专家解读|GB 2763.1-2022中农药残留限量配套检测方法修订情况
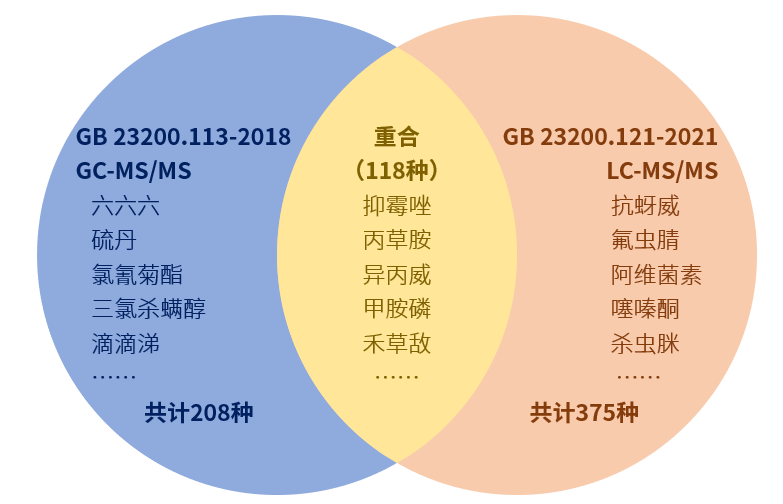
《食品安全国家标准 食品中农药最大残留限量》 (GB 2763) 是目前我国统一规定食品中农药最大残留限量 (MRLs) 的强制性国家标准。2022 年 11 月 11 日, 国家卫生健康委员会、农业农村部和国家市场监督管理总局联合发布《食品安全国家标准食品中 2, 4-滴丁酸钠盐等112 种农药最大残留限量》 (GB 2763. 1-2022) 标准 (以下简称增补版), 自 2023 年 5 月 11 日起正式实施。GB 2763. 1-2022是GB 2763-2021的 增补版,可以配套使用。为进一步强化农药残留限量标准宣贯,促进食品质量安全监管、检测人员和食品生产者及时全面了解国家最新农药残留限量标准。仪器信息网邀请到了农业农村部农药检定所罗媛媛老师,对GB 2763. 1-2022中农药残留限量配套检测方法进展进行了详细分析, 以便于标准使用者更好的理解和正确使用。一、农药残留限量配套检测方法 增补版标准共规定了 37 项配套检测方法标准, 其中, 根据农药残留标准制修订情况, 增补版标准相较于 GB 2763-2021 新增 GB 23200. 121 等4个检测方法 (表3)。 此外, 增补版标准中 32 种农药 62 项限量由于缺少配套的检测方法标准, 暂制定为临时限量。 对于阿维菌素等 60 种农药, 增补版标准与GB 2763-2021 对同一农药和食品类别推荐的配套检测方法存在一定差异, 但两个文本规定的检测方法均适用于相应参数的检测 (表 4)。 二、配套检测方法标准修订情况 基于检测方法适用性原则, 经第二届国家农药残留标准审评委员会第十二次全体会议审议, 对 GB 2763- 2021 中苯醚甲环唑等 3 种农药在相应食品类别上个别不适用的检测方法进行了修订 ,增补版标准中不再配套使用, 在整合新版 GB 2763 时相应修订文本。 目前, 检测机构在测定相关食品中农药残留时, 应按照标准中规范性引用文件的要求, 在配套检测方法中选择满足检测要求的方法进行检测。 三、配套检测方法变化相应调整限量类型由于增补版标准的发布, 新增推荐了检测方法标准, GB 2763-2021 中百菌清、 苯并烯氟菌唑、吡噻菌胺、 单氰胺、 氟啶虫胺腈、 氟噻草胺、氟吗啉、 氟唑菌酰胺、 精喹禾灵、 螺虫乙酯、氯虫苯甲酰胺、 亚胺唑、 依维菌素等 13 种农药在药用植物等 5 种食品类别上的 208 项限量由临时限量调整为正式限量。由于方法适用性问题, 删除了三氯吡氧乙酸在谷物上GB / T 20769 检测方法, GB 2763 - 2021 中此农药的稻谷和糙米两项限量由正式限量调整为临时限量。四、增补版标准的主要特点 1、优化了限量标准的配套性和可操作性增补版标准与 GB 2763-2021 相比, 规范性引用文件中新增了 4 种检测方法标准。 同时, 对阿维菌素等60 种农药的 7 种食品种类, 新增了部分配套检测方法, 为不同检测能力的机构采用适宜的方法提供了选择空间。 由于新增推荐检测方法, 百菌清等 13 种农药的 208 项限量由临时 限量调整为正式限量, 提高了标准的实用性。 2、跟踪评估并修订了韭菜中腐霉利残留限量近年来, 韭菜中腐霉利残留超标问题备受关注,为此, 农业农村部组织开展了韭菜中腐霉利最大残留限量的专项跟踪评价。 根据农药登记产品标签和生产用药实地调研情况, 开展了腐霉 利在韭菜上的残留验证试验, 基于膳食风险评估结果, 并综合考虑韭菜例行监测数据和日韩等周边国家残留限量等因素, 同步研究并对腐霉利在韭菜中最大残留限量和相关已登记农药标签中最多使用次数、 安全间隔期等合理使用技术提出了修改建议, 分别提交国家农药残留标准审评委员会、全国农药登记评审委员会执行委员会议审议通过, 将腐霉利在韭菜上的最大残留限量值由0. 2 mg / kg 修订为5 mg / kg, 并结合韭菜用药实际同步变更了腐霉利在韭菜上的农药产品标签, 既从源头上指导农民科学合 理用药, 又能有效保障韭菜的食用安全。 作者简介:罗媛媛 农业农村部农药检定所残留审评处、国家农药残留标准审评委员会秘书处农艺师,主要从事农药登记管理、农药残留风险管理和农药合理使用准则制定等工作。主要负责组织农药最大残留限量标准及农药检测方法国家标准的立项、起草、征求意见、送审、报批等工作。先后参与起草2019版、2021版和2022版《食品安全国家标准 食品中农药最大残留限量》(GB 2763),参与起草《农作物中农药残留试验准则》《畜禽中农药残留试验准则》《畜禽中农药代谢残留试验准则》等多项残留试验准则。






























 我要推广仪器
我要推广仪器
 下载APP
下载APP