
http://ng1.17img.cn/bbsfiles/images/2015/09/201509211106_566940_2410902_3.jpg做二硫化碳中的苯系物 二硫化碳拖尾严重 苯峰在拖尾峰上 求教高手该怎么办 程序升温 进样垫新换的 柱子重新戒掉进口段重新安装 结果还是那样
做苯系物二硫化碳峰和苯峰分不开,不知道什么原因,色谱柱用的FFAP柱炉120℃氢焰250℃毛细柱230℃。(为了看清所以是自己配的一个较大浓度的样,0.962的那个是苯)
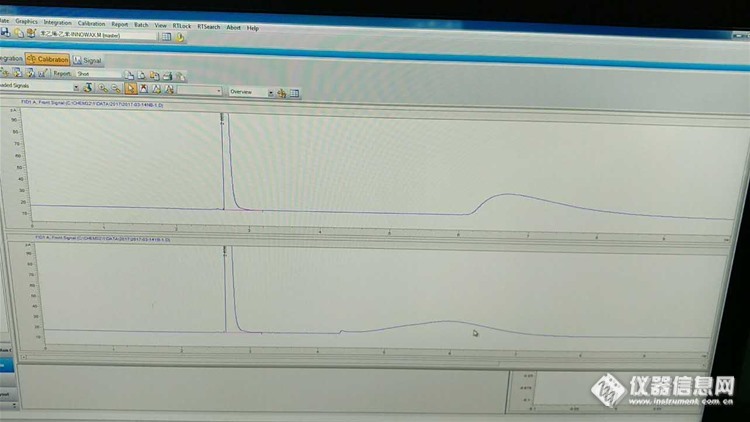
内标发测二硫化碳中苯乙烯,乙苯,正十二烷是内标,先进单标确定出峰时间,溶剂出峰还正常,可目标峰很宽,不知道是为什么,打给客服,建议重新安装一下柱子,安完还是一样。HP-INNOWAX柱,峰型如图,请问各位老师是为什么啊。http://ng1.17img.cn/bbsfiles/images/2017/03/201703141629_01_3052775_3.jpg
做环境空气中苯系物的时候,用二硫化碳解吸,苯的峰分成两半了,有时候不分,有时候分,标液(二硫化碳中苯系物)走出来也是这样,有没有大佬分析下是什么情况[img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108100926585253_264_5343586_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2021/08/202108100926588522_8346_5343586_3.png[/img]
在样品中可能含有间苯二胺和间苯二胺硫酸盐,在相同的流动相下请问间苯二胺硫酸盐与间苯二胺在同一个位置出峰吗?
这几天用岛津的碳18分析氨基酸,用的异硫氰酸苯酯为衍生剂。 用水代替氨基酸标准品,会出现很多杂峰,所以想应该是衍生过程引进的。衍生的方法是 标准品+三乙胺乙腈溶液+异硫氰酸苯酯乙腈溶液 反应一个小时候用正己烷萃取各位大虾有更好的衍生方法么?本实验室没有氮吹仪,没法吹干哦
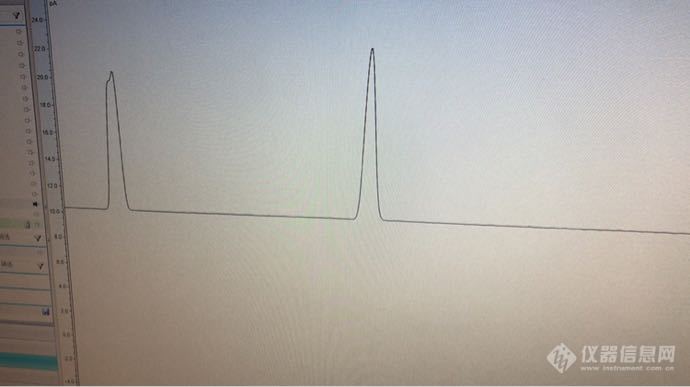
二硫化碳中苯系物中苯 分叉了。其余峰型都很尖锐 这个什么问题 前面一个是苯 第二个图前面2个是苯[img=,690,386]https://ng1.17img.cn/bbsfiles/images/2020/01/202001021344174008_6068_2990176_3.png[/img][img=,690,386]https://ng1.17img.cn/bbsfiles/images/2020/01/202001021344268683_9451_2990176_3.png[/img]
柱流量1.0ml/min。程序升温,50℃5min,5℃/min-90℃2min,其他六个峰都有出峰,峰型很好,就是没有苯峰。柱流量、柱温、分流比都改变过,就是不出苯峰,标准溶液也换过
仪器条件:进样口250℃柱温箱100℃(恒温)ECD:350℃标准品:异辛烷中六氯苯(坛墨)问题:不会分分辨溶剂峰和六氯苯的峰(出了四个峰)[img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212071657430429_8466_3515148_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/12/202212071657429145_3057_3515148_3.png[/img]
用安捷伦做二硫化碳中苯系物,10ug/ml的苯系物的峰面积有十几,而thermo的仪器的峰面积就有3万多,看看大家的信号值都是什么样的?
18883-2022中附录C.2中二硫化碳解析苯,曲线所有浓度点都没有问题,采样进行热脱附有峰,但是二硫化碳解析就不出苯峰,二硫化碳解析后溶剂峰没问题,但是苯系物都没有?
版友问题,做乙醇甲苯残留溶剂顶空进样不出峰,做其他的直接进样正常,会有什么原因呢?
NPD非极性柱,请问偶氮苯、马拉硫磷、异辛烷的出峰顺序,谢谢
请问elite-5分离测定二硫化碳中的苯系物时是苯乙烯先出峰还是邻二甲苯??两个峰挨在一起,我们没有单标,没法进行确认。

第一峰是溶剂峰,然后峰后面尾巴的一点点是苯。剩下六苯系物峰都很好。我柱稳是60摄氏度恒温,流量1ml/min,分流比20:1[img=,690,516]http://ng1.17img.cn/bbsfiles/images/2017/09/201709211747_01_2987409_3.png[/img]
请教一个问题,我在GCMS顶空分析样品是否残留苯时,SIM模式下找到78的位置,在全扫描下,该位置正好和水峰重叠,从面积看,水含量很高,怀疑可能覆盖了苯的信号,请问有没有做过类似分析的老师,有什么办法能消除水峰的影响,谢谢
GCMS计量中,用的10ng/微升的六氯苯,峰面积重复性30%多,峰形很好,没有拖尾,但是部分峰面积呈2倍关系,我们是不分流进样,进样口温度250℃,升温程序:60保持0min,以10℃/min升至250℃,保持5min。初步排除了进样口压力、进样针、方法、隔垫,衬管、微流平板、方法。排查过程中每次进样基本部分峰面积呈2倍关系。请问一下各位知道大概是什么情况吗?
气质6890-5973联用仪,走样时甲苯峰很高,换过衬管后,依然很高,不知为啥。而且,用二硫化碳溶剂进样时,会出现断链烷烃如庚烷等。进溶剂空样时,也是这样。在300度时,会出现一系列特征离子峰为94的峰,走溶剂空样时,未发现,但感觉不像是自己样里的东西。不知大家是否见过此类情况,谢谢!
柱温恒温65,进样150,检测器250。用福利的坐584苯系物的时候,二氧化硫拖尾严重。怎么办?什么影响峰拖尾?求大神帮忙,谢谢![img]https://ng1.17img.cn/bbsfiles/images/2019/01/201901211117430577_1656_3527580_3.png[/img]
各位大佬,有没有用安捷伦7890b,柱子是DB-FFAP的柱子做二硫化碳中苯系物的。按照国标走50ug/ml的那个标准曲线点,峰面积才50左右,0.5低浓度点都不出峰,这样我做方法验证检出限都没法做。进样口我也维护了。为啥仪器灵敏度这么低,还有什么方法可以改进么[img=,690,388]https://ng1.17img.cn/bbsfiles/images/2021/11/202111041801574668_4075_5372015_3.png[/img]
检验氟化苯残留时,使用艾科和阿拉丁家的氟化苯出峰时间都在6分钟左右,而最近因更换厂家导致该厂家氟化苯在同一仪器、色谱柱、方法下出分时间在9分钟,感觉与仪器无关是试剂问题,有对氟化苯了解的大佬,这是什么情况吗?这三家的氟化苯分子式,cas都是同一个
如题,gcms要做校准,今天按照检定规程来设置参数,3个样品出来的谱图都是一个斜坡状的,各提取特征离子,均没找到,柱子是强极性的,极限温度是280,30m长。接着降低进样口温度为200,传输线250,降低柱箱初温,出来的谱图都是几个 ’几‘字型,各提取特征离子,八氟萘和硬脂酸甲酯没找到峰,六氯苯找到几个连在一起的不规则的峰,但最高丰度的特征离子碎片是88,不是284.无论我如何设置升温程序,任何时间点的最高离子碎片的质荷比都是88,当然溶剂峰除外。是不是我的柱子有问题?
聚苯硫醚(PPS)纤维与其它纤维混纺产品定量分析方法研究Study on the Quantitative Analysis Method of Polyphenylene Sulfide ( PPS) Fiber and Other Blended Fiber文/戴颖摘要:本文确定了聚苯硫醚纤维在不同溶剂和不同试验条件下的质量修正系数,探讨了聚苯硫醚纤维与其它纤维混纺产品的定量化学分析方法。关键词:聚苯硫醚纤维;纤维含量;定量;溶解;修正系数1 引言聚苯硫醚,全称为聚亚苯基硫醚,英文名称为Polyphenylene sulfide,简称PPS。聚苯硫醚纤维分子结构比较简单,分子主链由苯环和硫原子交替排列,大量的苯环赋予聚苯硫醚纤维以刚性,大量的硫醚键又为其提供了柔顺性,使纤维的分子结构对称,易于结晶,无极性,电性能好,不吸水。因此,聚苯硫醚纤维是一种高性能特种纤维,具有优异的耐化学性和热稳定性以及抵抗恶劣环境、阻燃、绝缘、防辐射等功能,在高温、化学腐蚀环境等领域正得到广泛应用。伴随着聚苯硫醚纤维应用的增加,势必会产生相应的检测检验需求。本文结合目前广泛使用的纤维定量方法,得出PPS纤维与其他纤维混纺时,定量检验所采用的方法,并得出各种试剂对其他纤维产生的修正系数,为PPS纤维的定量检验提供了依据。 2 试验2.1 试验试剂乙醚、苯酚/四氯乙烷、75%硫酸、80%硫酸、1 mol/L碱性次氯酸钠、丙酮、80%甲酸、二氯甲烷、N,N-二甲基甲酰胺(DMF)、99%冰乙酸、20%盐酸等(试剂除标注浓度或含量的外,其它均为分析纯)。2.2 试样聚苯硫醚纤维(规格:20 tex),纯纤标准贴衬布(包括羊毛、棉、聚酯纤维、锦纶、腈纶、醋纤),氨纶。2.3 仪器索氏萃取器、恒温水浴锅、分析天平(精度为0.0001g)、具塞三角烧瓶等玻璃器皿。2.4 试样的预处理 取试样5 g左右,放在索氏萃取器中,用乙醚萃取1 h,每小时至少循环6次,待试样中的乙醚挥发后,把试样浸入冷水中,浸泡60 min,再在(65士5)℃的水中浸泡1 h,水与试样体积之比为100 : 1,不断搅拌溶液,然后抽吸或离心脱水、晾干。 2.5 质量修正系数的确定聚苯硫醚纤维与其它纤维混纺产品的定量分析方法参照GB/T 2910-2009《纺织品 定量化学分析》、FZ/T 01095-2002《纺织品氨纶产品纤维含量的测试方法》所规定的试验条件,每种试验条件下进行10个样品的重复性试验,求出质量修正系数的平均值。质量修正系数试验结果见表1。表1聚苯硫醚纤维的质量修正系数序号试剂试验条件质量修正系数d值温度/℃时间/min1苯酚/四氯乙烷40±5101.00275%硫酸50±5601.00380%硫酸25±5151.0041mol/L碱性次氯酸钠20401.005丙酮25±530+15+151.00680%甲酸25±5151.007二氯甲烷25±5301.008二甲基甲酰胺(DMF)90~9560+301.009冰乙酸25±520+20+201.001020%盐酸25±5151.0011
PE-GCMS外校时测六氯苯(10ng/ul)用开始温度150度,10度每分钟升到250度,不分流测样,出峰情况跟溶剂一样,5分钟后就是一条直线没峰出来,什么情况?
各位老师帮我分析下,我做22338国标,甲砜霉素 氯霉素 氟苯尼考时,用的是乙腈+0.1%甲酸水单独走每个物质都有出峰,唯独甲砜霉素响应值低,其他两个峰不错,但当我把他们方法合在一起,就不醒了,我看了下合在一起只是驻留时间变小了,其他无变化,驻留时间能影响这么大吗?我觉得不应该啊,还是因为他们的母离子太接近了,而且出峰时间有差不多,在扫描时发生混乱了???求助
请问分析甲醇中的苯系物要用什么做固定相的色谱填充柱?我们公司先买了一根适合分析二硫化碳中的苯系物,一作甲醇中的苯系物就只有溶剂峰。
对苯二胺硫酸盐、间苯二胺硫酸盐、邻苯二胺硫酸盐,用液相色谱C18柱能用吗?应配怎样的流动相? 它们的稀水溶液PH值在2-3左右。我用硫酸调流动相PH到2左右出两个峰。 又用三乙胺调PH到7多点还是两个

发现做三苯时候苯峰被干扰的问题,特发一帖,与各位分享。昨天在做三苯样品时候发现空白二硫化碳中含有少量苯,如下图一:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091717_531833_2780210_3.png一开始以为是二硫化碳被污染,本人遂再开一瓶,进样后发现仍然有苯峰,如下图二:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531834_2780210_3.png为再次确定结果,正好手头GCMS空闲,上GCMS上走Scan以及SIM后发现没有苯的定量离子,所以我得出的结论为,二硫化碳未被污染,而是GC问题。立即动手,更换耗材,老化柱子,老化检测器。老化过程中发现基线有一些杂峰出来,让我更加坚信是GC端的问题。当我开开心心的再次走三苯样品的时候,悲剧出现了,苯峰没有被完全消除!如下图三:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531835_2780210_3.png今天接了一个要分将对/间二甲苯分开的单,所以拿出好久没用的DB-WAX的柱子,在另外一台GC上老化后直接上曲线走样,在做空白的时候惊讶的发现万恶的苯峰又来了,我判断问题仍然处在二硫化碳上。可是同一瓶二硫化碳走过MS,MS的灵敏度总不会低于FID吧?在思考的时候,样品走到1ppm浓度的三苯标液上,我发现苯峰分叉了!如下图四,图五:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531836_2780210_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531837_2780210_3.jpg证明空白中的那个被当做苯的杂质并不是苯,是一种跟苯有着相同保留时间的另一种物质!突然想起又一次其他实验室的过我们实验室来交流的时候有说过,正己烷有可能干扰到苯的测定。回想一下,早上更换装WAX柱子GC的洗液时正好看洗液是做有机氯农药时候用的正己烷!立即拿着刚测定有苯的空白进GCMS走了一个Scan,如下图六:http://ng1.17img.cn/bbsfiles/images/2015/01/201501091718_531838_2780210_3.png正己烷 57 43 的离子果然在!终于发现问题所在了!各位,要小心正己烷冒充苯啊!刚刚很多老师建议说极性柱子正己烷出峰应该在前面,这的确是我没想到的地方,找个空闲时间进一针正己烷试试,谢谢各位老师提醒!刚刚我用相同的条件进了一针正己烷,出峰时间在二硫化碳后面一点!看来还是我考虑得不周到!

今天在做检测时,看到标准有一个试剂:新蒸馏的苯酚。不知要如何蒸馏?蒸馏的苯酚与未蒸馏的苯酚有什么区别?感谢“四季风”版友提供的图片及资料:把有结晶的苯酚放置100度C的水浴中溶解,然后倒入蒸馏烧瓶中,放入玻璃珠数粒,瓶口放一支带胶塞200度C的水银温度计,塞紧瓶口(见以下草图)。接好冷凝管,放一接收瓶,在电炉上先加热蒸馏至沸点182度C后约2---3分钟左右,拿去第一个接收瓶,另放一接收瓶接收沸点182度C的苯酚蒸馏液(冷却后即为纯苯酚结晶)。http://ng1.17img.cn/bbsfiles/images/2011/09/201109282355_320046_1641058_3.jpg
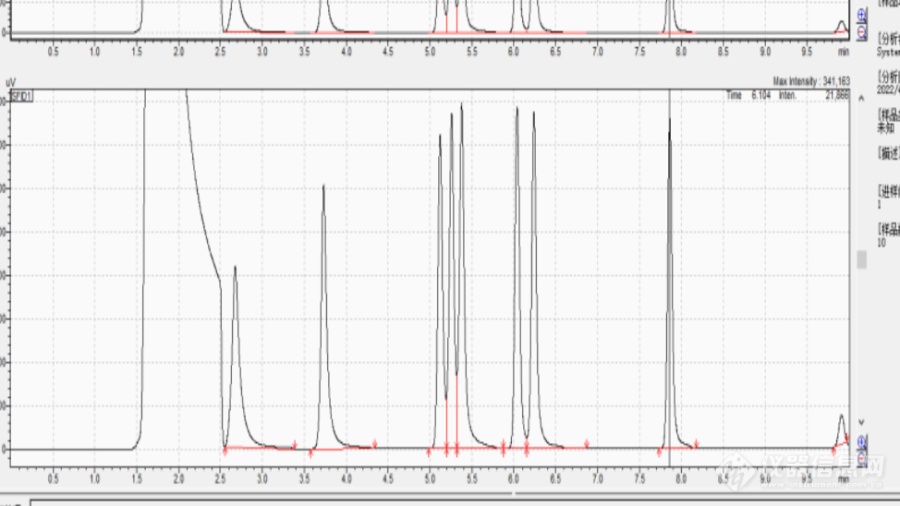
1.昨天在做HJ584-2010环境空气苯系物的测定时,当时CS2溶剂峰跟苯的峰靠的很近,溶剂峰有淹没苯峰的趋势。2.当时分析的仪器条件是:岛津GC-2014C,色谱柱是DB-WAX 30*0.25*0.25,进样口设置的是150℃,检测器220℃,升温程序为,50℃保持5min以5℃/min速率升至80℃保持2min,色谱柱流量1.5mL/min,进样量1.0μL,二硫化碳用的是天津科密欧的无苯级色谱纯二硫化碳。3.以下谱图是走的50μg/mL的8种苯系物的混标。[img=,690,387]https://ng1.17img.cn/bbsfiles/images/2022/04/202204221912557354_6419_3764668_3.png!w690x387.jpg[/img]







 我要推广仪器
我要推广仪器
 下载APP
下载APP