不合格 |NMPA发布20批不合格药品 大多在流通环节查出
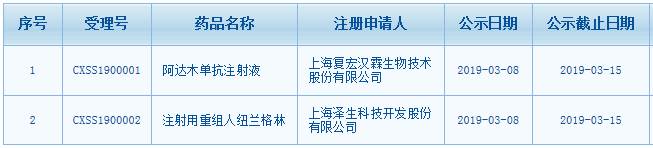
p style=" text-align: justify text-indent: 2em " 国家药监局网站今日发布了《关于20批次药品不符合规定的通告(2020年 第57号)》。通告中列出了20批次药物不合格的厂家、批次与不合格项目。 /p p style=" text-align: justify text-indent: 2em " 品种包括中成药、化药、中药饮片;剂型涵盖传统的中药大蜜丸(多厂家生产的石斛夜光丸)、颗粒剂、缓释片、注射剂等。其中不合格的项目主要有性状、鉴别、干燥失重、澄清度、装量差异等。主要的查处环节在 span style=" color: rgb(255, 0, 0) " strong 经营 /strong /span ,有3个批次来自 span style=" color: rgb(255, 0, 0) " strong 生产 /strong /span 。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 278px " src=" https://img1.17img.cn/17img/images/202009/uepic/72662e46-f0be-4ab8-9fb7-df489282847d.jpg" title=" 1.png" alt=" 1.png" width=" 600" vspace=" 0" height=" 278" border=" 0" / /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 249px " src=" https://img1.17img.cn/17img/images/202009/uepic/34cd4a2d-7bea-4cb6-a969-d000874de4bd.jpg" title=" 2.png" alt=" 2.png" width=" 600" vspace=" 0" height=" 249" border=" 0" / /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 274px " src=" https://img1.17img.cn/17img/images/202009/uepic/6bbd3ea7-7644-4de4-a270-9666948c17dc.jpg" title=" 3.png" alt=" 3.png" width=" 600" vspace=" 0" height=" 274" border=" 0" / /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 327px " src=" https://img1.17img.cn/17img/images/202009/uepic/a4e00453-6180-42b1-8b03-1a943e5856c9.jpg" title=" 4.png" alt=" 4.png" width=" 600" vspace=" 0" height=" 327" border=" 0" / /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 273px " src=" https://img1.17img.cn/17img/images/202009/uepic/604eec9d-2c21-40e8-b030-dec514896b0c.jpg" title=" 5.png" alt=" 5.png" width=" 600" vspace=" 0" height=" 273" border=" 0" / /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 600px height: 139px " src=" https://img1.17img.cn/17img/images/202009/uepic/cd169a59-dee7-403e-9b74-6f6ef68a03d3.jpg" title=" 6.png" alt=" 6.png" width=" 600" vspace=" 0" height=" 139" border=" 0" / /p p style=" text-align: justify text-indent: 2em " strong 原文如下: /strong br/ /p p style=" text-align: justify text-indent: 2em margin-top: 10px " 经广西壮族自治区食品药品检验所等7家药品检验机构检验,标示为吉林省通化博祥药业股份有限公司等10家药品生产企业生产的20批次药品不符合规定。现将相关情况通告如下: /p p style=" text-align: justify text-indent: 2em margin-top: 15px " 一、经广西壮族自治区食品药品检验所检验,标示为吉林省通化博祥药业股份有限公司、吉林市双士药业有限公司、吉林省天泰药业股份有限公生产的12批次 span style=" color: rgb(0, 112, 192) " strong 石斛夜光丸 /strong /span 不符合规定,不符合规定项目为 span style=" color: rgb(255, 0, 0) " strong 鉴别 /strong /span 。 br/ 经山西省食品药品检验所检验,标示为海南普利制药股份有限公司生产的1批次 span style=" color: rgb(0, 112, 192) " strong 克拉霉素缓释片 /strong /span 不符合规定,不符合规定项目为 span style=" color: rgb(255, 0, 0) " strong 干燥失重 /strong /span 。 br/ 经福建省食品药品质量检验研究院检验,标示为山西仟源医药集团股份有限公司生产的1批次 span style=" color: rgb(0, 112, 192) " strong 注射用阿洛西林钠 /strong /span 不符合规定,不符合规定项目为溶液的 span style=" color: rgb(255, 0, 0) " strong 澄清度 /strong /span 。 br/ 经内蒙古自治区药品检验研究院检验,标示为河北国金药业有限责任公司、广州花城药业有限公司生产的2批次 span style=" color: rgb(0, 112, 192) " strong 复方金银花颗粒 /strong /span 不符合规定,不符合规定项目为 span style=" color: rgb(255, 0, 0) " strong 装量差异 /strong /span 。 br/ 经深圳市药品检验研究院和中国食品药品检定研究院分别检验,标示为安徽省泽华国药饮片有限公司生产的1批次 span style=" color: rgb(0, 112, 192) " strong 红景天 /strong /span 和1批次 span style=" color: rgb(0, 112, 192) " strong 制川乌 /strong /span 不符合规定,不符合规定项目均为 span style=" color: rgb(255, 0, 0) " strong 性状 /strong /span 。 br/ 经大连市药品检验检测院检验,标示为安国市彤康药业有限公司生产的1批次 span style=" color: rgb(0, 112, 192) " strong 前胡 /strong /span 不符合规定,不符合规定项目为 span style=" color: rgb(255, 0, 0) " strong 性状、鉴别 /strong /span 。 br/ 经山西省食品药品检验所检验,标示为湖南省自然堂中药饮片有限公司生产的1批次 span style=" color: rgb(0, 112, 192) " strong 秦艽 /strong /span 不符合规定,不符合规定项目为 span style=" color: rgb(255, 0, 0) " strong 性状 /strong /span 。 /p p style=" text-align: justify text-indent: 2em " 二、对上述不符合规定药品,药品监督管理部门已要求相关企业和单位采取 span style=" color: rgb(255, 0, 0) " strong 暂停销售使用、召回 /strong /span 等风险控制措施,对不符合规定原因开展调查并切实进行整改。 /p p style=" text-align: justify text-indent: 2em " 三、国家药品监督管理局要求相关省级药品监督管理部门依据《中华人民共和国药品管理法》,组织对上述企业和单位生产销售假劣药品的违法行为立案调查,并按规定公开查处结果。 /p p style=" text-align: justify text-indent: 2em " 特此通告 /p p style=" text-align: right text-indent: 2em " 国家药监局 /p p style=" text-align: right text-indent: 2em " 2020年9月2日 /p p style=" text-align: justify text-indent: 2em margin-top: 10px " 药品质量安全是保证药效的重要因素之一,只有合格的药品才能在医师和药师的正确指导下保证药物达到最佳疗效。2020年版《中国药典》即将实施,很多更加严格的标准要适用。对于药品的质量给予了更大的保障;与此同时,对于药品生产企业和检测机构来说,是对于生产环节和检验环节的更大挑战。 /p p style=" text-align: center" img style=" max-width: 100% max-height: 100% width: 660px height: 50px " src=" https://img1.17img.cn/17img/images/202009/noimg/4601c91a-0c71-4fb2-badf-4ae3a07324ff.gif" title=" 七夕情人节爱心动态分割线分隔符.gif" alt=" 七夕情人节爱心动态分割线分隔符.gif" width=" 660" vspace=" 0" height=" 50" border=" 0" / /p p style=" text-align:center" a href=" http://instument1999.mikecrm.com/lGWNMkR" target=" _blank" img style=" max-width: 100% max-height: 100% width: 550px height: 179px " src=" https://img1.17img.cn/ui/bimg/SH100000/special/w920h3002020ChP.jpg" title=" " alt=" " width=" 550" vspace=" 0" height=" 179" border=" 0" / /a /p p style=" text-align: justify text-indent: 2em margin-top: 10px " 仪器信息网将特别推出“ span style=" color: rgb(255, 0, 0) " strong 2020年版《中国药典》变化盘点 /strong /span ”专题,盘点通则增修、药典仪器以及相关资讯。敬请广大读者关注! span style=" color: rgb(0, 112, 192) " strong 【点击图片进入专题】 /strong /span /p p style=" text-align: center " a href=" https://www.instrument.com.cn/zt/2020ChP-changes" target=" _self" img style=" max-width: 100% max-height: 100% width: 550px height: 94px " src=" https://img1.17img.cn/17img/images/202009/uepic/1a99183e-131f-46da-b578-e18bff0eb239.jpg" title=" w640h1102020ChP.jpg" alt=" w640h1102020ChP.jpg" width=" 550" vspace=" 0" height=" 94" border=" 0" / /a /p






















 我要推广仪器
我要推广仪器
 下载APP
下载APP