mg的无水碳酸钠。根据本液的消耗量与无水碳酸钠的取用量,算出本液的浓度,即得。盐酸滴定液(0.5mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.8g。每1ml盐酸滴定液(0.5mol/L)相当于26.50mg的无水碳酸钠。盐酸滴定液(0.2mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.3g。每1ml盐酸滴定液(0.2mol/L)相当于10.60mg的无水碳酸钠。盐酸滴定液(0.1mol/L) 照上法标定,但基准无水碳酸钠的取用量改为约0.15g。每1ml盐酸滴定液(0.1mol/L)相当于5.30mg的无水碳酸钠。如需用盐酸滴定液(0.05 mol/L、0.02 mol/L或0.01 mol/L)时,可取盐酸滴定液(1mol/L或0.1mol/L)加水稀释制成。必要时标定浓度。7、计算公式: 盐酸滴定液(1 mol/L) F=M/(V×0.053)盐酸滴定液(0.5 mol/L) F=M/(V×0.0265)盐酸滴定液(0.02mol/L) F=M/(V×0.0106)盐酸滴定液(0.1 mol/L)F=M/(V×0.0053)式中:M无水碳酸钠的质量(g) V滴定所耗盐酸滴定液的体积(ml) 8、注意事项反应致近终点时,应加热煮沸2分钟,以赶走溶解于水中的CO2,排除CO2对反应的干扰。

【作者】邓婕,梅虎,杨胜喜,刘振德,肖玉梅,周素容,李志良【单位】(1.重庆大学化学化工学院,药物化学研究所生物医学工程重庆市重点实验室,重庆400044;2.重庆大学生物工程学院生物力学与组织工程教育部重点实验室,重庆400030;3.重庆人本药物研究院。重庆400040)【摘要】:建立盐酸帕洛诺司琼中有机残留溶剂的测定方法。毛细管气相色谱顶空进样法。色谱条件为:DM一624,mmlD,膜厚度为3.0Ixm的毛细管柱;柱温为80。C;顶空进样瓶的平衡温度为80。C,平衡时间为80℃下rain;进样1:3温度250"C,分流10:l;检测器温度250T;,载气:氮气。恒定压力,27cm/sec(80。C),H2流速:mL/min;空气流速:400mL/min。甲醇在40.4—202pg/mL,异丙醇在66.92—334.6斗g/mL,二氯甲烷在8.12—IXg/mL,乙酸乙酯在67.08—335.4雌r/mL,四氢呋喃在9.52—47.6pg/mL,甲苯在11.84—59.2斗g/mL,正己pg/mL范围内线性关系良好。甲醇、异丙醇、二氯甲烷、乙酸乙酯、四氢呋喃、甲苯及正己烷的检Ixg/mL。各溶剂的平均回收率为98.5%一102.1%。本方法操作简单,结果准确,是控制盐酸帕洛诺司琼中残留溶剂的可靠方法。【关键词】:盐酸帕洛诺司琼;毛细管气相色谱;顶空进样;有机溶剂残留量http://ng1.17img.cn/bbsfiles/images/2012/08/201208071047_382151_2352694_3.jpg







 我要推广仪器
我要推广仪器
 下载APP
下载APP