改性固体材料表面的磷酸根形态分析?拉曼可以吗?是碳铁改性材料,吸附了水里面的磷酸根磷酸都应该在表面的,和材料表面官能团形成了不同的物质,有可能以磷酸二氢根,磷酸氢根,磷酸根形式存在。各位大侠有什么高招吗?帮帮忙[img]http://simg.instrument.com.cn/bbs/images/default/em09512.gif[/img]
需要观察含水煤样的表面形态,但是不知道SEM 是否可以观察含水煤样。请求帮助,非常感谢!
1、当时培训时老师说超过微米级的表面用AFM就没什么意义了,为什么啊?(除了针容易断之外)2、在进行AFM测试前通常会问样品表面的平整程度如何(粗糙度),怎样用其它手段确定表面的平整度啊?比如压片的高分子材料。
不知道你们发现没有 如果样品表面贴有标签 上表面压在标签上 等于隔了一层绝缘层 但样品一样能激发;再如喷过漆的钢板,有机漆层的厚度在0.2MM左右,一样可以激发;大家讨论讨论为何绝缘层没能阻止电流通过,这个绝缘层的存在会对数据有多大影响?
金属样品的表面能如何测量?
处理重金属检测样品,样品粉碎前应去掉样品表面污物并用蒸馏水清洗,然后擦干表面多余水分。这句话是错的,错在哪里,正确的做法是什么?请赐教
对于小比表面积样品,如电池材料、有机材料、生物材料、金属粉体、磨料等空隙度微小的材料,由于吸附量微小,静态法测试的结果较含有风热助脱装置和检测器恒温装置的高精度动态法仪器误差大。对静态法为什么在小比表面样品测试方面精度难以保证,原因如下: 以比表面积1m2/g的样品为例,该样品0.5g对氮气的吸附量在BET分压范围内在标况下约0.1ml,在测试过程中的吸附环境液氮温度下的体积约0.03ml;样品管装样部分的剩余体积(也就是背景体积)约在3-5ml左右,要在3-5ml的样品管体积中准确定量出0.03ml的总吸附量且保证精度达到3%以内,可以算出要求压力传感器的精度要达到0.03%以上;但目前进口最好的压力传感器的精度只有0.1%,而且通常比表面及孔径分析仪用的压力传感器精度为0.15%,也就是说目前最高精度的压力传感器,即使温度场理想测定,液氮面理想恒定,环境温度理想准确条件下,对吸附量确定量的不确定度也只能达到0.003ml,即不确定度达到10%;若对于比表面再小或堆积密度小也就是装样量也难以很大的样品,其准确度就可想而知了。 但对于中大比表面样品,一般吸附量不会那么微小,静态法的精度很容易保证在2%甚至1%以内便不是问题; 所以在小比表面样品的测试方面,静态法只能通过增加装样量来降低误差,常见的是静态一般都会为小比表面积样品配备大容量样品管,但由于背景体积(吸附腔体积)也随之增大,所以准确度提高也是有限的;而有些厂家宣称静态法小比表面测试下限可以达到0.0001m2/g,是不负责任的; 对具有风热助脱、检测器恒温、低温冷阱的高精度动态法仪器,其相对不具有该装置的标准动态法比表面仪,其精度得到明显提高;动态法比表面仪,与其它分析仪器类似,其精度和灵敏度 大小主要取决于信噪比;也就是要提高精度和灵敏度,就需要从提高信号强度、抑制背景噪声、消除外界干扰三方面来控制。增加信号强度的方法一般有增加称样量、增加检测器电流,但增加 检测器电流一般噪声也会同时增大,所以检测器电流会有个最佳范围;所以在抑制噪声、消除外界干扰方面可做的工作就比较多了;其源于仪器自身的误差来源主要有:检测器温漂,信号锐度 ;以检测器恒温装置来抑制温漂,风热助脱装置可以提高信号锐度,其对于比表面1m2/g的样品0.5g对氮气的吸附量在分压0.2左右时脱附峰面积与背景可以保证在2%以内的误差; 所以对于小比表面样品,对具有风热助脱、检测器恒温、低温冷阱的动态法仪器,其灵敏度和分辨率的优势就体现出来了;但对中大比表面样品,由于信号强,普通动态法比表面积仪和静态 法比表面积仪都可以保证精度;这点就像万分之一分析天平和千分之一天平的区别; 但绝大多数含有微孔、介孔等空隙的材料,比表面不会很小;要是很小比表面的材料,其空隙度的研究价值就有限了; 综上, 一、对于小比表面样品(10m2/g以下)优先选择采具有风热助脱及检测器恒温装置的用动态色谱法比表面仪器,利用其分辨率、灵敏度高的优势; 二、对于中大比表面样品,若只测试比表面积,动态法和静态法没有明显的优劣势,动态法由于具有固体标样参比法,具有快速测定比表面的优势,静态法具有BET多点法较省时液氮消耗 小的优势; 三、需要测比表面及孔径分布的样品,建议采用静态容量法的比表面及孔径分析仪;
样品表面与周围衬底表面不一致时会引起峰的偏移,是整体迁移还是某些峰迁移?还是会多出某些峰?原因是什么?请假各位大侠?
本人最近在做EBSD。以前都是先做XRD,再处理样品,做EBSD,前一阵某次实验颠倒了下顺序。EBSD对样品的表面质量要求是非常高的,我用做完EBSD的样品再去做XRD,发现XRD的峰强有的甚至能高达100000,而之前一般处理的样品峰强顶多只有几千。做的虽然是取向多晶,但是有的整个图谱里面甚至只有一个峰,真的是只有一个峰,根本没有什么三强峰。难道横截面内取向这么强?自己都有点开始怀疑了。样品是三元金属间化合物,切成8*10*2的薄片,一般会砂纸磨到1500,做个XRD。之后再经过高目砂纸,金刚石抛光膏,硅乳胶抛光,再去做EBSD。样品表面质量对峰强,甚至相对峰强都有这么大的影响。。。感觉以后再有做取向多晶的XRD,样品表面真的需要好好处理啊!不知道大家有什么看法?
玻璃样品表面镀了一层膜,但不知道组成,只能确定是无机膜。EDS能对样品表面膜层进行全元素分析吗,找出含有什么元素吗?
我用的日本理学Ultima IV在制备xrd样品过程中1金属样品表面要达到什么程度能够得到准确谱图?比如粗糙度程度、是否电解都有什么要求?2粉末样品含量过少得到的结果准确么?3橡皮泥固定样品时为什么有时候出现橡皮泥峰,怎么避免?
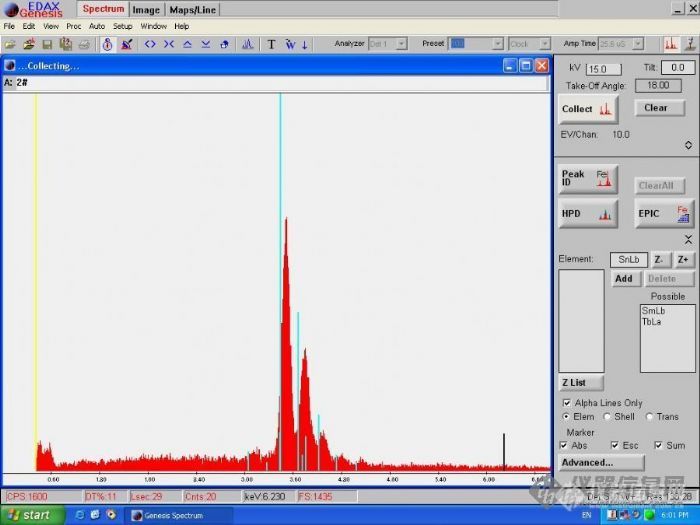
前几天在做无铅锡块的分析,取了2个样,第一个样品比较疏松,表面不是狠光滑;另外一个样品表面很光滑。分析的时候第一个样品多处获取的Sn峰都发生偏移,而第二个样品却比较吻合。请各位发表一下看法,是不是样品的形态的影响?还是别的?谢谢~~另外,对Sn的逃逸峰该如何处理?[img]http://ng1.17img.cn/bbsfiles/images/2005/11/200511291614_10978_1628456_3.jpg[/img][img]http://ng1.17img.cn/bbsfiles/images/2005/11/200511291615_10979_1628456_3.jpg[/img]
[size=4]表面状况对样品的结合能位置有哪些影响? 峰形呢?表面状况包括表面化学修饰或化学吸附、表面污染或覆盖有其他物种。比如 吸附有水 ,会有哪些影响? 等等[/size]
制作样品时能用水对样品表面进行清洗吗?
手持式光谱仪检测样品,必须要使样品的表面有一定光洁度,才能进行分析,手持式光谱仪对样品表面的处理要求到底有多高?
原因一:处理后比表面变小,可能是样品在处理过程中温度过高局部发生相变而损失了部分表面结构而造成;这点可能性最大,也最常见;原因二:有可能就是样品经过处理后化学结构发生变化,表面能降低,吸附能力减弱,也就是破坏了材料的化学性能;原因三:样品表面吸附杂质气体后相对不吸附时对氮气分子的吸附能力强,也就是杂质气体有助于吸附氮气分子;这点比较少见。
我用化学方法去除热镀铝板表面的镀层后,感觉表面还有一层薄膜,做金相没做出来,想做XRD, 但我担心膜太薄会被击穿影响结论。我最终的目的是想了解表面是否还有铝,以什么形态存在,含量有多少,我应该做哪些测试。
我这有个样品要测量他的表面粗糙度,这个样品比较特殊,是做特种合金的,这个样品又非常小 而且针头还带有螺纹,现要测它螺纹表面的粗糙度与螺纹槽的粗糙度,RA值要求在0.1-0.8mm。请问大家知道哪个地方有嘛?我已知讯好多地方都没有。都是符合不了我要求
请问: 1.样品表面是曲面可以进行扫描电镜观察吗? 2.样品表面经抛光后很光亮是否可以用SEM观察到清晰影像?衬度对比是否会明显?本人刚接触电镜,用词可能不太专业,如有不当之处,还请大侠们指正,谢谢
对于表面积只有几个平方米的样品,其吸附量很小,吸附引起的系统压力变化过小,因而测量的精确度变差。因此,气体吸附法测量低表面积固体不但需要选择在实验温度下饱和蒸汽压较小的吸附质,而且还要提高压力测量的灵敏度。在液氮温度下,氪和氙的饱和蒸汽压较低,现在常用来作测量低表面积固体的吸附质。
激发样品时,为什么激发后样品表面都没有变化呢,是什么原因导致的,
请问FIB工作时样品表面的温度最高达到多少?
我们是康塔的测试仪,球形样品管,装入我们的催化剂测试后,样品管容易产生静电,下次装入样品时,样品都被吸附在球的上表面,如何能消除,请高手帮助!!
对于已经制备好的金属材料透射样品,样品内的位错会在多久时间逸出样品表面?

大家好,最近做氧化锌的时候,加了点油酸做稳定剂(原来体系里有胺),结果在样品表面出现鱼纹状的条纹,请问那会是油酸或者副产物酯吗?谢谢了!http://ng1.17img.cn/bbsfiles/images/2011/10/201110110823_322840_1682649_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/10/201110110823_322841_1682649_3.jpg
原因一:处理后比表面变小,可能是样品在处理过程中温度过高局部发生相变而损失了部分表面结构而造成;这点可能性最大,也最常见;原因二:有可能就是样品经过处理后化学结构发生变化,表面能降低,吸附能力减弱,也就是破坏了材料的化学性能;原因三:样品表面吸附杂质气体后相对不吸附时对氮气分子的吸附能力强,也就是杂质气体有助于吸附氮气分子;这点比较少见。
激发光源是一小束光射入样品池中,并没有覆盖整个样品池表面,会对荧光强度有影响吗?
现在研究应力测试,想了解下,应力测试用盲孔法做测试是否需要对样品进行表面处理?
请问大家,BET法测定比表面时,仅仅把它们磨碎行不行?还需要把所有样品过同一目数的筛子吗?
样品进行比表面测试时间过长是什么原因







 我要推广仪器
我要推广仪器
 下载APP
下载APP