2017上半年新增环境标准、规范汇总!
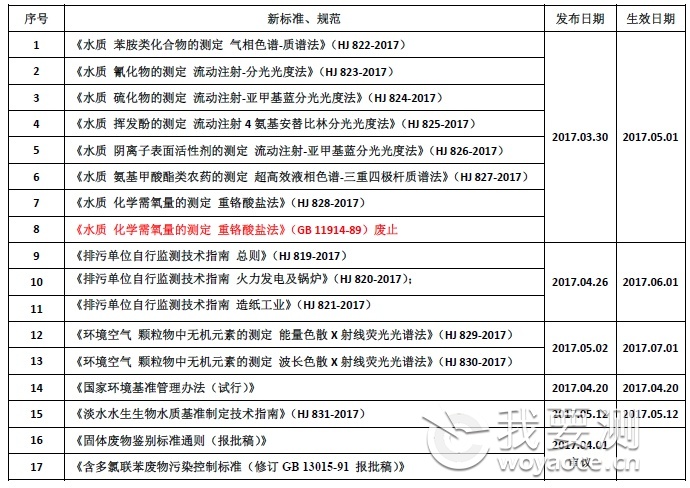
p style=" text-align: justify line-height: 1.5em margin-bottom: 10px margin-top: 5px " span style=" color: rgb(0, 0, 0) " 我国目前已形成 span style=" color: rgb(0, 112, 192) " strong 两级五类 /strong /span 的环保标准体系,分别为国家级和地方级标准,类别包括环境质量标准、污染物排放(控制)标准、环境监测类标准、环境管理规范类标准和环境基础类标准。 /span /p p style=" text-align: justify line-height: 1.5em margin-bottom: 10px margin-top: 5px " span style=" line-height: 1.5em " span style=" line-height: 24px text-align: justify " /span 截至“十二五”末期,累计发布国家环保标准1941项(其中“十二五”期间发布493 项),废止标准244 项,现行标准1697 项。在现行环保标准中,环境质量标准16 项,污染物排放(控制)标准161 项,环境监测类标准1001 项,管理规范类标准481 项,环境基础类标准38 项。 /span /p p style=" text-align: justify line-height: 1.5em margin-bottom: 10px margin-top: 5px " 《国家环境保护标准“十三五”发展规划》指出,未来将发布约800 项环保标准,包括质量标准和污染物排放(控制)标准约100 项,环境监测类标准约400 项,环境基础类标准和管理规范类标准约300 项,支持环境管理重点工作。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " span style=" color: rgb(0, 112, 192) " strong 2017上半年环境保护部发布新标准、规范汇总如下: /strong /span /p p style=" text-align: center margin-bottom: 10px line-height: 1.5em " img src=" http://img1.17img.cn/17img/images/201706/wycimg/56815293-17f0-4510-b30d-a08b45f7df3d.jpg" title=" 微信截图_20170615180242.png" width=" 600" height=" 423" border=" 0" hspace=" 0" vspace=" 0" style=" width: 600px height: 423px " / /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " span style=" color: rgb(0, 112, 192) " strong 环境监测类标准 /strong /span /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " 根据《大气十条》《水十条》《土十条》环境管理需求和监测技术进展,“十三五”期间将着力构建支撑质量标准、排放标准实施的环境监测类标准体系,贯彻《生态环境监测网络建设方案》(国办发〔2015〕56 号)中建立统一的环境质量监测网络的要求。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " “十三五”期间,水、大气、土壤环境监测方面将着重修订重金属、农药、持久性有机污染物、挥发性有机物、恶臭污染物等环境监测分析方法标准;完善地表水与污水、空气与废气、土壤环境监测技术规范体系,开展生态环境、生物、环境振动、环境噪声等监测技术规范制修订;加强主要污染物及重金属、挥发性有机物的监测监控,制修订环境自动监测系统技术要求;重点加强挥发性有机物、多氯联苯、多溴联苯醚、农药类、重金属类等环境监测和科研急需的标准样品研制,健全环境标准样品体系。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " span style=" color: rgb(0, 112, 192) " strong VOCs排放控制标准 /strong /span /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " 近年来,VOCs作为新型大气污染物逐渐进入公众视野。2013年以来,国家陆续发布了《挥发性有机物污染防治技术政策》、《重点行业挥发性有机物削减行动规划》、《挥发性有机物排污收费试点办法》等多项政策措施。此外,“十三五”规划纲要首次将VOCs新纳入总量控制指标,提出在重点区域、重点行业推进VOCs排放总量控制,全国排放总量下降10%以上。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " 截至2016年底,已有15个省市开展VOCs排放收费,北京、上海、安徽、江苏、河北、湖南、四川、辽宁、天津、浙江等已建立明确VOCs排污收费标准。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " 4月12日,为推动京津冀区域大气环境质量改善,北京、天津、河北三地共同制定了《建筑类涂料与胶粘剂挥发性有机化合物含量限值标准》,并将于9月1日起同步实施。这是京津冀三地在环保领域发布的首个统一标准。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " 国家质量监督检验检疫总局、国家标准化管理委员会于2016年12月30日批准了《儿童房装饰用水性木器涂料》国家标准,明确将于今年7月1日开始实施。儿童漆标准严格限定了VOC、游离甲醛、苯、甲苯、乙苯、二甲苯总量等有害物质的限定值。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " 北京、江苏、河北、河南、陕西等多省发布挥发性有机物排放控制标准。《陕西省挥发性有机物排放控制标准》(DB61/T1061-2017)自2018年2月10日起执行,江苏省《化学工业挥发性有机物排放标准》(DB32/3151-2016)、《表面涂装(家具制造业)挥发性有机物排放标准》(DB32/3152-2016) 自2017年2月1日起实施。 /p p style=" margin-bottom: 10px text-align: justify line-height: 1.5em " a href=" http://www.woyaoce.cn/news/212824.html" target=" _self" title=" " style=" color: rgb(255, 0, 0) text-decoration: underline " span style=" color: rgb(255, 0, 0) " 当前全国多地各行业VOCs排放最新标准一览 /span /a br/ /p p style=" margin-bottom: 10px text-align: right line-height: 1.5em " 编辑:我要测网 徐娜娜 /p


























 我要推广仪器
我要推广仪器
 下载APP
下载APP