了解不同时间药物在血浆或血清中的浓度,对于计算一种药物的代谢动力学很有必要;反之,药物动力学也是药物吸收、分布、代谢和排泄过程的一部分。准确了解药物在体内吸收、分布、代谢和排泄的规律,便于精确地计算所需药物剂量,既能保持有效的药物浓度,同时避免用药过量致毒。预先对多屏深孔Solvinert(MultiScreen Deep Well Solvinert )和多屏Solvinert滤板进行了验证,进行血浆或血清中蛋白质的板内沉淀,以便展开总药物分析。在滤板上可以快速、细致并完整地转移滤液,这样就可以在进行总药物分析之前为样品制备提供一个自动化兼容的平台。Solvinert滤板过滤的滤液中不含蛋白质,这与质谱分析法和紫外线分析法的结果一致。使用多屏深孔和多屏Solvinert滤板可产生有复验性的结果,它是一个稳定且可靠的平台。血清中的蛋白质被这些滤板过滤并沉淀之后,得到的样本中基本上不含蛋白质,回收率很高,便于萃取。药物动力学特性可以让新药开发商更了解药物的有效性和安全性,而这在新药的注册审批中是必要的。为了更好地了解候选药物的代谢动力,金斯瑞( GenScript)建议用动物来做药物分布及其代谢的研究,分析在不同时间段、不同组织或血清中,药物及其代谢物的情况。金斯瑞进行精确的药物和药物代谢动力学研究,涉及两个主要方面:药物分布及其代谢动力研究和抗体药物的代谢动力研究。群体药代动力学研究的是个体之间药物浓度变异来源及其相关性,这些个体是指按临床上相关剂量接受候选药物的目标患者人群。患者的某些人口统计学特征、病理生理特征以及治疗方面的特征,比如体重、排泄和代谢功能、以及接受其他治疗,都能够有规律地改变药物剂量-浓度关系。例如,主要由肾脏排除的药物,在接受同样剂量的情况下,在肾功能衰竭患者体内的稳态浓度,通常高于肾功能正常的患者体内的稳态浓度。群体药代动力学的研究目的就是找出那些使剂量-浓度关系发生变化的、可测定的病理生理因素,确定剂量-浓度关系变化的程度,当这些变化与临床上有意义的治疗指数改变相关的情况下,能够恰当地调整剂量。在药品开发中使用群体PK方法,使获得完整的药代动力学资料有了可能,不但能从来自研究受试者的相对稀疏的数据中获取资料,而且还能从相对密集的数据或从稀疏数据和密集数据的组合中获取资料。群体PK方法能够分析来自各种不均衡设计的数据,也能分析因为不能按常用的药代动力学分析方式分析而通常被排除的研究数据,比如从儿科患者和老年患者获取的浓度数据,或在评价剂量或浓度与疗效或安全性之间的关系时所获取的数据。传统药代动力学研究的受试者通常是健康的志愿者或特别挑选的患者,一组成员的平均情况(即平均血浆浓度-时间曲线)一直是关注的主要焦点。许多研究将个体之间药代动力学的变异作为一个需要降到最低的因素进行观察,通常是通过复杂的研究设计和对照方案,或通过有严格限制的入选标准/排除标准,将其降到最低。事实上,这些资料对在临床应用期间可能会出现的变异至关重要,但是却被这些限制所掩盖。而且,传统药代动力学研究只关注单个变量(例如肾功能)的作法,还使其难以研究变量之间的交互作用。
极性大的药物进行药物代谢的研究,怎样进行生物样品前处理啊?
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=111832]GLP药物研究与GMP药物生产规范 [/url]pdf格式。GLP药物非临床研究规范&GMP_药物生产规范电子文本,具有一定的参考价值
想研究一种药物,别人已经研究过它的的组分,自己还有研究价值吗?不知道该如何下手,是不是考虑从方法和条件等方面创新
手性药物药学研究的基本要求如下:在原料药制备工艺研究时应根据手性中心的引入方式,采取有效的过程控制手段,严格控制产品的光学纯度;在结构确证时,需结合其制备工艺、结构确证用对照品及文献数据等已有的研究基础,选择合适的方式来证明该药物的立体构型;制剂的处方与工艺研究过程中应注意保证手性药物立体构型的稳定;质量研究时应结合工艺确定需研究控制的立体异构体杂质,并注意验证各手性分析方法的立体专属性,在制订质量标准时从各个方面控制产品的光学特性与光学纯度;在稳定性研究时,应设立灵敏的光学纯度质控指标,以监测立体构型的稳定性。 药物的研发一般分为三个不同的专业:药学、药理毒理及临床,在研究的过程中,这三个专业之间是紧密联系、相互印证的。即使在药学专业内部的各项研究间也是如此,在各项研究的过程中需要随时参考其它研究的结果,才能使我们的研究工作更为全面与准确。下面分别论述各药学研究间的关系:

[align=center][b][img=,600,336]https://ng1.17img.cn/bbsfiles/images/2019/09/201909121439522763_1873_932_3.jpg!w690x387.jpg[/img][/b][/align][b]质量研究与质量标准质量研究[/b]🔥 原料药的质量研究合成多肽原料药的质量研究除参考一般化学药物的研究思路进行常规项目的研究外,还应根据合成多肽的结构特征、制备工艺特点和生物学特点等进行针对性的研究,研究项目一般包括:外观性状、理化常数、鉴别、氨基酸组成分析、水分、反离子含量、纯度、有机溶剂和反应试剂残留量、生物学安全性检查、含量和/或活性效价测定等。检测方法研究和验证的基本思路和要求与已颁布的相关技术指导原则相一致。对于合成多肽药物,除常规项目外,理化常数一般需要关注其比旋度、等电点(pI)、溶解性(主要为水和缓冲液中)等。一般而言,多肽药物的常规检查项目与其它化学药物相同。此外,与多肽药物的结构及合成特点相关的一些检查项目,例如氨基酸组成分析、反离子(例如三氟醋酸或醋酸根)含量、反应试剂残留量(例如从树脂上裂解多肽使用了氢氟酸,需要检查氟化物残留量)等,则需要在原料药质量研究中予以重视。相关肽检查(或称有关物质检查)是反映多肽化学纯度的重要指标之一,根据多肽的理化性质、分子大小,可选择合适的色谱、电泳等方法进行。短肽可参考一般化学药品有关物质检查的研究思路选用适宜的方法;长肽的有关物质检查方法除常见的RP-HPLC外,还可考虑使用高效离子交换色谱(HPIEC)、毛细管电泳技术等,非解离条件下的高效分子排阻色谱(HPSEC)、聚丙烯酰胺凝胶电泳(PAGE)以及激光散射粒度测定等技术可用于聚合体/低聚体的检查。有关物质检查的方法学验证应能证明所采用的方法可以有效分离目标多肽与工艺杂质(例如缺失肽等)、降解产物(例如二硫键交换或氧化产物等)、聚合物等。一般应考察两种以上不同原理的方法,高效液相色谱法至少应包括一种梯度洗脱方法,并采用多肽粗品和强制降解试验等对方法的专属性等进行考察、对比,此外还应注意研究多波长检测的结果并选择合适的检测波长等。合成多肽因结构特征不同于通常的小分子化学药品,纯度检查有时难以从根本上有效控制产品安全性,需要进行必要的生物学安全性检查(如过敏试验、降压物质、升压物质、异常毒性等)以全面控制产品质量、保证安全性。此外,根据产品具体情况,对于长肽,有时尚需进行免疫原性或抗原活性等生物特性的研究。含量测定是评价多肽质量的重要指标之一,理化方法测定其含量时称为“含量测定”,生物学方法或酶化学方法测定其效价时称为“效价测定”。对于短肽,理化方法测得的含量可以反映其有效程度时,首选简单、通用的含量测定方法;对于具有一定空间结构才能发挥其活性的多肽,需进行生物学方法或酶化学方法测定药物活性(效价)的研究,包括含量与活性的关系、相应的方法学验证等。🔥 制剂的质量研究合成多肽制剂的质量研究基本思路和要求可参照《化学药物质量标准建立的规范化过程技术指导原则》、《化学药物制剂研究基本技术指导原则》等相关内容,根据合成多肽的具体特点,在原料药质量研究的基础上,结合剂型特点、处方工艺以及临床使用特点,重点研究所用辅料和制剂工艺对产品质量的影响、制剂辅料和制剂产生的降解产物对检测方法的影响以及与剂型相关的质量要素。研究项目一般亦应包括性状、鉴别、检查(安全性、均一性、纯度要求与有效性指标等)、含量或效价测定等几个方面。[b]质量标准[/b]合成多肽药物质量标准的制订原则、要求与《化学药物质量标准建立的规范化过程技术指导原则》是一致的。即,在系统的质量控制研究基础上,充分考虑药品安全、有效、质量可控的要求,以及生产、流通和使用等环节的影响,确定能够揭示、控制药物内在品质的检测项目、分析方法和限度要求,如原料药质量标准应包括氨基酸组成、等电点、中长肽的肽图等。合理可行的质量标准应能有效控制产品质量以保证临床用药的安全性和有效性,并有效地控制药品批间质量的一致性。相关质控项目的限度确定也应参考相关的指导原则,例如对于有关物质检查限度的确定可以参考《化学药物杂质研究的技术指导原则》、仿制品种同时还可参考《化学药品仿制研究技术指导原则》等的原则性要求,并结合产品本身的特性及临床使用情况,视具体情况而定。随着药物研发进程的深入,研究数据积累的不断丰富、方法学研究的完善和药物研究技术的不断发展,质量标准在不同研究阶段需要不断修订和完善。[b]稳定性研究[/b]合成多肽药物稳定性研究的基本原则应遵循《化学药物稳定性研究技术指导原则》的一般性要求。与一般化学药物相比,多肽药物的稳定性较差。引起多肽药物不稳定的原因主要有水解、氧化、外消旋化、二硫键的断裂及重排、β消除、凝聚、沉淀、吸附等。当多肽处于溶液中或高湿下保存时,其降解或聚合的速度会比干燥条件下大为增加。因此,稳定性研究应根据多肽药物稳定性的特点合理选择试验条件、考察项目。加速试验和长期留样试验的试验条件应依据药物对温度、湿度和光照等条件的敏感程度的考察(影响因素试验)基础上选择;考察项目除常规项目(例如原料药的比旋度、有关物质和含量等)外,根据具体情况,可能还需要考察其生物活性的变化。与其他化学药物不同,多肽药物可能具有一定程度的表面活性,有与直接接触药品的包装材料和容器发生吸附等相互作用的可能,从而引起制剂效价、生物活性下降。例如有些多肽分子能够与玻璃表面的硅醇基发生相互作用。因此,在包装材料的选择方面需注意其与多肽药物相互作用的研究,有些情况下可选择特殊处理后的包装容器,如表面经硅烷化处理的容器等。[b]名词解释非天然氨基酸:[/b][color=#717070]除自然界生物体中存在的氨基酸外,其它由人工合成制备的氨基酸。[/color][b]反离子:[/b][color=#717070]和多肽形成离子对的带有相反电荷的离子。[/color][b][b][/b][/b]参考文献1.Guidance for Industry for the Submission ofChemistry,Manufacturing,and Controls Information for Synthetic Peptide Substances,FDA,1994。2.合成多肽专题研讨会会议纪要,药品审评中心,2001。3.多肽药物分析方法研究进展,叶晓霞,俞雄,中国医药工业杂志,2003,34(7)。[b]著 者《合成多肽药物药学研究技术指导原则》课题研究组。[/b]
一、概述药物中的残留溶剂系指在原料药或辅料的生产中、以及在制剂制备过程中使用或产生而又未能完全去除的有机溶剂。根据国际化学品安全性纲要,以及美国环境保护机构、世界卫生组织等公布的研究结果,很多有机溶剂对环境、人体都有一定的危害,因此,为保障药物的质量和用药安全,以及保护环境,需要对残留溶剂进行研究和控制。本指导原则是在参考人用药物注册技术要求国际协调会(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)颁布的残留溶剂研究指导原则,美国药典(the United States Pharmacopoeia,USP)、英国药典(British Pharmacopoeia, BP)、欧洲药典(European Pharmacopoeia,EP)、中国药典(Chinese Pharmacopoeia, ChP)相关内容的基础上,结合我国药物研发的特点,通过分析、研究残留溶剂问题与药物的安全性、有效性及质量可控性之间的内在关系而制定的。本指导原则总结了对残留溶剂问题的一般认识,旨在帮助药物研发者科学合理的进行残留溶剂方面的研究,也为药物评价者提供参考。考虑到残留溶剂研究涉及的范围比较广泛,本指导原则主要对原料药的残留溶剂问题进行讨论,并以此为基础,探讨和总结药物研究过程中对残留溶剂问题的一般性原则。药物研发者可参考本指导原则对制剂和辅料的残留溶剂问题进行研究。考虑到药物研究开发的阶段性,本指导原则适用于药物研发的整个过程。

药物代谢研究的实践与展望中国科学院上海药物研究所药物代谢研究中心钟大放第一部分 LC/MS/MS方法与药代动力学研究第二部分 9-硝基喜树碱的体内代谢产物鉴定第三部分 CYP2C9 多态性对氯诺昔康临床药代动力学的影响部分内容截图:http://ng1.17img.cn/bbsfiles/images/2010/11/201011211738_260910_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211739_260911_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211741_260912_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211741_260913_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211742_260914_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211742_260915_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211743_260916_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211743_260917_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211743_260918_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211743_260919_1637626_3.jpghttp://ng1.17img.cn/bbsfiles/images/2010/11/201011211743_260920_1637626_3.jpg
[back=transparent]质谱成像是以质谱技术为基础的可视化方法,通过质谱离子源直接扫描生物样本,可以在一张组织切片上同时分析数百种分子的空间分布特征,已成为精确解析药物分子及其代谢产物组织空间分布的关键技术之一,[back=transparent]质谱成像[/back]应用于药物ADME的研究。[/back]一般在生活中肾脏是药物排泄的主要器官。但是药物排泄过程的正常与否关系到药效强度、药效维持时间以及毒副作用。所以,这是我们必须要借助一些科学例如高分辨质谱技术来助力药物。近年来,高分辨质谱成像技术的诞生为定位药物组织分布研究提供了全新的技术和思路。本文将主要介绍TransMIT AP-SMALDI 10高分辨率质谱成像系统如何一步步揭秘伊马替尼在小鼠肾脏组织中的空间分布特征。TransMIT AP-SMALDI 10质谱成像系统是目前少有的集高空间分辨率和高质量精度于一体的质谱成像系统。该系统采用常压基质辅助激光解吸电离技术,通过先进的准直光束聚焦实现了5μm的成像分辨率;质谱端搭载Thermo Scientific? Q Exactive?系列质谱仪,保证了离子分析的高质量分辨率和高质量精度。综上所述,研究成功的揭示了伊马替尼在重要排泄器官肾脏中的组织分布特征,同时也获取了组织中各种内源性化合物的空间分布信息,为研究药物分子的累积和排泄机制提供了可靠的科学依据。TransMIT AP-SMALDI 10质谱成像系统集高空间分辨率、高质量分辨率和高质量精度于一身,不仅成为了药代动力学研究的利器,也应用于肿瘤标志物研究、植物次生代谢物研究、药用植物药效成分研究、微生物和单细胞研究等。未来,期待TransMIT AP-SMALDI 10质谱成像系统为我国药物研发人员和各领域科研工作者带来更多的惊喜,加快研究进程,加速成果转化。
来源:石家庄日报日前,华北制药集团新药研发公司承建的微生物药物国家工程研究中心项目,通过了省发改委组织的验收。这标志着微生物药物国家工程研究中心正式落户华药。 微生物药物国家工程研究中心2004年经国家发改委批准建设,总投资约1.4亿元,是“十五”期间国家重点建设的23个国家工程研究中心之一,是我国在微生物药物方面唯一由国家命名的工程研究中心。 按照国家对国家级工程研究中心的定位,国家工程研究中心是代表我国相关领域内国家水平的专业性研究开发机构,具备原始创新、集成创新、对引进技术消化吸收和再创新的能力,形成从产品源头开发、中试到产业化的一整套技术体系。该中心依托于华北制药集团新药研究开发公司, 采用基因工程等现代生物技术,开展微生物菌种的选育、发酵、提取分离等方面的工艺研究,为微生物制药产业发展提供技术保障。 目前,该中心建成了27000株菌的微生物菌种资源库,54000种微生物产物的化合物库,微生物药物高通量筛选及活性评价技术平台、菌种选育技术平台、微生物药物工程化生产验证平台等,形成了规范化、专业化的微生物药物研发和工程化验证技术体系。建设期间,成功开发了9个微生物药物新产品,完成了柔性系统集成技术验证,初步具备了向行业转移相关技术的能力。 目前,华北制药已经成为河北省唯一拥有国家级企业技术中心、国家863高技术成果产业化基地、国家工程研究中心三个国家级重要称号的单位。
国外一些研究新药开发的科学家和大学教授写的一本关于质谱在药物代谢研究中的应用的参考书,希望对研究新药及药物检测的同仁有所参考价值。[~120736~]
对于固体药物,其理化性质通常包括:性状(如外观,颜色,物理状态);熔点或沸点;比旋度,溶解性,吸湿性,溶液pH, 分配系数,解离常数,除此之外,对于将用于制剂生产的药物,还需要了解其物理形态,如多晶型、溶剂化物或水合物等,那么对于多晶型和溶剂化物应该如何进行研究?或者说关于这一部分信息应该如何进行叙述?
《柳叶刀》杂志1月6日刊登一份研究报告,称全球大约两亿人使用非法药物。研究人员所参考资料包括联合国毒品和犯罪问题办事处公布的数字、各国调查报告以及关于毒品使用影响的报告。迷幻药、滥用处方药以及合成代谢类固醇不在报告统计范畴。报告说,2009年,全球1.49亿至2.71亿人使用非法药物,其中1.25亿至2.03亿人吸食大麻;1500万至3900万人吸食海洛因、吗啡等鸦片类药物以及苯丙胺和可卡因;1100万至2100万人注射毒品。报告写道:“大麻可能导致上瘾和精神紊乱,却似乎不会切实提高死亡率……过度使用和依赖非法鸦片类药物是致死的主要原因。”另外,人们注射毒品时可能共用针头,感染艾滋病病毒和肝炎病毒的风险颇高。(来源:新华网 刘红霞)
化学药物杂质研究的技术指导原则、化学药物残留溶剂研究的技术指导原则,从国家食品药品监督管理局下载,希望有用。[img]http://simg.instrument.com.cn/bbs/images/default/em09511.gif[/img]
化学药物残留溶剂研究的技术指导原则一、概述 药物中的残留溶剂系指在原料药或辅料的生产中、以及在制剂制备过程中使用或产生而又未能完全去除的有机溶剂。根据国际化学品安全性纲要,以及美国环境保护机构、世界卫生组织等公布的研究结果,很多有机溶剂对环境、人体都有一定的危害,因此,为保障药物的质量和用药安全,以及保护环境,需要对残留溶剂进行研究和控制。 本指导原则是在参考人用药物注册技术要求国际协调会(International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use,ICH)颁布的残留溶剂研究指导原则,美国药典(the United States Pharmacopoeia,USP)、英国药典(British Pharmacopoeia, BP)、欧洲药典(European Pharmacopoeia,EP)、中国药典(Chinese Pharmacopoeia, ChP)相关内容的基础上,结合我国药物研发的特点,通过分析、研究残留溶剂问题与药物的安全性、有效性及质量可控性之间的内在关系而制定的。本指导原则总结了对残留溶剂问题的一般认识,旨在帮助药物研发者科学合理的进行残留溶剂方面的研究,也为药物评价者提供参考。 考虑到残留溶剂研究涉及的范围比较广泛,本指导原则主要对原料药的残留溶剂问题进行讨论,并以此为基础,探讨和总结药物研究过程中对残留溶剂问题的一般性原则。药物研发者可参考本指导原则对制剂和辅料的残留溶剂问题进行研究。 考虑到药物研究开发的阶段性,本指导原则适用于药物研发的整个过程。 二、基本内容 (一)残留溶剂研究的基本原则 1、确定残留溶剂的研究对象 从理论上讲,药物制备过程中所使用的有机溶剂均有残留的可能,均应进行残留量的研究。但是,药物研发者可以通过对有机溶剂的性质、药物制备工艺等进行分析,提出科学合理的依据,有选择性的对某些溶剂进行残留量研究,这样,既可以合理有效的控制产品质量,又有利于降低药物研究的成本,避免不必要的浪费。因此,药物研发者在进行残留溶剂研究之前,需要首先对药物中可能存在的残留溶剂进行分析,以确定何种溶剂需要进行残留量的检测和控制。 2、确定残留溶剂时需要考虑的问题 原料药中有机残留溶剂与其制备工艺密切相关,同时也需要结合其制剂的临床应用特点来考虑如何对可能残留的溶剂进行研究。 2.1 原料药制备工艺 原料药制备工艺中可能涉及的残留溶剂主要有三种来源:合成原料或反应溶剂、反应副产物、由合成原料或反应溶剂引入。其中作为合成原料或反应溶剂是最常见的残留溶剂来源,本部分主要对此进行讨论。 影响终产物中残留溶剂水平的因素较多,主要有:合成路线的长短,有机溶剂在其中使用的步骤,后续步骤中使用的有机溶剂对之前使用的溶剂的影响,中间体的纯化方法、干燥条件,终产品精制方法和条件等等。 2.1.1 合成路线 由于有机化学反应及后处理工艺的复杂性,对于在得到终产物之前的第几步工艺中使用的溶剂可能在终产物中残留不可能有准确定论。但是,一般来说,后面几步中使用的溶剂的残留可能性较大,因此,对于较长路线的工艺,尤其需要关注后几步所使用的各类溶剂。 2.1.2 后续溶剂的影响 后续使用的溶剂对此前使用溶剂的影响是非常复杂的,取决于各溶剂的性质、后续反应中物料状态以及后续步骤除去溶剂的方法等。 2.1.3 中间体的影响 中间体的处理方法、纯化方法和干燥条件等影响中间体的残留溶剂情况,从而影响终产品的溶剂残留情况。 2.2 制剂及其临床应用特点 控制原料药的残留溶剂,最终目的是控制制剂的残留溶剂,使之符合规定。有时候根据制剂的一些特点,可能对原料药残留溶剂的研究和限度要求进行特殊性的考虑。需要注意,以下所列的因素并不是孤立的,在考虑下列因素时需要注意它们之间的相互影响。 2.2.1 剂型、给药途径 不同制剂发挥疗效的机理不同,对其残留溶剂的要求也可能有所不同。例如注射剂与某些局部使用局部发挥药效的皮肤用制剂相比,残留溶剂的要求就可能相对比较严格。 2.2.2 处方 辅料的残留溶剂也是制剂残留溶剂的组成部分。通过对处方中所使用辅料的残留溶剂水平的了解,可以估算原料药中所能允许存在的残留溶剂水平。 2.2.3 工艺 制剂的制备工艺可能引入新的溶剂,也可能使原料药和辅料中的残留溶剂水平降低。例如素片包衣可能引入新的残留溶剂,干燥工艺可能降低残留溶剂水平等。 2.2.4 适应症 出于治疗一些特殊疾病的考虑,有时候较高水平甚至超出安全值水平的残留溶剂也可能被允许,但需要进行充分的利弊分析。 2.2.5 剂量、用药周期 高剂量、长期用药的制剂,与低剂量、短期用药的制剂相比,对于残留溶剂的要求可能相对严格一些。 3、残留溶剂分类及研究原则 根据有机溶剂对人体及环境可能造成的危害的程度,分为以下四种类型进行研究: 3.1 第一类溶剂及研究原则 第一类溶剂是指人体致癌物、疑为人体致癌物或环境危害物的有机溶剂。因其具有不可接受的毒性或对环境造成公害,在原料药、辅料以及制剂生产中应该避免使用。当根据文献或其他相关资料确定合成路线,涉及到第一类溶剂的使用时,建议重新设计不使用第一类溶剂的合成路线,或者进行替代研究。 由于有机溶剂的选用是合成工艺中比较重要的一点,建议替代研究在工艺研究初期即开始进行,这样,有利于将由于溶剂替换对后续的结构确证、质量研究、稳定性考察的影响降至最低。但替代研究是一项比较复杂、耗时的工作,有时候由于条件、时间等的限制,替代研究工作在临床研究前可能无法充分进行。在严格控制残留溶剂量的前提下,可使药物进入临床研究。在临床研究期间、注册标准试行期间、注册标准转正后,仍可进一步进行替代溶剂的研究工作。 因为溶剂的改变可能导致产品物理化学性质以及质量的改变,因此如发生溶剂的替代,则需要进行溶剂改变前后的产品物理化学性质、质量的对比研究,必要时还需要进行结构对比确证,以说明产品的结构是否发生变化。 如果工艺中不可避免的使用了第一类溶剂,则需要严格控制残留量,无论任何步骤使用,均需进行残留量检测。 3.2 第二类溶剂及研究原则 第二类溶剂是指有非遗传毒性致癌(动物实验)、或可能导致其他不可逆毒性(如神经毒性或致畸性)、或可能具有其他严重的但可逆毒性的有机溶剂。此类溶剂具有一定的毒性,但和第一类溶剂相比毒性较小,建议限制使用,以防止对病人潜在的不良影响。 考虑到第二类溶剂对人体的危害以及所使用的溶剂在终产品中残留的可能性,建议对合成过程中所使用的全部第二类溶剂进行残留量研究,以使药物研发者全面掌握产品质量情况,为最终制定合理可行的质量标准提供数据支持。 3.3 第三类溶剂及研究原则 第三类溶剂是GMP或其他质量要求限制使用,对人体低毒的溶剂。第三类溶剂属于低毒性溶剂,对人体或环境的危害较小,人体可接受的粗略浓度限度为0.5%,因此建议可仅对在终产品精制过程中使用的第三类溶剂进行残留量研究。 3.4 尚无足够毒性资料的溶剂及研究原则 这类溶剂在药物的生产过程中可能会使用,但目前尚无足够的毒理学研究资料。建议药物研发者根据生产工艺和溶剂的特点,必要时进行残留量研究。 随着对这类溶剂毒理学等研究的逐步深入,将根据研究结果对其进行进一步的归类。 (二)研究方法的建立及方法学验证 在确定了需要进行残留量研究的溶剂后,需要通过方法学研究建立合理可行的检测方法。目前,常用的检测方法为气相色谱法(Gas Chromatography,GC),也有其他一些检测方法。 1、研究方法的建立 1.1 气相色谱法(GC法) GC法具有检测灵敏度较高,选择性较好的特点,采用此法所需的样品用量较少,基本可以满足所有残留溶剂测定的要求。采用GC法时,需要结合药物和所要检测的溶剂的性质,通过方法学研究确定合适的检测条件。由于通常要同时检测多种溶剂,为操作的可行性和简便性,建议尽量采用同样的检测条件控制尽量多种类的残留溶剂。 1.1.1 进样方法 GC法包括溶液直接进样和顶空进样两种进样方法。通常情况下,沸点低的溶剂建议采用顶空进样法,沸点高的溶剂可以采用溶液直接进样法,当样品本身对测定有影响时,也建议采用顶空进样法。 1.1.2 供试品溶液和对照品溶液的配制 对于固体原料药,如采用溶液直接进样法,需先用水或合适的溶剂使原料药溶解,以使其中的有机溶剂释放于溶液中,才能被准确测定。如采用顶空进样法,通常以水作溶剂;当药物不溶于水,但可溶于一定浓度的酸或碱液中时,可采用不挥发的酸或碱液为溶剂,但不能使用盐酸溶液或氨水;对于非水溶性药物,可采用合适的溶剂,如N,N—二甲基甲酰胺、二甲基亚砜等为溶剂。 不管采用何种进样法,所选择的溶剂应能够尽量同时溶解样品和待检残留溶剂,所
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=11223]药物稳定性研究[/url]
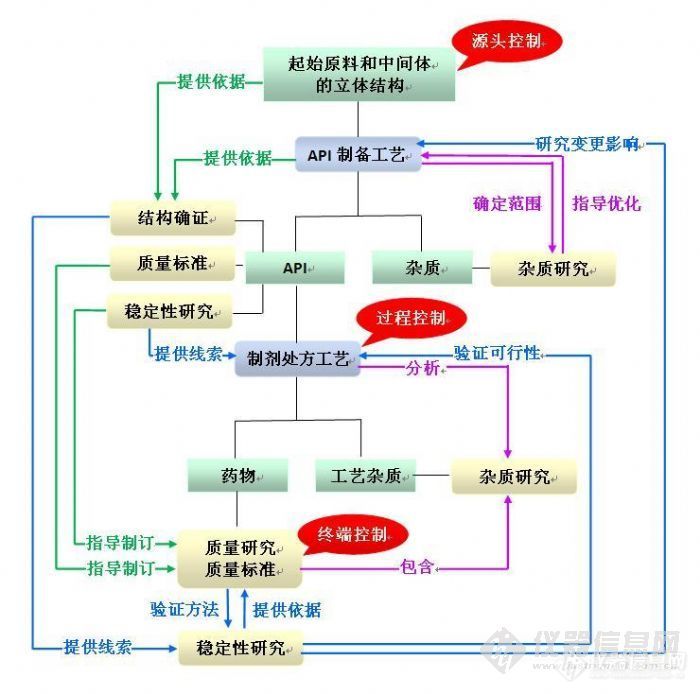
看过这篇电子刊物之后,觉得不光是对手性药物,对认识其他化学药物的药学研究也很有帮助,所以做了一份图解。既为加深理解,也为与大家分享。欢迎批评指正。声明:本人发贴的目的是分享和讨论,不是为赚取积分或奖品,因此会在仪器信息网、丁香园和SFDAIED论坛同时发,不参加原创大赛。转载的朋友就不必在以上论坛转载了,谢谢。http://ng1.17img.cn/bbsfiles/images/2011/12/201112080052_336496_1612179_3.jpg
药物杂质研究的基本思路及案例分析
化学药物制剂处方工艺的研究
药物临床研究标准操作规程新药申报用的谢谢!![img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=53469]药物临床研究标准操作规程资料[/url]
药物的构效关系,一般都用什么方法来研究啊?
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=70797]化学药物残留溶剂研究的指导原则[/url]化学药物残留溶剂研究的指导原则内容简介:(一)残留溶剂研究的基本原则1、确定残留溶剂的研究对象2、确定残留溶剂时需要考虑的问题3、残留溶剂分类及研究原则(二)研究方法的建立及方法学验证1、研究方法的建立2、方法学验证(三)研究结果的分析及质量标准的制定1、残留溶剂表示方法2、质量标准制定的一般原则及阶段性要求(四)需要关注的几个问题1、附录中无限度规定和未收载的有机溶剂2、未知有机挥发物3、多种有机溶剂综合影响4、中间体的残留溶剂5、制剂工艺对制剂残留溶剂的影响6、辅料残留溶剂的研究及对制剂的影响
12月10日“抗体药物研究与应用”网络研讨会,邀您免费报名参加!足不出户听领域专家分享学术最新进展,机会难得。点击链接免费参加:https://www.instrument.com.cn/webinar/meetings/Drug1210/[url=http:https://www.instrument.com.cn/webinar/meetings/Drug1210///]点击打开链接[/url]
药物非临床研究质量管理规范认证管理办法国家食品药品监督管理局:GLP认证新办法正式施行 为进一步规范药物非临床研究质量管理规范(简称GLP)认证管理工作,近日,国家食品药品监督管理局颁发了新修订的《药物非临床研究质量管理规范认证管理办法》,同时废止了2003年10月1日施行的《药物非临床研究质量管理规范检查办法(试行)》。 本次修订加强了对通过认证的药物非临床研究机构的监督管理,明确规定了药物非临床安全性评价研究机构对人员和设施的重大变更或可能影响GLP实施的严重事件的报告制度,明确规定对已通过GLP认证的机构将实施随机检查、有因检查和3年一次的定期检查,并规定了定期检查的程序要求。本次修订还明确规定了GLP认证申请机构的基本条件,要求申请机构应在申请前按照GLP的要求运行12个月以上,并按照GLP的要求完成申请试验项目的药物安全性评价研究。除此以外,本办法进一步规范了认证检查、审核、公告的程序和要求,并细化和完善了检查项目,提高了认证检查标准。 本次修订总结了2003年发布的《药物非临床研究质量管理规范检查办法(试行)》实施以来的经验,广泛听取意见,借鉴国内外相关的先进管理经验,参考了相关法律法规。此《办法》增强了检查标准的可操作性及评定方式的科学性和客观性,既有总体内容上的突破,同时兼顾具体条款的修缮,具有很强的现实意义。 修订后的《办法》自发布之日起施行。(2007.04.19) 药物非临床研究质量管理规范认证管理办法 第一章 总 则 第一条 为加强药物非临床研究的监督管理,规范药物非临床研究质量管理规范(以下简称GLP)认证管理工作,根据《中华人民共和国药品管理法》和《中华人民共和国药品管理法实施条例》及有关规定,制定本办法。 第二条 GLP认证是指国家食品药品监督管理局对药物非临床安全性评价研究机构的组织管理体系、人员、实验设施、仪器设备、试验项目的运行与管理等进行检查,并对其是否符合GLP作出评定。 第三条 国家食品药品监督管理局主管全国GLP认证管理工作,省级药品监督管理部门负责本行政区域内药物非临床安全性评价研究机构的日常监督管理工作。 第二章 申请与受理 第四条 拟申请GLP认证的药物非临床安全性评价研究机构可根据本机构的研究条件,申请单项或多项药物安全性评价试验项目的认证。申请GLP认证的机构,应在申请前按照GLP的要求运行12个月以上,并按照GLP的要求完成申请试验项目的药物安全性评价研究。 第五条 申请GLP认证的药物非临床安全性评价研究机构,应向国家食品药品监督管理局报送《药物非临床研究质量管理规范认证申请表》、申请资料(附件1、2)和电子版本。申请资料中有关证明文件的复印件应加盖申请机构公章。 第六条 国家食品药品监督管理局在收到申请资料之日起5个工作日内做出是否受理的决定,并书面告知申请机构和申请机构所在地省级药品监督管理部门。 第三章 资料审查与现场检查 第七条 国家食品药品监督管理局自受理之日起20个工作日内完成对申请资料的审查。 第八条 资料审查符合要求的,在20个工作日内制订检查方案,组织实施现场检查。资料审查不符合要求的,发给申请机构不予行政许可的通知,书面说明原因;需要补充资料的,应当一次性告知申请机构要求补充的全部内容。申请机构须在2个月内按要求一次性完成补充资料的报送,逾期未报的,视为自动放弃认证申请。 第九条 实施现场检查前,国家食品药品监督管理局提前5个工作日通知被检查机构和所在地省级药品监督管理部门现场检查安排。 第十条 实施现场检查时,被检查机构所在地省级药品监督管理部门应派分管药品研究监督管理的人员作为观察员参加现场检查。 第十一条 被检查机构应积极配合检查组工作,按检查组要求协助开展检查工作。 第十二条 现场检查工作由检查组组长负责组织实施。在检查开始前,应宣布检查纪律,提出检查要求,明确检查范围、检查方式和检查日程安排。 第十三条 检查组应按照检查方案和GLP认证标准(附件3)进行检查,详细记录检查的情况,对检查中发现的不符合GLP的事项如实记录,必要时应予取证。 第十四条 检查组在现场检查结束前应对检查中发现的问题进行评议汇总,撰写现场检查意见。检查组评议期间,被检查机构人员应回避。 第十五条 检查结束时,检查组应向被检查机构宣读现场检查意见。现场检查意见须由检查组全体成员和被检查机构负责人签字。 第十六条 被检查机构对现场检查意见有异议时,可向检查组说明,双方不能达成一致意见的问题,检查组须做好记录,经检查组全体成员和被检查机构负责人签字,由检查组提交国家食品药品监督管理局。 第十七条 检查组完成现场检查后, 应退还被检查机构提供的所有资料。 第十八条 现场检查时间一般为3至5天,根据检查工作的需要可适当调整。
[align=center]靶向抗癌药物的研究进展[/align][align=center]摘 要[/align][align=center] [/align][align=left]恶性肿瘤是威胁人类健康的常见病和多发病,其病死率极高,死亡率占所有疾病死亡率的首位,引起了人们的普遍关注。传统意义上的抗癌药物在体内无一定的特异性分布,导致对肿瘤细胞起作用的药物剂量无法达到要求,在杀死或抑制肿瘤细胞的同时也损伤相当数量的正常细胞,从而产生严重的器质性病变。随着人体内各种通路及肿瘤实体生理机制的阐明,出现了一种通过将药物或者相关蛋白直接导向肿瘤细胞的新型抗癌药物,即靶向抗癌药物。这类药物的特异性强,疗效显著。本文主要就近年来靶向抗癌药物的研究进展进行了综述,并对靶向抗癌药物发展前景进行了预测和展望。[/align][align=left] [/align][align=left]关键词:靶向;抗癌药物;研究进展[/align][align=center] [/align][align=center] [b]Progressin the research of anticancer drugs targeting [/b][/align][align=center][b]ABSTRACT[/b][/align][align=center][b] [/b][/align]Malignant tumor is aserious threat to human health of the common and frequently occurring disease,its mortality is very high, mortality accounts for all disease mortality in thefirst place, caused the widespread attention of people. Anticancer drug in thetraditional sense of no specific distribution in the body, leading to drug doseeffect not to meet the requirements of the tumor cells, damage to normal cellsin a considerable number of kill or inhibit tumor cell at the same time,resulting in serious pathological changes. With the elucidation of variouspathways and tumor entity and physiological mechanism of the human body, theemergence of a new anticancer drug or by direct guidance related proteins intumor cells, namely targeting anticancer drugs. This drug is of good effect. Thispaper mainly in recent years targeted anticancer drugs research progress were narrated,and the target anticancer drug development prospect is forecasted andprospected.[b]Keywords[/b]: Targeting anticancer drug research progress[b]一 引言[/b][align=left] 在癌症的治疗中,传统化疗药物细胞毒的特异性差,在杀死或抑制肿瘤细胞的同时也损伤相当数量的正常细胞,直接影响心、肺、肝、肾以及神经系统等功能。严重者必须中断化疗,导致化疗失败[sup][/sup]。并且,药物分子不能有效到达预期靶标部位,造成药物在靶部位浓度不足。因此,研究者在癌症治疗中开始寻求具有较好靶向性并具有较高释放效率[sup][/sup]的体系,即靶向抗癌药物。靶向抗癌药是指针对在肿瘤发生发展过程中具有关键作用的特定靶标进行治疗的药物[sup][/sup],近年来这类药物被相继开发出来。例如,肿瘤新生血管生成抑制剂阿瓦斯丁等已经开始在临床应用;拓扑异构酶Ⅰ抑制剂及其同类物最近对其机理的研究也取得了新的进展;另外基于肿瘤信号转导机制的药物如蛋白激酶抑制剂等在肿瘤治疗中的研究也成为新的热点[sup][/sup]。[/align][b]二 肿瘤新生血管生成抑制剂[/b]任肿瘤细胞的生长与转移很大程度上依赖于新血管的生成。肿瘤会诱导其周围血管的快速生成,导致血管内皮细胞组织形态的改变,内皮细胞不断迁移、增殖,从而使新血管逐渐生成[sup][/sup]。开发和研究能够破坏或抑制血管生成、有效地抑制肿瘤生长和转移的药物,是新型抗肿瘤药物研究的活跃领域之一。与抑制肿瘤生长的传统治疗方式相比,靶向新生血管生成的治疗模式可能意味着更高的特异性,更低的毒性,以及有利于克服肿瘤的耐药性,而且还可广泛用于多种肿瘤转移的治疗。抗肿瘤新生血管药物的主要类型有:以血管内皮细胞生长因子为靶点的单克隆抗体,如贝伐单抗(阿瓦斯丁);以血管内皮细胞生长因子受体为靶点的多靶点小分子酪氨酸激酶抑制剂,如索拉非尼、舒尼替尼;作用于血管内皮细胞靶点的血管生成抑制剂,如重组人血管内皮抑制素(恩度)、沙利度胺(反应停)。以近几年来研究热门的蛋白酪氨激酶(PTK)抑制剂为例,对其作用机制和上市情况进行阐述。PTK是一组酶系,能催化ATP上的磷酸基转移到许多重要的蛋白质的酪氨酸残基上,使其残基磷酸化,从而激活各种底物酶,通过一系列反应影响细胞的生长、增殖和分化[sup][/sup]。多数肿瘤细胞PTK活性异常升高,因此PTK是一个非常重要和有价值的抗肿瘤靶点。2005-2010 年间,此类药物累计申报新化合物总量已经达到 27 个,占所有类别抗肿瘤新药的50%[sup][/sup]。这些药物大多数为me-too药,如盐酸埃克替尼,该产品是在2004年获得 FDA批准上市的厄洛替尼基础上进行结构改造得到,是一种表皮生长因子受体( EGFR) 酪氨酸激酶抑制剂,针对同样靶点的上市产品还有吉非替尼[sup][/sup]。[b]三 微管蛋白抑制剂[/b]一直以来,肿瘤细胞的微管都被视为抗肿瘤药物的良好靶点,微管蛋白抑制剂被作为最有效的抗肿瘤临床一线药物。许多化合物都能干扰微管蛋白的功能,主要是与微管作用以抑制其聚合,使细胞分裂停止于有丝分裂的中期或者促进微管聚合,抑制微管解聚进而影响细胞分裂[sup][/sup]。较为经典的如长春碱类和鬼臼霉素类可与微管蛋白结合,阻滞微管蛋白聚合成微管,影响微管蛋白稳定性,干扰纺锤体的形成,阻滞有丝分裂的进行,使细胞分裂停滞在中期[sup][/sup]。近年来,细胞微管相关的抗肿瘤药物在原有基础上得到了进一步的发展,长春花生物碱类长春氟宁、多拉司他汀、罗米地新、艾立布林等药物已经相继进入二到三期临床试验,有的甚被作为某些肿瘤的二三线临床用药[sup][/sup]。这些药物在原有基础上增加了药物作用的特异性,并在一定程度上减少了不良反应。相信在不久的将来,以细胞微管为靶点的抗癌药物能为我们带来更多的惊喜,成为临床上更有效的抗癌药物。[b]四 叶酸介导的抗肿瘤药物[/b][align=left]在叶酸是核酸生物合成的代谢物,叶酸缺乏时白细胞减少,因此叶酸拮抗物可用于治疗急性白血病。某些以二氢叶酸还原酶为靶点的叶酸拮抗剂可以不可逆的抑制二氢叶酸还原酶的活性。这样通过抑制二氢叶酸还原酶,从而抑制DNA和RNA的合成,阻碍肿瘤细胞的生长[sup][/sup]。此类药物如甲氨喋呤,临床用于急性白血病和绒毛膜上皮癌,常与亚叶酸钙合用降低毒性[sup][/sup]。[/align][align=left]此外,叶酸受体在大部分人体肿瘤细胞表面过度表达,而在正常细胞表面则很少表达,甚至不表达。这就使得利用叶酸介导的抗肿瘤药物靶向作用于叶酸呈阳性的肿瘤细胞成为可能,从而减少传统抗癌药物对正常细胞的毒副作用。此类药物如EC0225,Endocyte 公司于2007 年 3 月开始对其进行Ⅰ期临床试验用于治疗顽固性或病灶转移性肿瘤[sup][/sup]。[/align][b]五 DNA拓扑异构酶抑制剂[/b] DNA 的不间断复制是肿瘤细胞不断增生的关键所在。如果有效的抑制DNA 引物酶,阻断引物的合成,DNA制将会受到限制,肿瘤细胞将不再增生,进而抑制了肿瘤的生长[sup][/sup]。DNA引物酶作为理想靶点为抗肿瘤药物的研究提供了线索,当前抑制DNA复制主要以 DNA拓扑异构酶为靶点展开的。作为一种独特酶,DNA拓扑异构酶存在于真核生物和原核生物细胞中,用以调节DNA空间构型动态变化,参与DNA的不断复制、翻译、重组和修复等过程,在形成染色体结构及染色体分离与浓缩方面起到了主导作用[sup][/sup]。[b]六 端粒酶抑制剂[/b]在基因端粒酶是一种RNA 聚合酶,能以本身RNA 为模板,在染色体末端合成六聚脱氧核苷酸TrAGGG 的重复序列,以补偿细胞分裂时的染色体末端缩短,解决“末端复制问题”[sup][/sup]。正常人体细胞的端粒酶活性较低,但大多数的肿瘤细胞的端粒酶活性显著升高。因此,检验端粒酶活性是癌诊断的重要方法,是抗癌药物需要研究的靶点之一。端粒酶在肿瘤细胞被过度表达,它是肿瘤细胞增殖所必需的。抑制端粒酶的活性,肿瘤细胞进入静止状态,最后产生细胞凋亡。端粒酶抑制剂是一类潜在的高选择性的抗肿瘤药物,在恶性肿瘤的基因治疗中有重要作用。已发现的端粒酶抑制剂有AZT、AZGT及其衍生物、异噻唑啉衍生物TMPI、类黄连素、小的非核苷合成复合物BIBR等。[align=center]七 总结与展望[/align][align=left]近年来,抗癌药物的研究已从利用和改进毒性较大的传统抗癌药物发展向靶向药物设计和研发转变。随着科学技术的发展和对肿瘤发病机制认识的深入,临床抗癌药物的靶向性越来越集中。靶向抗癌药物已凭其特异性、针对性和有效性较强,患者耐受性较好而毒副反应相对于细胞毒药物较低等特点,在肿瘤治疗中取得很大成功。与此同时,靶向抗癌药物研发中仍存在很多需要解决的科学与技术问题。例如:单一靶点的小分子类药物治疗范围窄,且易产生耐药性;即使是使用抗肿瘤靶向药物治疗非常成功的适应证,由于肿瘤细胞具有高度变异的性质,患者的肿瘤仍可能在初始治疗成功后复发;有些肿瘤靶点精确性不够,它们也存在于某些正常细胞,所以会导致相应毒性;多数靶向抗癌药物不良反应多等。[/align][align=left]这些问题都有待于解决。并且,要清醒的认识到靶向抗肿瘤药物开发近几年呈现爆炸式增长,可以想象在未来 5-10年市场将相当拥挤,此类药物的开发立项中需要趋于冷静和理性,需要药企和研究者之间的合作,探索更具有创新性思维的临床研究模式。[/align][align=left]无论如何,根据特定导向的靶点的需要来设计制备疗效确切的抗癌药物,将是靶向抗癌药物的发展方向,靶向抗癌药物必将随着其疗效最优化的进步而在各类肿瘤中得到更广泛的应用。在靶点治疗理念的指导下,在合理的研究设计、全面的临床前评价和严谨合理的临床试验的基础上,抗肿瘤药物新靶点研究前景变得更加广阔,也能开发出更多新靶点特异性抗肿瘤新药。[/align][align=left]另一方面,根据患者的基因和蛋白资料实施给药方案,并“量体裁衣”式地对患者合理用药,以提高药物的疗效,降低药物的毒副反应,同时减轻患者的痛苦和经济负担,利用基因导向个体化用药;能否通过对靶位基因的筛查达到早期诊断并提示预后,甚至可以通过干预这些基因的异常表达达到治疗的目的。个人认为,这将是我们努力的方向。[/align][b]参考文献[/b] EavanG1 Prolififeration,elleyeleandaPotosisineaneer.Nature,2004,411(6835):342~348. Cho K J,Wang X,Nie S M. Clin.CancerRes.,2008,14: 1310—1316 Sawyers C. Targeted cancer therapy .Nature, 2004,432(7015): 294-297 梁岩,何珩,孙翠萍.抗肿瘤用药的应用及进展.中国医疗前沿,2009,4(21):17~18 林健,卜一珊.分子抗癌靶向药物的研究进展.天津药学,2005,17(12):61~62 郑晓克.抗肿瘤药物的研究进展.中山大学研究生学刊,2008,4(29):7~12 DuffaudF, Blay JY. Gastrointestinal stromal tumors and ncatmcnl . Oncology, 2003,65 (3):187-197 陈晓媛,张虹,高晨燕.小分子靶向抗肿瘤药物临床研究策略探讨及案例分析.新药申报与审评技术,2013,22(3):269~273 孙西洋,任常山,曹心珂.抗肿瘤药物的靶向给药系统研究进展.中国医药生物技术,2008,3(6):461~464 Ng SS,Figg WD T.antiangiogenesisin vitroinfluence of formulation vehicles and binding proteins.Cancer Res,2004,64(3):821-824 Bellmunt J,ThéodoreC,Demkov T.Phase III trial of plus best supportive care compared with alone after aplatinum-containing regimen in patients with advanced transitional cellcarcinoma of the tract.Clin Oncol,2009,27(27):4454-4461 王祎,刘燕,丁秀云.抗肿瘤药物的研究进展.包头医学,2012,36(3):129~132 曹胜利,郭燕文,王先波.抗叶酸剂类抗肿瘤药物的研究进展.中国新药杂志,2007,16(10):747~753 梁旭华,孙洋,谭春雷.叶酸受体介导的抗肿瘤靶向前药研究进展.中国新药杂志,2012,21(22):2647~2653 卿晨.恶性肿瘤化疗耐药及克服耐药的研究.昆明医科大学学报,2013,34(1):1~3 王丽鸿.分子靶向抗癌药物的研究进展.牡丹江医学院学报,2013,34(2):81~82 李嘉,包春波.抗肿瘤化学药物研究进展.华西药学杂志,2009,4(21):1~5
药物研究选择剂型时,是很据什么来选定的呢?
关于研究药物构效关系的一些参数,都有哪些呢?
随着新版《药品注册管理办法》的实施,对药品注册的相关技术提出了新的要求,特别是抗生素类高风险产品,目的是全面提升注册上市药品的质量和品质。 杂质研究是药物质量控制研究的重要项目。对抗生素而言,由于其多为半发酵、半合成产品,所含的杂质种类与杂质含量都比普通合成化学药物复杂;同时由于国内抗生素使用范围较广,面临的安全性问题更为突出,因此,杂质研究和杂质控制更是抗生素质量控制研究的关键项目。 对于仿制国内外已上市抗生素的品种,根据仿制药的基本技术要求,应选择被仿药物进行系统的质量对比研究,以保证其质量的一致性。 在杂质研究方面,根据相关技术要求,结合我国抗生素生产和研发的历史以及现实情况,提出如下要求:
之前发了肿瘤药物的分析,有版友要求多发些此方面的于是看到有人需要,就发出来分享一下找了好久啊[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=172577]2008_抗高血压药物市场研究报告[/url]ps:[URL=http://www.instrument.com.cn/bbs/shtml/20090918/2116028/]2009肿瘤药物行业分析[/URL]
最新发现与创新 中国科技网讯 南开大学药物化学生物学国家重点实验室在药物传输载体研究方面取得重要进展,其研究成果“基于蛋白—多肽特异性结合的小分子水凝胶”,近日发表在《德国应用化学》上。 据课题组介绍,药物传输是实现药物疗效不可或缺的重要环节。利用现代生物化学技术开发的新型多肽/蛋白质、抗体、疫苗及基因等新型药物在环境及人体内极易失活和降解,从而导致生物利用度低。而先进的药物载体和传输技术是提高药物的生物利用度、增加药物疗效、降低其毒副作用和改善病人耐受性的主要手段。从20世纪90年代开始,外表类似果冻的小分子水凝胶作为一种新颖的生物材料,在药物传输方面展现了良好的应用前景。如何在温和条件下制备水凝胶用于药物传输,一直是科学家力求达到的目标。 南开大学杨志谋、龙加福教授课题组结合各自在相应研究领域的积累,提出利用蛋白质和多肽特异性结合的特点制备新型蛋白—多肽杂化水凝胶。该体系利用蛋白—多肽的特异性结合来增强多肽自组装纤维之间的结合力,从而形成三维网络结构以及形成性质更为优异的水凝胶。他们针对抗肿瘤药物、多肽/蛋白质药物及基因药物,重点以嵌段共聚物、超分子化合物、小分子凝胶及高分子水凝胶等材料为基础,研发出生物相容性高的可注射局部药物传输系统。该类新型药物传输系统由蛋白质和多肽组成,生物相容度高。 同时,该类水凝胶能包裹各类药物,可局部注射于病灶,起到局部长期缓释药物的效果,提高病人耐受性,减轻毒副作用。(通讯员 周兴龙 韦承金 记者 冯国梧) 《科技日报》(2012-7-15 一版)







 我要推广仪器
我要推广仪器
 下载APP
下载APP