化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检测方法1 适用范围 本方法规定了用高效液相色谱法定性检测化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的方法。 本方法适用于化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的定性测定。2 方法提要 样品在经过提取后,经高效液相色谱仪分离,二极管阵列检测器检测,经与平行操作的补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮对照品及补骨脂对照药材比较,以保留时间和紫外光谱图定性,鉴别补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的存在。本方法对补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检出限和取样品0.5 g时的检出浓度见表1。 表1 4种补骨脂特征成分的检出限和检出浓度化合物检出限(ng)检出浓度(μg/g)补骨脂素0.30.6异补骨脂素0.30.6新补骨脂异黄酮0.30.6补骨脂二氢黄酮0.30.63 试剂和材料 除另有规定外,所用试剂均为分析纯,水为实验室用一级水。3.1 乙腈,色谱纯。3.2 补骨脂素,纯度≥99%。3.3 异补骨脂素,纯度≥99%。3.4 新补骨脂异黄酮,纯度≥98%。3.5 补骨脂二氢黄酮,纯度≥99%。3.6 补骨脂,中国食品药品检定研究院,供鉴别用。3.7 补骨脂特征性成分混合标准溶液(=0.1 μg/mL):分别称取补骨脂素(3.2)、异补骨脂素(3.3)、新补骨脂异黄酮(3.4)、补骨脂二氢黄酮(3.5)对照品各5 mg(精确到0.1 mg),置500 mL量瓶中,用甲醇溶解并稀释至刻度,摇匀,配制成质量浓度各为10 μg/mL的标准溶液。精密量取各标准溶液0.1 mL置10 mL量瓶中,加甲醇稀释至刻度,摇匀,即得0.1 μg/mL的混合标准溶液。3.8 补骨脂标准储备溶液:取补骨脂对照药材0.2 g,置50 mL三角瓶中,加30 mL 70%乙醇回流提取1h,滤过,滤液置100 mL量瓶中,加70%乙醇稀释至刻度,摇匀,即得。4 仪器4.1 高效液相色谱仪:具二极管阵列检测器。4.2 分析天平:感量为0.1 mg。4.3 移液器。4.4 涡旋振荡器。4.5 超声波清洗仪(功率不低于200W)。4.6 高速离心机:转速不小于10000 r/min。
[size=5]超临界流体色谱法测定补骨脂中补骨脂素和异补骨脂素含量[/size] 来源: 作者:陆峰,刘荔荔,李玲,吴玉田摘要目的:建立超临界流体色谱法用于测定补骨脂中补骨脂素和异补骨脂素的含量,并研究其影响因素。方法:用改性的超临界C02萃取中药补骨脂,超临界流体色谱法测定其中的香豆素成分(补骨脂素和异补骨脂素)含量。色谱条件:15 cm×1mm×3μm氨基柱,流动相为含5%甲醇的CO2,柱温40℃,柱头压27.6MPa,UV 247 nm检测。结果:在选定固定相条件下.流动相对组分的洗脱和选择性影响最大,柱温、柱压次之。补骨脂素的回收率为96.9%(RSD=1.8%),异补骨脂素的回收率为95.I%(RSD=I.6%)。培论:与超临界流体萃取法联用,本法可用于香豆素类化合物的分离分析。关键词 超临界流体色谱法;补骨脂素;异补骨脂素;超临界流体萃取法材料和方法 药品和试剂:补骨脂药材由上海长海医院提供,并由本院生药教研室李彬鉴定;补骨脂素(psoralen)、异补骨脂索(isopsoralen)对照品购自中国药品生物制品检定所;SFE和SFC所用CO2购自上海BOC气体公司;蒽(内标物)和氯仿为分析纯;甲醇为HPLC级。 仪器:SFX 2-10萃取器,model 100DX/DM 注射泵,泵控制器. MWD检测器(ISCO.USA),eppendof CH.30色谱柱加热器。 SFE条件:萃取溶剂为CO2,加入氯仿0.06 ml作改性剂,压力为38.5 MPa,温度为70℃:,静态萃取1 min,动态萃取7ml,限流管温度80℃,甲醇作吸收溶剂。 SFC条件:用SpheriorbNH2 柱(150mm×lmm×3μm,A1ltech,USA),流动相为含5%甲醇的CO2.柱头压27.6 MPa,柱温4O℃ ,进样量为0.5, 流速为0.10ml/min,采用50cm×15μm石英毛细管作限流管,检测器AUFS 0.05,247 nm检测。结果与讨论1 固定相/流动相对保留的影响 以超临界C02(含少量改性剂)为基本流动相的SFC是正相色谱,一些极性较弱的化合物在C18柱上几乎不保留 本实验中补骨脂素和异补骨脂素很快从Cl8柱上洗脱,几乎没有分离效果,所以未用C18柱而改用正相NH2柱。 改性剂浓度对组分的保留影响很大。在NH2柱上.用纯C02作流动相时,补骨脂素和异补骨脂素在60min内未洗脱;而在高浓度改性剂时,因两者结构较相似,几乎同时洗脱。本实验用5% 甲醇时洗脱效果良好.2 温度、压力对SFC的影响 温度和压力对保留也有影响。本实验在20.7~ 34.5 MPa压力范围和3O~6O℃ 温度范围内考察了这两个因素的影响。保留时问随压力增大而缩短;随温度升高而延长.3 标准曲线 在上述最佳色谱条件下.补骨脂素、异补骨脂素、内标蒽保留时间分别为6.37,5.00和4.00 min。 用甲醇分别配制对照品溶液和内标溶液:补骨脂素0.423mg/ml,异补骨脂素0.17mg/ml,蒽1.343 mg/ml。分别精密吸取对照品溶液25,50.100.200,400和600μl于10m1量瓶中,加入内标溶液5Oμl,定容,进行SFC分析。以对照品峰与内标峰面积之比(Y)对各自浓度(x,μ/m1)回归.得回归方程。 补骨脂素:Y=23.49X+1.37×10-4,r =0.999; 异补骨脂素:Y:20.04X+0.016.r =0.999。4 方法回收率 精密量取补骨脂素、异补骨脂素溶液各150.250和350μl,加入酸洗硅藻土50mg,挥干溶剂.按SFE条件萃取;甲醇吸收液再加入内标溶液50μl,定容、稀释,各进祥3次,按SFC步骤渊定,计算上述3种浓度的回收率,补骨脂素的回收率为96.9%( r=9.RSD=1.8%);异补骨脂素的回收率为95.1% ( r=9,RSD:1.6% )。5 样品测定 分别取3批干燥药材粉碎.过1OO目筛.精密称取粉末50mg按上述条件萃取;在甲醇吸收液中加入适量内标液,稀释后每份溶液进样3次.按上述色谱条件测定,代入标准曲线,得3批药材中的补骨脂素、异补骨脂索含量分别为0.77%.0.70% ;0.86% .0.82% :0.65% ,0.53% 。 以上结果表明:流动相组成是影响分离的最重要因素,加入改性剂可大大改善中等极性化合物的洗脱分离。系统的温度和压力对分离也有影响。与SFE联用,SFC可以较方便地用于某些香豆素类化合物的分离分析。
化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检测方法
[b][size=16px]药用补骨脂[/size][/b][size=16px][/size][b][size=16px]《药性论》:“主男子腰疼,膝冷囊湿,逐诸冷痹顽,止小便利,腹中冷。”《本草纲目》:“治肾泄,通命门,暖丹田,敛精神。”《本草求真》:“能敛神明,使心包之火与命门之火相通,因而元阳坚固,骨髓充实。”《本草经疏》:“能暖水脏,阴中生阳,壮火益土之要药也。”[/size][/b][size=16px][/size][size=16px]补骨脂单用有效,也常与杜仲、核桃仁、熟地黄、五味子、菟丝子等药材同用,以增强补肾壮阳、固精缩尿的效果。[/size][size=16px][/size][size=16px]如用于治疗由肾阳不足引起的阳痿遗泄、尿频、遗尿等症,常配伍仙灵脾、菟丝子等同用。治虚冷泄泻,常与肉豆蔻等同用。肾气不足,摄纳无权,引起喘促,补骨脂温肾而纳气平喘,常与胡桃肉配伍以治虚寒气喘。[/size]
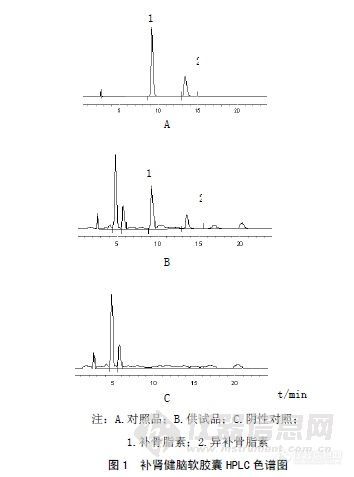
作者:徐晓明;徐大勇;邢俊波;(梅河口市卫生职工中等专业学校;梅河口市医院爱民医院药剂科;中国人民解放军总后卫生部药品仪器检验所;)摘要:目的建立补肾健脑软胶囊中补骨脂素和异补骨脂素的含量测定方法。方法采用HPLC法,色谱柱为Diamonsil-C18(250mm×4.6mm,5μm),流动相为甲醇-水(48∶52),流速为1.0mL/min,检测波长为246nm,柱温40℃。结果补骨脂素和异补骨脂素线性范围分别为0.01008~0.252μg(r=0.9998)、0.00914~0.2285μg(r=0.9999)。平均回收率分别为98.91%(RSD=1.74%)、99.93%(RSD=1.27%)。结论本法分离度好,快速、简便,可作为该产品的质量控制方法。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208131425_383490_1606903_3.jpg

作者:程龙琼, 周晓英(成都市食品药品监督检测中心,四川 成都 610045)摘要:目的采用HPLC法测定青娥丸中补骨脂的含量。方法色谱柱为钻石C18柱(200 mm×4.6 mm,5μm)。流动相为甲醇-水(55:45);流速1.0 ml.min-1;检测波长246 nm;柱温为室温。结果补骨脂素的线性范围是0.1004~1.5060μg(r=0.9998)。平均加样回收率为97.8%,RSD=1.92%(n=6)。异补骨脂素的线性范围是0.0830~1.2450μg(r=0.9993)。平均加样回收率为98.5%,RSD=1.60%(n=6)。结论所建方法可用于测定青娥丸中的补骨脂。谱图:http://ng1.17img.cn/bbsfiles/images/2012/07/201207161015_377764_1606903_3.jpg
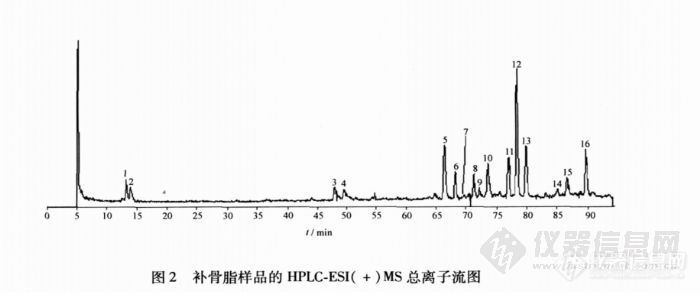
【作者】 刘亚男; 王跃飞; 韩立峰; 潘桂湘; 王虹;【Author】 LIU Ya′nan,WANG Yuefei,HAN Lifeng,PAN Guixiang,WANG Hong(Engineering Research Center of Modern Chinese Medicine Discovery and Preparation Technique Traditional Chinese MedicineResearch Centre of Tianjin University of Traditional Chinese Medicine,Tianjin 300193,China)【机构】 天津中医药大学中医药研究院现代中药发现与制剂技术教育部工程研究中心;【摘要】 目的:建立补骨脂药材中化学成分的高效液相色谱-电喷雾质谱分析方法。方法:采用DiamonsilTMC18(4.6 mm×250 mm,5μm)色谱柱;流动相0.05%甲酸水溶液-乙腈,梯度洗脱;流速1 mL.min-1;柱温30℃;DAD扫描范围190~400nm;检测波长246 nm。Finnigan电喷雾离子阱多级质谱仪;正离子检测模式;ESI喷雾电压4 500 V;鞘气(N2)流速60个单位;辅助气(N2)流速20个单位;毛细管温度350℃;毛细管电压19 V,扫描范围m/z90~800。结果:补骨脂中化学成分获得了较好的分离和检测,共鉴定出2个香豆素苷,3个香豆素,8个黄酮和1个单萜酚类成分。结论:该方法灵敏度高、分离度好,适用于补骨脂药材中化学成分的快速定性鉴定。 http://ng1.17img.cn/bbsfiles/images/2012/07/201207301609_380605_2379123_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/07/201207301609_380606_2379123_3.jpg
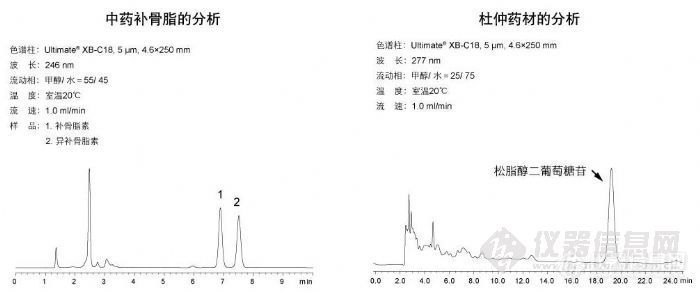
中药 中药补骨脂的分析和杜仲中松脂醇二葡萄糖苷的分析http://ng1.17img.cn/bbsfiles/images/2009/10/200910311054_179312_1896702_3.jpg
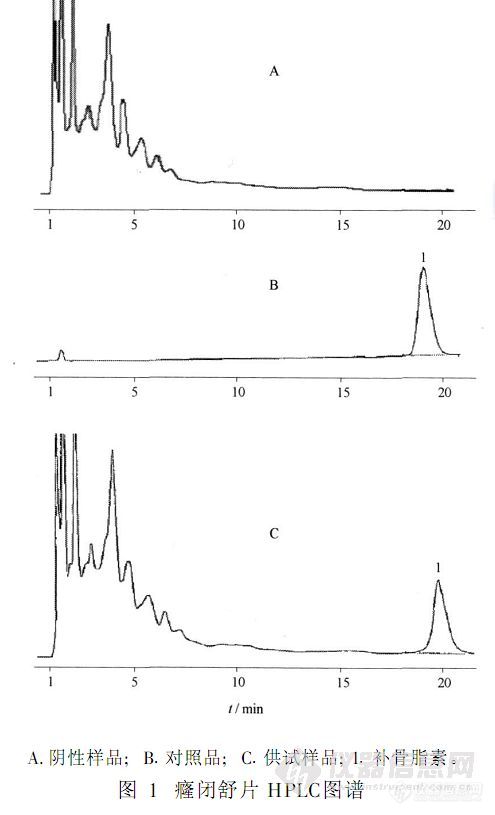
【作者】 叶英响;【机构】 浙江省义乌市中心医院; 温州医学院附属义乌医院中药科;【摘要】 目的:建立癃闭舒片中补骨脂素的含量测定方法;方法:采用高效液相色谱法,色谱柱Diamonsil C18(4.6 mm×250 mm,5μm),流动相甲醇-1%冰醋酸水溶液(45∶55),检测波长297 nm;结果:补骨脂素回归方程为Y=41 651X+9 228,r=0.999 4,线性范围为3.52~17.6μg.mL-1,平均回收率101.1%,RSD 2.1%。结论:本方法简单、稳定,测定准确。 更多还原http://ng1.17img.cn/bbsfiles/images/2012/08/201208131339_383473_2379123_3.jpg
[size=4][color=#DC143C][font=黑体]同位素比质谱方法检测内源性类固醇雄烯二酮[/font][/color][/size]=========================================在所使用的禁用物质中,类固醇激素是较为普遍使用的一类药物。人体自身能合成与分泌的类固醇激素称为内源性类固醇激素,如攀酮。由于在检测方法上有一定难度,一些运动员选择使用内源性类固醇制剂以逃避兴奋剂检测。目前,兴奋剂检测实验室应用同位素比质谱分析方法检测内源性类固醇来源。13C和12C是碳元素在自然界中的天然同位素。有机化合物的来源不同,其同位素比(如13C与12C的比值)也不同。人体自身分泌的类固醇与相同化学结构的类固醇制剂的同位素比不同。应用同位素比质谱分析技术可以测定化合物13C与12C的比值,同位素比用δ(‰)值表示。根据仪器的分析流程和组成部分,本文方法称为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比质谱([url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS)方法。该方法在兴奋剂检测中的应用时间较短,文献方法较为繁琐,本文建立了快速灵敏的[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析方法。-------------------------------------------------试验材料与方法1. 试剂和对照品试剂:β-葡萄糖醛酸酶,Sigma;其余均为国产分析纯试剂。对照品:睾酮(缩写T)、雄酮(缩写An)、本胆烷醇酮(缩写Etio)、5α-雄烷-3α,17β-二醇(缩写5α-diol)、5β-雄烷-3α,17β-二醇(缩写5β-diol)、孕二醇(缩写PD)购自Sigma公司。2. 样品两名健康志愿者,一名男性40岁,尿样为sample 1;一名女性38岁,尿样为sample 2,均没有服用任何药物。收集其晨尿为阴性对照尿。阳性尿样为世界反兴奋剂机构水平考试所用尿样来自兴奋剂检测中心。3. 仪器[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]/燃烧炉/同位素比值质谱仪(HP6890/DELTA PLUS,Finnigan);高效液相色谱仪(Waters2796,检测器:Waters2996 PAD,自动收集器:Waters Fraction Collector Ⅲ);Anilent 5973i[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-质谱联用仪。4. 方法4.I [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS操作条件色谱柱:HP5毛细管色谱柱,25m×0.2mm i.d.×0.3μm;柱流速:1mL/min(室温);升温程序:60 ℃(2min)一50 ℃/min→255℃一2.5℃/min→280℃(6.5min);进样口温度:260℃;燃烧炉温度:960℃;质谱离子源:EI;参考气:CO2,1.8V。4.2 高效液相色谱仪操作条件色谱柱:ZQRBAX SB-C18(4.6mm×250mm,5μm);流动相:水-乙睛,梯度洗脱(0-18min:乙睛从30%→100%);流速1mL/min;柱温:室温。4.3 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD操作条件色谱柱:HP-1毛细管色谱柱,25 m×0.2mm i.d.×0.11μm;柱流速:1 mL/min (室温);升温程序:150 ℃(1min一5℃/min→200℃一30℃/min→310℃(10min)。4.4 样品预处理取尿样2mL,加人1Ml0.2mol pH=7.0的磷酸盐缓冲液和10μLβ-葡萄糖醛酸酶(5000 IU)混匀,在55℃恒温水浴中培养3h,取出后加pH=8.8的碳酸盐固体缓冲剂约100mg 和5mL叔丁基甲醚,振荡萃取,离心后,取出上层有机溶液,在加热的情况下,用氮气吹干,加人50μL甲醇溶解残渣,备用。将上述甲醇溶液置HPLC仪上,依前述色谱条件分离,分段收集流出液,确定收集时间程序。分别将流出组分用氮气吹干,加人50μL环己烷,备用。4.5 对照品溶液的制备分别配制对照品睾酮、雄酮、本胆烷醇酮、5α-雄烷-3α,17β-二醇、5β-雄烷-3α,17β-二醇,孕二醇的甲醇溶液,浓度为1mg/mL。4.6 样品测定4.6.1 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD 分析依上述[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/MSD仪器操作条件,取样品溶液及对照品溶液进行全扫描,扫描范围m/z 20~450,选择待测物的特征离子,获得SIM图。经对样品中与对照品有相同保留时间的峰进行质谱分析,及与标准品质谱图的对比,确定待测样品的组成。4.6.2 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析依上述CC/C/IRMS仪器操作条件,对处理后的样品进行[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]/C/IRMS分析,测得内源性类固醇激素的δ值。
[em06] 如题菲罗门柱250*4.6,1.0ml/min,246nm 其中对照品浓度为:补14.32ug/ml,异补10.064ug/mlyy000021 对照品 柱温35度 进样20ul 两峰分离度 2.41 补塔板数4023 甲醇:水(43:57)yy000022 对照品 35度 进样20ul 两峰分离度 2.37 补塔板数4015 甲醇:1%醋酸(43:57)yy000024 对照品 40度 进样5ul 两峰分离度 3.19 补塔板数8117 甲醇:1%醋酸(43:57)yy000025 中药复方制剂的醇提液 35度 进样10ul 两峰分离度 3.13 补塔板数7415 甲醇:1%醋酸(43:57)
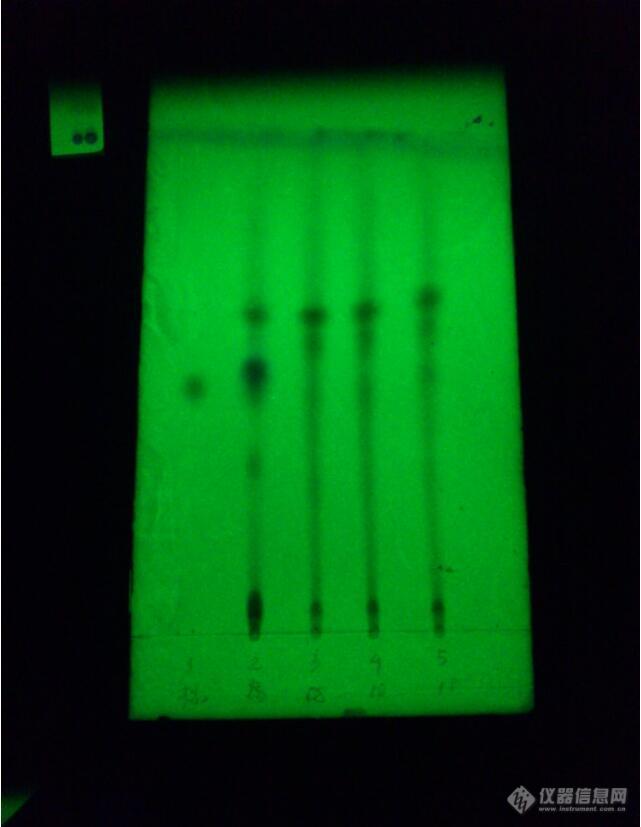
按照药典规定的,展开剂:氯仿-醋酸乙酯-丙酮-甲酸=6:2.5:2.5:0.21为原儿茶酸对照品2药材对照品3、4、5均为制成的中药制剂供试品。我已经增大浓度试过了,色谱图无差异[img=,641,827]https://ng1.17img.cn/bbsfiles/images/2019/08/201908140947125804_8826_1791505_3.jpg!w641x827.jpg[/img]

中药制剂中木瓜的薄层鉴别:供试品与对照药材处相应的点颜色不同是为什么?狗脊的薄层鉴别也是一样,提取时处理方法是一样的图1前三供试品(绿色),中二木瓜对照药材(蓝色),后二阴性对照图2前二供试品(亮的点蓝色),中二狗脊对照药材(绿色),后二阴性对照不知道该怎么办,诚请各位大师解答,谢谢[img=,690,459]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329352099_2969_1816508_3.jpg!w690x459.jpg[/img][img=,380,362]https://ng1.17img.cn/bbsfiles/images/2019/08/201908061329533546_2463_1816508_3.jpg!w380x362.jpg[/img]
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7681612594 七氟醚杂质C Sevoflurane Related Compound C 对照品/标准品1612572 七氟醚杂质 B Sevoflurane Related Compound B 对照品/标准品1612550 七氟醚杂质 A Sevoflurane Related Compound A 对照品/标准品1612540 七氟醚 Sevoflurane 对照品/标准品1612539 盐酸舍曲林 Sertraline Hydrochloride 对照品/标准品1612528 盐酸舍曲林杂质A Sertraline Hydrochloride Related Compound A 对照品/标准品1612517 盐酸舍曲林消旋体混合物 Sertraline Hydrochloride Racemic Mixture 对照品/标准品1612506 L-丝氨酸 L-Serine 对照品/标准品1612426 芝麻油杂质B Sesame Oil Related Compound B 对照品/标准品1612415 芝麻油杂质A Sesame Oil Related Compound A 对照品/标准品1612404 芝麻油 Sesame Oil 对照品/标准品1612029 番泻苷 B Sennoside B 对照品/标准品1612018 番泻苷 A Sennoside A 对照品/标准品1612007 番泻苷 Sennosides 对照品/标准品1611955 硒 蛋氨酸 Selenomethionine 对照品/标准品1611900 盐酸司来吉兰 Selegiline Hydrochloride 对照品/标准品1611004 司可巴比妥 CII Secobarbital CII 对照品/标准品1610090 东莨菪亭 Scopoletin 对照品/标准品1610001 氢溴酸东莨菪碱 Scopolamine Hydrobromide 对照品/标准品1609831 沙奎那韦杂质A Saquinavir Related Compound A 对照品/标准品1609829 甲磺酸沙奎那韦 Saquinavir Mesylate 对照品/标准品1609807 双水杨酯 Salsalate 对照品/标准品1609625 沙美特罗杂质B Salmeterol Related Compound B 对照品/标准品1609614 沙美特罗杂质A Salmeterol Related Compound A 对照品/标准品1609603 昔美酸沙美特罗 Salmeterol Xinafoate 对照品/标准品1609501 水杨酸片 Salicylic Acid Tablets 对照品/标准品1609024 水杨酸杂质B Salicylic Acid Related Compound B 对照品/标准品1609013 水杨酸杂质A Salicylic Acid Related Compound A 对照品/标准品1609002 水杨酸 Salicylic Acid 对照品/标准品1608000 水杨酰胺 Salicylamide 对照品/标准品1607506 连翘粉状贯叶提取物 Powdered St. John's Wort Extract 对照品/标准品1607040 糖精钠 Saccharin Sodium 对照品/标准品1607029 糖精钙 Saccharin Calcium 对照品/标准品1607007 糖精 Saccharin 对照品/标准品1606503 芦丁 Rutin 对照品/标准品1606208 硝酚胂酸 Roxarsone 对照品/标准品1605523 罗哌卡因杂质B Ropivacaine Related Compound B 对照品/标准品1605512 罗哌卡因杂质A Ropivacaine Related Compound A 对照品/标准品1605500 盐酸罗哌卡因 Ropivacaine Hydrochloride 对照品/标准品1604916 罗库溴铵合剂峰的识别 Rocuronium Peak Identification Mixture 对照品/标准品1604905 罗库溴铵 Rocuronium Bromide 对照品/标准品1604870 利凡斯的明杂质B Rivastigmine Related Compound B 对照品/标准品1604869 利凡斯的明杂质A Rivastigmine Related Compound A 对照品/标准品1604814 利托那韦杂质混合物 Ritonavir Related Compounds Mixture 对照品/标准品1604803 利托那韦 Ritonavir 对照品/标准品1604701 盐酸利托君 Ritodrine Hydrochloride 对照品/标准品1604665 利培酮系统适用性试验用混合物 Risperidone System Suitability Mixture 对照品/标准品1604654 利培酮 Risperidone 对照品/标准品1604643 利塞膦酸杂质C Risedronate Related Compound C 对照品/标准品1604632 利塞膦酸杂质B Risedronate Related Compound B 对照品/标准品1604621 利塞膦酸杂质A Risedronate Related Compound A 对照品/标准品1604610 利塞膦酸钠 Risedronate Sodium 对照品/标准品1604600 利美索龙 Rimexolone 对照品/标准品1604508 盐酸金刚乙胺 Rimantadine Hydrochloride 对照品/标准品1604348 利鲁唑杂质A Riluzole Related Compound A 对照品/标准品1604337 利鲁唑 Riluzole 对照品/标准品1604202 醌式利福平 Rifampin Quinone 对照品/标准品1604009 利福平 Rifampin 对照品/标准品1603800 利福布丁 Rifabutin 对照品/标准品1603108 核糖 Ribose 对照品/标准品1603006 维生素B2 Riboflavin (Vitamin B2) 对照品/标准品1602706 利巴韦林 Ribavirin 对照品/标准品1602003 间苯二酚 Resorcinol 对照品/标准品1601849 二类残留溶剂-二甲苯 Residual Solvent Class 2 - Xylenes 对照品/标准品1601827 二类残留溶剂-三氯乙烯 Residual Solvent Class 2 - Trichloroethylene 对照品/标准品1601805 二类残留溶剂-甲苯 Residual Solvent Class 2 - Toluene 对照品/标准品1601780 二类残留溶剂-四氢萘 Residual Solvent Class 2 - Tetralin 对照品/标准品1601770 二类残留溶剂-四氢呋喃 Residual Solvent Class 2 - Tetrahydrofuran 对照品/标准品1601769 二类残留溶剂-二氧噻吩烷 Residual Solvent Class 2 - Sulfolane 对照品/标准品1601747 二类残留溶剂-吡啶 Residual Solvent Class 2 - Pyridine 对照品/标准品1601725 二类残留溶剂-硝基甲烷 Residual Solvent Class 2 - Nitromethane 对照品/标准品1601703 二类残留溶剂-N-甲基吡咯烷酮 Residual Solvent Class 2 - N-Methylpyrrolidone 对照品/标准品1601689 二类残留溶剂-甲基环己烷 Residual Solvent Class 2 - Methylcyclohexane 对照品/标准品1601667 二类残留溶剂-甲基丁基酮 Residual Solvent Class 2 - Methylbutylketone 对照品/标准品1601645 二类残留溶剂- 2-甲氧基乙醇 Residual Solvent Class 2 - 2-Methoxyethanol 对照品/标准品1601623 二类残留溶剂-甲醇 Residual Solvent Class 2 - Methanol 对照品/标准品1601601 二类残留溶剂-己烷 Residual Solvent Class 2 - Hexane 对照品/标准品1601587 二类残留溶剂-甲酰胺 Residual Solvent Class 2 - Formam
大家好!我正在用氨基柱分离单糖,之前都好好的,就是分离度较叉。昨天突然发现所有对照品的峰都有了分叉。开始我以为是因为我进的样品针是酸性的缘故,于是我用10倍柱体积的乙腈-0.5%的氨水(50-50)洗了柱子,然后用正常流动相洗了柱子。今天进对照品时发现还是老样子,没有改善。有人说是柱头塌陷了,有这么严重么?还有没有其它原因啊?请有心人帮帮忙啊!一个新手碰到了这样的问题真的很急很担心啊!请帮帮忙!谢谢!!!!

[size=4][b] 小卢推荐:一种标定二级对照品的方法[/b][/size]对照品作为实验室(制药行业)一种常用的、重要的试剂,根据其类型,可分为:一级对照品,即为从中国药品生物制品检定所(简称:中检所)购买后直接使用的对照品;二级对照品,由一级对照品标定原料药得到的对照品。由于一级对照品的规格小、价格高、购买周期长的缺点,对于实验室对照品用量大的企业来说,使用二级对照品成了实验室的首选。现在,我就介绍一种标定二级对照品的方法,供大家参考一下。[b]第一,选定样品[/b]一般来说,选择自己生产的原料药价格便宜,不需要外购,且取用方便,是我们的首选。如果我们的生产工艺不好、稳定相差,最好选择外购知名企业的原料药。但要注意,要选择作为对照品的原料药一定是相对其他批次各检验项目都比较好的同一批原料药。[b]第二,标定方法[/b]现以高效液相测定法检测含量为例,来表述其测定方法。由于要严格保证所标定原料药的含量,因此采用3人、3份样品的方法进行测定,即:每个人称取2份对照品、3份样品进行测定;共有3人进行测定。如果有条件,3个人可以选择3台不同的液相色谱仪进行实验。在这里要求2份对照品共进样5针,计算校正因子,并求RSD应小于0.5%,3份样品各进1针,求平均值。方法和要求如下表:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182733_1622024_3.jpg[/img]按照表格内容,由3人得到的3个不同的含量,最后求得均值,即得样品(二级对照品)的含量,并要求3者的RSD≤1%。[b]第三,分装[/b]使用抗生素瓶分装,装量按照每次使用量(如60mg,则装入80-100mg即可)为标准,即使每个抗生素瓶中的对照品只使用一次。这样既能避免对照品被污染,又能使其少吸潮。如果为了节省抗生素瓶,采用大装量,即一瓶中的对照品可以使用多次,那么,建议在使用3-6次后就报废本瓶对照品。因为每次打开瓶口称取对照品都是对该瓶对照品的一次污染,尤其是空气中水分对它的影响,这样会是对照品的含量发生变化,原来的标定也就失去了意义。分装环境:建议在层流罩下进行,严格控制温湿度(建议温湿度:18-24℃,45-65%)。封口步骤:分装后,用橡胶盖盖紧,再用封口膜封好后,用铝盖压实即可。[b]第四,制定有效期[/b]一般比较稳定的样品制定2年,不是很稳定的样品制定1年。但是这个有效期不能超过该样品本身法定的有效期。[b]第五,贴签[/b]制定好了有效期就可以把样品(二级对照品)的标签贴上去了,标签格式如下:[img]http://ng1.17img.cn/bbsfiles/images/2009/11/200911090939_182734_1622024_3.jpg[/img][b]第六,储存[/b]不管原来样品法定的存储温度是多少,都建议保存的温度最好在2-10°,即冰箱中的冷藏温度。根据资料研究,2°是药品的最佳保存温度,因为这个温度下药品的降解速度最慢。[b]第七,复核[/b]我们制定了有效期后,并不是就完成了所有的工作。我们要在有效期的一半时,对二级对照品进行复核,检验方法同本法中第二步骤,所取样品则是从原标定的二级对照品中抽取。如果复核结果没有变化,则继续使用;如果复核结果发生了变化,那就按照复核的含量,从新贴签标示。通过以上7步就完成了对照品的标定工作,大家有什么看法可以回帖说明,我们共同讨论![em09505][em09505](全文完!)
对照品溶液的制备 取补骨脂素对照品和异补骨脂素对照品适量,精密称定,加甲醇制成每1ml各含2ug的溶液,即得。供试品溶液的制备 取本品水蜜丸,研细,取0.5g,精密称定,置研钵中,加硅藻土2g,研匀,转移至具塞锥形瓶中,精密加入甲醇25ml,密塞,称定重量,超声处理(功率250W,频率50kHz)30分钟,放冷,再称定重量,用甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。问题1、对照品溶液制备的过程中,补骨脂素对照品和异补骨脂素对照品是称好放同一量瓶中溶解稀释更好,还是分别配好呢?2、这个供试品的制备中,精密称定后,“置研钵中,加硅藻土2g,研匀,转移至具塞锥形瓶中”,我觉得这个精密称定后,加硅藻土置研钵中研匀会造成样品的损失吧,你再怎么倒也倒不干净吧。。。。我是直接称好放入锥形瓶中,再加2g硅藻土进去,拿个玻璃棒搅一下,不知道这样可不可以???
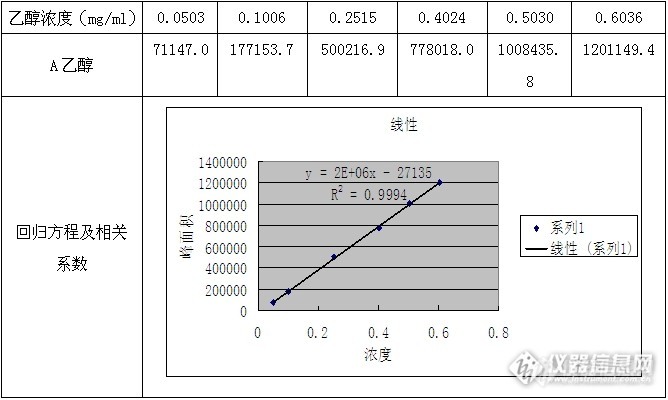
因工作需要,需要对酮咯酸氨丁三醇中的残留有机溶剂乙醇和1,2-二氯乙烷进行方法学研究,乙醇为三类溶剂,药典规定限度为0.5%,1,2-二氯乙烷为一类溶剂,药典规定限度为0.0005%,因为1,2-二氯乙烷的限度较低,在FID检测器下很难检测,故需要用到ECD检测器检测1,2-二氯乙烷。 方法学研究为,方法一,乙醇的检测;方法二,1,2-二氯乙烷的检测。1.1方法概述应用GC外标法对酮咯酸氨丁三醇中的残留有机溶剂乙醇进行定量分析。载气:氮气;检测器:FID。1.2对照品及样品名 称来源批号酮咯酸氨丁三醇样品某医药企业120201乙醇西陇化工股份有限公司11070111.3仪器和仪器参数气相色谱仪型号:岛津公司GC-2010天平型号:梅特勒公司XS105顶空进样器型号:DANI公司 HSS86.50色谱柱类型:DB-624 规格30m×0.53mm×3.0µm 载气:氮气 柱温:50 ℃检测器:FID检测器温度: 250℃;进样口温度: 200℃;流速: 3.0 ml/min;进样量: 1.0ml;分流比: 10:1样品盘平衡温度: 80℃;定量环温度: 90℃;传输线温度: 100℃;样品盘平衡时间: 30min1.4溶液配制对照溶液:准确称取乙醇50mg于100ml容量瓶中,用水稀释定容至刻度,摇匀,精密移取3ml置于20ml顶空瓶中,密封即得对照溶液。准确称取样品0.3g,置于20ml顶空瓶中,加水3.0ml,密封即得供试品溶液。1.5验证内容及结果1.5.1系统适用性试验方法:取酮咯酸氨丁三醇溶残对照溶液,依法连续进样5次,记录乙醇峰面积的相对标准偏差(RSD%)。乙醇峰面积的相对标准偏差RSD应不大于10%,乙醇的理论塔板应不小于10000,乙醇的拖尾因子应不大于1.5。结果:序号12345RSD%A乙醇[/si
http://www.greenherbs.com.cn/bbs/dispbbs.asp?boardid=2&Id=7691601521 二类残留溶剂-1,4-二氧;六环 Residual Solvent Class 2 - 1,4-Dioxane 对照品/标准品1601500 二类残留溶剂-N,N-二甲基酰胺 Residual Solvent Class 2 - N,N-Dimethylformamide 对照品/标准品1601485 二类残留溶剂-N,N-二甲基乙酰胺 Residual Solvent Class 2 - N,N-Dimethylacetamide 对照品/标准品1601463 二类残留溶剂-1,2-二甲氧基乙烷 Residual Solvent Class 2 - 1,2-Dimethoxyethane 对照品/标准品1601441 二类残留溶剂-二氯甲烷 Residual Solvent Class 2 -Methylene Chloride 对照品/标准品1601420 二类残留溶剂- 1,2- 二氯乙烯 Residual Solvent Class 2 - 1,2-Dichloroethene 对照品/标准品1601408 二类残留溶剂-环己烷 Residual Solvent Class 2 - Cyclohexane 对照品/标准品1601383 二类残留溶剂-氯仿 Residual Solvent Class 2 -Chloroform 对照品/标准品1601361 二类残留溶剂-氯苯 Residual Solvent Class 2 - Chlorobenzene 对照品/标准品1601340 二类残留溶剂-乙腈 Residual Solvent Class 2 -Acetonitrile 对照品/标准品1601306 二类残留溶剂-混合物 C Residual Solvent Class 2 - Mixture C 对照品/标准品1601292 二类残留溶剂-混合物 B Residual Solvents Class 2 - Mixture B 对照品/标准品1601281 二类残留溶剂-混合物 A Residual Solvents Class 2 Mixture A 对照品/标准品1601226 一类残留溶剂- 1,1,1- 三氯乙烷 Residual Solvent Class 1 -1,1,1 对照品/标准品1601204 一类残留溶剂- 1,1- 二氯乙烯 Residual Solvent Class 1 -1,1-Dichlo 对照品/标准品1601180 一类残留溶剂- 1,2- 二氯乙烷 Residual Solvent Class 1 -1,2-Dichlo 对照品/标准品1601168 一类残留溶剂-四氯化碳 Residual Solvent Class 1 -Carbon Tetrachloride 对照品/标准品1601146 一类残留溶剂-甲苯 Residual Solvent Class 1- Benzene 对照品/标准品1601102 一类残留溶剂混合物 Residual Solvents Mixture Class 对照品/标准品1601000 利血平 Reserpine 对照品/标准品1600846 瑞格列奈杂质C Repaglinide Related Compound C 对照品/标准品1600835 瑞格列奈杂质B Repaglinide Related Compound B 对照品/标准品1600824 瑞格列奈杂质A Repaglinide Related Compound A 对照品/标准品1600813 瑞格列奈 Repaglinide 对照品/标准品1600121 瑞鲍迪甙 A Rebaudioside A 对照品/标准品1599500 红车轴草提取粉 Powdered Red Clover Extract 对照品/标准品1599000 萝芙碱 Rauwolfia Serpentina 对照品/标准品1598802 树莓酒 Raspberry Alcohol 对照品/标准品1598700 雷尼替丁杂质C Ranitidine Related Compound C 对照品/标准品1598609 雷尼替丁杂质B Ranitidine Related Compound B 对照品/标准品1598507 雷尼替丁杂质A Ranitidine Related Compound A 对照品/标准品1598450 雷尼替丁分离度用混合物 Ranitidine Resolution Mixture 对照品/标准品1598405 盐酸雷尼替丁 Ranitidine Hydrochloride 对照品/标准品1598347 雷米普利杂质D (二酮哌嗪雷米普利)Ramipril Related Compound D 对照品/标准品1598338 雷米普利杂质C Ramipril Related Compound C 对照品/标准品1598323 雷米普利杂质B Ramipril Related Compound B 对照品/标准品1598314 雷米普利杂质A Ramipril Related Compound A 对照品/标准品1598303 雷米普利 Ramipril 对照品/标准品1598201 盐酸雷洛昔芬 Raloxifene Hydrochloride 对照品/标准品1598008 3- 奎宁环基 3-Quinuclidinyl Benzilate 对照品/标准品1597504 奎宁酮 Quininone 对照品/标准品1597005 硫酸奎宁 Quinine Sulfate 对照品/标准品1596807 二水合盐酸奎宁 Quinine Hydrochloride Dihydrate 对照品/标准品1595509 硫酸奎尼丁 Quinidine Sulfate 对照品/标准品1595000 葡萄糖酸奎尼丁 Quinidine Gluconate 对照品/标准品1594506 金鸡纳酸 Quinic Acid 对照品/标准品1594007 喹乙宗 Quinethazone 对照品/标准品1593423 喹那普利杂质 B Quinapril Related Compound B 对照品/标准品1593412 喹那普利杂质 A Quinapril Related Compound A 对照品/标准品1593401 盐酸喹那普利 Quinapril Hydrochloride 对照品/标准品1593004 盐酸米帕林 Quinacrine Hydrochloride 对照品/标准品1592409 槲皮素 Quercetin 对照品/标准品1592227 夸西泮杂质 A Quazepam Related Compound A 对照品/标准品1592205 夸西泮CIV Quazepam CIV 对照品/标准品1592001 恩波吡维铵 Pyrvinium Pamoate 对照品/标准品1589109 丙酮酸 Pyruvic Acid 对照品/标准品1589007 乙胺嘧啶 Pyrimethamine 对照品/标准品1588004 马来酸吡拉明 Pyrilamine Maleate 对照品/标准品
关于印发化妆品中氢化可的松等禁用物质或限用物质检测方法的通知 国食药监保化13号 2012年01月18日 发布 各省、自治区、直辖市食品药品监督管理局(药品监督管理局): 为规范化妆品中禁用物质和限用物质检测技术要求,提高化妆品质量安全,化妆品中氢化可的松等禁用物质或限用物质的检测方法已经国家食品药品监督管理局化妆品标准专家委员会审议通过,现予印发。 附件:1.化妆品中氢化可的松等7种禁限用物质的检测方法 2.化妆品中水杨酸的检测方法 3.化妆品中酮麝香的检测方法 4.化妆品中巯基乙酸的检测方法 5.化妆品中8种邻苯二甲酸酯的检测方法 6.化妆品中4-氨基偶氮苯和联苯胺的检测方法 7.化妆品中苯并芘的检测方法 8.化妆品中4-氨基联苯及其盐的检测方法 9.化妆品中间苯二酚的检测方法 10.化妆品中32种禁限用染料成分的检测方法 11.化妆品中苯扎氯铵的检测方法 12.化妆品中羟基喹啉的检测方法 13.化妆品中过氧化氢的检测方法 14.化妆品中苄索氯铵、劳拉氯铵和西他氯铵的检测方法 15.化妆品中颜料橙5等5种禁用着色剂检测方法 16.化妆品中呋喃香豆素类(三甲沙林、8-甲氧基补骨脂素、5-甲氧基补骨脂素)和欧前胡内酯的检测方法 17.化妆品中补骨脂特征成分补骨脂素、异补骨脂素、新补骨脂异黄酮和补骨脂二氢黄酮的检测方法 国家食品药品监督管理局 二○一二年一月十六日
[size=3][color=#ff6600][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法检薄荷桉油含片中薄荷脑的含量,仪器条件和操作方法如下[/color]:[/size][size=3][b][font=宋体]色谱条件与系统适用性试验[/font][/b][font=宋体] 采用弹性石英毛细管柱(30m×0.32mm×0.25[/font][/size][font=宋体][size=3]um )(PEG)聚乙二醇为固定液,进样口温度220℃,检测器温度为250℃。分流进样。[/size][/font][font=宋体][size=3]程序升温,初始90℃,保持1分钟,每分钟5℃升至170℃。理论塔板数按薄荷脑峰计[/size][/font][font=宋体][size=3]算应不低于10000。薄荷脑与内标物质峰的分离度应大于4。[/size][/font][size=3][b][font=宋体]校正因子测定[/font][/b][font=宋体] [/font][font=宋体]取水杨酸甲酯适量,加丙酮稀释成每1ml含1.0mg的溶液,摇匀,作为内标溶液。另取薄荷脑对照品适量,加丙酮稀释成每1ml含1.0mg的溶液。精密取内标溶液与对照品溶液各5ml,置25ml量瓶中,加丙酮至刻度,摇匀,精密吸取1μl注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],计算效正因子。[/font][/size][size=3][b][font=宋体]测定法[/font][/b][font=宋体] [/font][font=宋体]取本品20片,精密称定,研细,取粉末适量(约10片量),精密称定,置具塞瓶中,精密加入丙酮25ml,振摇30分钟,滤过,精密量取续滤液10ml与内标溶液5ml,置25ml量瓶中,加丙酮至刻度,摇匀,精密吸取1μl注入[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],测定,计算,即得。我按上述方法操作,用的机子是岛津GC-14C,柱子是0.53×30的PEG-20M,各气体流量也正常,但在做时发现只有水杨酸甲酯的内标峰出来,保留时间大约在7分钟的样子,而进薄荷脑对照品怎么没有薄荷脑的峰呢?进样品同样没有薄荷脑的峰,只有内标峰,请大家帮帮忙分析一下原因出在哪里?[/font][/size]
关于HPLC主成分自身对照法检查有关物质时检测波长确定的讨论审评二部张玉琥有关物质检查,包括对产品中残留合成原料、中间体、副产物及可能的降解产物的检查,是控制药品质量的重要指标,目的是检查药品中所含的上述杂质是否符合安全性的要求,同时也是药品稳定性评价中需重点考察的项目。有关物质检查常用的方法之一是HPLC主成分自身对照法(紫外检测器),即将HPLC色谱图中杂质峰面积与主成分自身对照液峰面积进行比较,以确定杂质限度是否合格。采用此方法时确定的检测波长是否合理直接影响到方法的可行性,因此检测波长的选择是方法学研究的重要内容。在审评中发现一些申报单位在采用HPLC主成分自身对照法检查有关物质时直接或间接地以主成分的最大吸收波长作为检测波长,由于有关物质检查的对象是杂质,若将主药的最大吸收波长确定为检测波长,则杂质在此波长下的吸收可能偏低,某些杂质甚至无吸收,这样会造成对杂质含量的低估甚至漏检,从而不能反映产品的真实质量,影响了对品种质量可控性及稳定性的评价。在有关物质检测波长确定方面,申报资料中比较常见的做法有:1.直接将主药的最大吸收波长选作检测波长。2.简单地套用含量测定的色谱条件。在HPLC法进行含量测定时,为提高方法的灵敏度,降低干扰,往往选用主成分的最大吸收波长作为检测波长。若套用含量测定的色谱条件,实际仍是以主药的最大吸收波长作为有关物质检测波长。3.以样品进行破坏性试验(酸、碱、热、光照、氧化等)后的溶液做紫外扫描,将扫描图谱中最大吸收波长确定为有关物质的检测波长。因破坏性试验后溶液中存在尚未破坏的主药、降解产物、辅料等,此溶液的紫外吸收为各成分紫外吸收的加和,并不能反映降解产物的紫外吸收特性。由于未破坏主药所占比例较大,故破坏性试验后溶液的最大吸收波长一般仍为主药的最大吸收波长。采用HPLC主成分自身对照法检查有关物质,其前提之一是需检查的杂质与主成分在确定的检测波长下应有相近的紫外吸收(响应值接近),选择检测波长时需对产品中可能存在的杂质(合成原料、中间体、副产物以及降解产物)的紫外吸收特性进行研究。已知杂质的紫外吸收特性可采用对其流动相溶液直接进行扫描的方法考察,未知杂质(如未知降解产物等)可通过二极管阵列检测器考察其紫外吸收情况,根据各主要杂质及主成分的紫外吸收特性,选取响应值基本一致的波长作为有关物质的检测波长。若对不同杂质难于找到均适宜的检测波长,可考虑选择在不同波长下分别测定,也可考虑采用加校正因子的主成分自身对照法。只有经试验研究确认主成分的最大吸收波长符合有关物质检查对测定波长的要求时,为方便操作,可选作有关物质的检测波长,以与含量测定的色谱条件一致。另外,HPLC主成分自身对照法检查有关物质比较适用于对微量杂质总量的控制,也可用于单个杂质的限度(一般不超过0.5%)控制。对于具有明确归属的已知杂质,建议采用杂质对照品法进行检查。对于有毒有害杂质,更应采用质对照品法单独测定,并制定严格的限度。
问题1:内毒素日常检查中:在加入鲎试剂前的供试品对照溶液(0.1ml)药典规定是含内毒素浓度2λ的。假如MVD=2,C=10mg/ml,λ=0.25EU/ml。那么应该用1EU/ml(也就是4λ)的内毒素溶液对半稀释浓度为10mg/ml的供试品溶液,终浓度才是含量为0.5EU/ml(2λ)内毒素、供试品为MVD浓度的供试液的供试品对照液!!! 问题2:还有如果供试品为注射用水,假如L=0.25,那么鲎试剂的灵敏度最少要用λ=0.125的;如果用λ=0.25的,因为供试品液(0.1ml)加入鲎试剂(0.1ml)后,稀释了1倍,假如结果为+,那么说明供试品的内毒素限制大于0.5EU?而不是大于0.25EU吧?
【摘要】 目的 研究麝香祛痛搽剂中人工麝香质量的控制方法。方法采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法对处方中的人工麝香药材进行定性鉴别 用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法对处方中人工麝香的有效成分麝香酮进行定性测定。结果供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高,同时更能保证检测结果的真实性、准确性、重复性,阴性无干扰,专属性强。结论该方法可用于麝香祛痛搽剂中麝香酮质量的控制。【关键词】 麝香祛痛搽剂 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法 麝香酮麝香祛痛搽剂原收载于《中国药典》2005年版Ⅰ部,根据2010年版《中国药典》计划任务表要求,本文将处方中的麝香修订为人工麝香,因为人工麝香为贵重药材,所以应对麝香祛痛搽剂中人工麝香制订质量控制方法,本文采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法对处方中的人工麝香药材进行定性鉴别,供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高,同时更能保证检测结果的真实性、准确性、重复性,阴性无干扰,专属性强,结果较满意。1 仪器与试药[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]Agilent6890型(FID检测器) Agilent色谱工作站 HP-5弹性石英毛细管柱(柱长30 m,内径0.25 mm,膜厚度0.25 μm) 聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm) 麝香祛痛搽剂样品: 武汉马应龙药业集团股份有限公司产品(批号080402,080403,080413),襄樊隆中药业有限责任公司产品(批号 080101,080201,080401),绍兴华通制药有限公司产品(批号C03A0753,C03A0809),李时珍医药集团有限公司产品(批号 2009020001)。麝香酮对照品,中国药品生物制品研究所产品(批号110719-200512) 无水乙醇为分析醇。2 方法与结果2.1 色谱条件色谱柱为HP-5弹性石英毛细管柱(柱长30 m,内径0.25 mm,膜厚度0.25 μm) 聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm) 柱温:程序升温:初温130℃,保持5 min后,每分钟升高0.8℃至180℃,保持2 min后,再以每分钟升高20℃至220℃保持5 min 检测器为FID检测器 检测器温度:250℃ 进样口温度220℃ 流速1.0 ml/min 理论板数按麝香酮计算应不低于20 000。2.2 色谱条件的确定2.2.1 色谱柱的确定取供试品(绍兴华通制药有限公司,批号:C03A0753)按供试品制备方法制备得供试品溶液,用聚乙二醇(PEG)-20M毛细管柱(柱长 30 m,内径0.32 mm,膜厚度0.5 μm)进样。结果表明,用聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm)能收获较好的分离效果,得到准确高效的分析结果。故选用:聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm)。2.2.2 程序升温条件的确定将“2.3.1”项下制得供试品溶液按程序升温:初温130℃,保持3 min后,每分钟升高1.5℃至180℃,再以每分钟升高20℃至220℃保持5 min 流速1 ml/min。结果表明,在该方案下,供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高。通过试验,确定色谱条件为:色谱柱:聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm) 检测器为FID检测器 进样器温度:220℃ 检测器温度:250℃ 分流进样(分流比1∶1) 程序升温:初温130℃,保持5 min后,每分钟升高0.8℃至180℃,保持2 min后,再以每分钟升高20℃至220℃保持5 min 流速1 ml/min 进样量:1 μl。2.3 溶液的制备2.3.1 供试品溶液的制备针对麝香酮的脂溶性,采用石油醚(30~60℃)提取的方法,这样组分的转移率较高,同时操作简便。但是因为本品麝香酮的处方量过低 必须进行富集的过程。因为样品为50%的乙醇制剂,与石油醚(30~60℃)会发生混溶,因此加入4倍量的水,再置于分液漏斗中用石油醚(30~60℃)提取2次,100 ml/次,石油醚(30~60℃)液自然挥干,可使麝香酮成分不会损失。取本品50 ml,加水200 ml,摇匀。置于分液漏斗中,用石油醚(30~60℃)提取2次,100 ml/次。合并石油醚,自然挥干。残渣用无水乙醇2 ml溶解,取上清液作为供试品溶液。2.3.2 对照品溶液的制备取麝香酮对照品,加无水乙醇制成每毫升含0.1 mg的溶液,作为对照品溶液。2.3.3 阴性对照溶液的制备按处方量制备不含麝香酮的阴性样品,按供试品溶液制备方法制备阴性对照溶液。2.4 专属性考察取对照品溶液、供试品溶液、阴性对照溶液,分别依法测定。在该色谱条件下,在与麝香酮对照品保留时间相同的位置不出现色谱峰,表明其他成分对麝香酮的测定无干扰。结果见图1~3。2.5 样品测定按正文所述方法,对各厂家各批次样品进行检验,结果均呈正结果。3 讨论因为麝香酮具有脂溶性,采用石油醚(30~60℃)提取的方法,这样组分的转移率较高,同时操作简便。但是因为本品麝香酮的处方量过低,必须进行富集的过程。因为样品为50%的乙醇制剂,与石油醚(30~60℃)会发生混溶,因此加入4倍量的水,再置于分液漏斗中用石油醚(30~60℃)提取,可使麝香酮成分不会损失。但是富集后导致杂质峰较多,用平温或较快的升温速率均达不到较好的分离效果,因此经过摸索将升温速率定为每分钟升高0.8℃,在该方案下,供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高,同时更能保证检测结果的真实性、准确性。【参考文献】[1] 国家药典委员会.中国药典,Ⅰ部[s].北京:化学工业出版社,2005.[/s]
[B][center]药物中杂质的来源及杂质限量检查[/center] [/B]药物只有合格品与不合格品;一般化学试剂分为4个等级(基准试剂、优级纯、分析纯、化学纯) [B]药物中一般杂质检查 [/B][B]氯化物为一指示性杂质。[/B] 通过对氯化物的控制,可同时控制与氯化物结合的一些阳离子以及某些同时生成的副产物。可从氯化物检查结果显示药物的纯度,间接考核生产、贮藏过程是否正常。 1. 原理 药物中微量的氯化物在硝酸酸性条件下与硝酸银反应,生成氯化银的胶体微粒而显白色浑浊,与一定量的标准氯化钠溶液在相同条件下产生的氯化银浑浊程度比较,判定供试品中氯化物是否符合限量规定。 Ag+ + Cl- → AgCl ↓ [B]硫酸盐检查法 [/B] 1. 原理 药物中微量的硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成硫酸钡的微粒而显白色浑浊,与一定量的标准硫酸钾溶液在相同条件下产生的硫酸钡浑浊程度比较,判定供试品中硫酸盐是否符合限量规定。 [B]铁盐检查法 [/B]硫氰酸盐法 巯基醋酸法 砷盐检查法 1. 古蔡氏法 1. 原理 金属锌与酸作用产生新生态的氢,与药物中微量砷盐反应生成具挥发性的砷化氢,遇溴化汞试纸产生黄色至棕色的砷斑,与同条件下一定量标准砷溶液所生成的砷比较斑,判断砷盐的含量。 [B]硒、氟及硫化物检查法 [/B]1. 氧瓶燃烧法 适用于以共价键结合的卤素、硫、硒的有机药物。 本法系将有机药物防入充满氧气的密闭燃烧瓶中进行燃烧,将燃烧所产生的欲测组分吸收于适当的吸收液中,然后根据欲测组分的性质,选用合适的分析方法进行鉴别、检查或含量测定。 [B]注意事项及讨论 [/B]1. 根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 2. 测定氟化物时应改用石英燃烧瓶。 1. 硒检查法 (1). 操作方法 样品与对照品液,调节Ph2.0±0.2,加盐酸羟胺,二氨基萘,比色。 [B]硫化物检查法 [/B] 方法同砷盐检查第一法,不装醋酸铅棉花,以醋酸铅试纸代替溴化汞试纸。 标准液取1ml 5/ml [B]澄清度检查法 [/B]将一定浓度的供试品溶液与浊度标准液分别置于配对的比浊用玻璃管,同置黑色背景上,在漫射光下观察。浊度标准液 硫酸肼与乌洛托品溶液混合分五个等级,未超过0.5等级即为澄清。BP98规定未超过1等级即为澄清。 [B]溶液颜色检查法 [/B]CHP2000 [B]1. 比色法[/B] 色调标准贮备液 黄色液 重铬酸钾液(BP98用氯化铁) 红色液 氯化钴液 蓝色液 硫酸铜液 配成各种色调色号标准比色液共50种。 [B]2. 分光光度法 [/B] [B]易碳化物检查法 [/B]检查药物中含有的遇硫酸易碳化或易氧化而呈色的有机杂质。 对照品液 样品液 加硫酸5后,加供试品。 [B]炽灼残渣检查法[/B] 取供试品1.0~2.0g或个药品项下规定的重量,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全碳化,放冷至室温;除另有规定外,加硫酸使湿润,低温加热至硫酸蒸气除尽后,在700~800炽灼使完全灰化,移至干燥器内,放冷至室温,精密称定,再在700~800炽灼至恒重,即得。残渣限量一般为0.1~0.2% 一般应使炽灼残渣量为1~2mg 若需将炽灼残渣留作重金属检查时,炽灼温度必须控制在500~600。 [B]干燥失重测定 [/B]1. 常压恒温干燥法 2. 干燥剂干燥法 3. 减压干燥法 [B]水分测定法 [/B][B]费休氏法 [/B] 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。 [B]甲苯法[/B] 在加热状态下,甲苯夹带着水分蒸出,收集蒸出的水分测定。 [B]药物中特殊杂质检查 [/B] [B]一、物理法 [/B] [B]二、化学反应法 [/B](一)容量分析法 (二)重量分析法 (三)比色法和比浊法 [B]三、色谱法 [/B]1.纸色谱法 薄层色谱法 TLC是药典中最常用的特殊杂质限量检查方法。 1.在一定供试品及检查条件下,不允许有杂质斑点存在 2.以待测杂质对照品检测 3.将供试品稀释到适当浓度作为杂质对照品溶液 4.选用质量符合规定的与供试品相同的药物作为杂质对照品 [B]高效液相色谱法 [/B] [B][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 [/B] 1.面积归一化法 2.主成分自身对照法 3.内标法测定 4.内标法加校正因子法 5.外标法 有机溶剂残留量测定法 [B]分光光度法 紫外分光光度法 比色法 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/B]
做一个甾体皂苷的含量测定,已经做第4次了,问题始终没解决,遂请教。具体要求如下:色谱条件要求:C8的柱子;流动相是乙腈-水(30:70);检测波长210nm。样品处理:取一定量,加75%乙醇超声10分钟,放冷,补足减失重量,滤过,即可。 对照品也是用75%乙醇溶解。测试经过如下:有同一厂家同一品种不同批号两批样品,用C8的柱子做,计算了下,含量合格,但峰实在太难看。怕峰形太差影响结果的准确性,但只有一根这C8的柱子,只好换根C18的柱子,调整流动相试试;峰形很好,进了一针其中一批的样品,计算了也合格,就编个序列继续做下去了。第四天才发现另一批不合格,只好第五天返工。第二次,问题出现了。返工时,同样的仪器,色谱柱,色谱条件,用原来配的对照品溶液,重新处理样品,不合格的那一批还是不合格,结果比上次测的还低一点,合格的那批也不合格了。又另配了对照品溶液,与之前配的浓度相差不大,但峰面积小多了。但是之前配的对照品峰面积反而变大了,估计是因为流动相微调,把之前没分开的小峰分开了,不至于斜着积分的原因吧。怀疑过对照品峰面积小,是不是冷冻(对照品要求冷冻保存哈)后没放至室温,直接称,导致称不准。样品怀疑过的原因 A:是不是不合格那一批取样量大了,浓度高了,导致过饱和,未全溶 B:样品是不是未混匀,取样代表性不够 C:超声时间,功率是不是有问题,是不是超声发热导致样品降解(对照品要冷冻保存嘛)E:是不是第二次配的溶样溶剂有问题第三次做,更蹊跷了。之前怀疑的样品的原因都排除了。减少,增加取样量;混匀样品;超声不同时间,用不同超声设备(不同功率);超声中换水,防止温度升高,水浴上回流处理样品;重配75%乙醇几次,用无水乙醇配也试了,还换用甲醇,样品溶液居然都没有目标峰。但之前两次配的对照品溶液峰高变化不大,高的还是高,低的还是低。第四次,就是今天。又重配了对照品溶液,换甲醇(色谱纯)溶解,居然对照品也没峰了,用75%乙醇溶解也是。之前配的对照品峰高还是老样子。将第一次配的对照品溶液跟这次提取的测不到目标峰的样品溶液混合,也能出目标峰。这根柱子换走另一个对照品测试,峰好得很。再换另一根柱,走样品和对照品,还是老样子,之前能出的还出,出不了的还是还是出不了。总结:一共配了三次对照品,浓度差异不大,但峰面积在变小,第三次就没了。样品也是,第一次合格的,第二次提取也不合格了,以后干脆目标峰也不出了。 分析:应该不是目标物不稳定的原因,因为第一次配的对照品,隔了一周,峰高依然变化不大,更何况新配的对照品溶液。 几次都是我操作,对照品溶液都超声助溶过,应该也是溶了的。而且用紫外扫过,稀释后吸收值会降低,虽然最大吸收波长在约205nm。其实用甲醇比用75%乙醇溶解要好些,几乎振摇就能溶,用75%乙醇必须稍超声一下。[/fo
今天我把昨天的两份对照品储备液分别稀释成对照品溶液,顶空进样,但是第一份对照品溶液待测组分未出峰,第2份对照品溶液出峰正常,我想问下隔24小时以后再用待测组分都会挥发完全吗?为什么第二份正常出峰呀?对照品溶液里面的组分分别是乙醇,乙酸乙酯,丙酮,二氯甲烷,水为溶剂
标准品:即标准物品,作为一种衡量标准。分类:化学计量标准品、冶金标准品、药检标准品。 对照品:指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。国家标准品及生物参考品系指用于鉴别、检查含量或效价测定的标准物质,其制备与标定应符合“生物制品国家标准物质制备和标定规程”要求,并由国务院药品监督管理部门指定的机构分发。企业工作标准品或参考品必须经国家标准品或参考品标化后方能使用。在中国药典凡例中是这样定义的:标准品是用微生物方法测定含量时参照的标准,而对照品是由仪器分析或其它分析方法测定含量的标准,二者都是由国家指定部门中国生物制品检定所提供的。
日本取消对中国产养殖杂色鲍中呋喃唑酮的强化监控检查2014年8月25日,日本厚生劳动省医药食品局食品安全部监视安全课发布食安输发0825第1号:取消对中国产养殖杂色鲍中呋喃唑酮、羽衣甘蓝中六氯苯、冬瓜中甲霜灵和精甲霜灵的强化监控检查。 根据2014年度进口食品等的监控检查计划,按2014年3月28日发布的食安输发0328第10号(最终修正:2014年8月6日发布的食安输发0806第5号),对食品实施监控检查。 此次,根据过去一年的检查结果,取消对中国产养殖杂色鲍中呋喃唑酮、羽衣甘蓝中六氯苯、冬瓜中甲霜灵和精甲霜灵。
植物成分标准品、对照品、单体、http://hi.baidu.com植物提取物http://hi.baidu.com植物提取物标准品1加兰他敏、石蒜碱,丹皮酚 Paeonol、光甘草定、丹参酮系列(丹参酮ⅡA、隐丹参酮、丹参酮Ⅰ),齐墩果酸,白黎芦醇(RESV),叶黄素、红景天苷、原花青素B2 Procyanidin B2金丝桃苷,金丝桃素、辣椒素、Asiaticoside(积雪草苷)Astragaloside IV(黄芪甲苷)系列等等。 联系方式:jiehua0501@yahoo.com.cn 刘 推荐,请告电话联系方式。1到2天回复。 qq37144588(请注明事由)。MSN:jiehuahua0501@hotmail.com 13482587565 植物提取物:单体白黎芦醇、绿原酸、加兰他敏、石蒜碱、盐酸青藤碱,二十八烷醇,丹皮酚,、丹参酮ⅡA,葛根素,番茄红素、莽草酸、5-HTP(五羟色氨)、青蒿素、二氢杨梅素、獐牙菜苦苷,鬼臼毒素,冬凌草甲素,熊果酸等等。以上产品提供20%-99%的产品,大量供应,包装大小根据您的需要。提取物:葡萄籽提取物(原花青素opc95%)、茶多酚(tp),红景天(甙)提取物,枇杷叶提取物,锯叶棕提取物,葛根提取物等,以及各种比例提取物。 标准品2木犀草素,甘草酸单铵,异欧前胡素,Vindoline(文多灵),Rosmarinic acid(迷迭香酸),Sailkosaponins D(柴胡皂苷D),Imperatorin(欧前胡素),Isoimperatorin(异欧前胡素),Vinblastine sulfate(硫酸长春碱),肉苁蓉苷A,芦荟大黄素,β-谷甾醇,秦皮甲素,Cichoric acid(菊苣酸),Mangiferin(芒果苷),α-Cyperone(α-香附酮),1-Deoxynojirimycin (1-脱氧野尻霉素),Sarsasapogenin(知母皂苷元),Nitidine Chloride(氯化两面针碱),10-Deacetylbaccatine III,Buddleoside(蒙花苷),Silybin(水飞蓟宾),6-Gingerol(6-姜酚),Catharanthine(长春质碱),Syringin(紫丁香苷),人参皂苷Rb3,三七皂苷R1,柴胡皂苷A,五味子丙素,佛手柑内酯,蛇床子素,白花前胡甲素,羽扇豆醇,Praeruptorin A,柴胡皂苷C,白头翁皂苷B4,积雪草苷,豆腐果苷,五味子酯甲,五味子甲素,大黄酸,五味子乙素,五味子醇甲,苍术素, Pseudohypericin(伪金丝桃素), 苍术素醇,安五脂素,细辛脂素,苦杏仁苷,Polydatin(虎杖苷),3,29-二苯甲酰栝蒌仁三醇,大黄素甲醚,薄荷醇,细辛脂素,鬼臼毒素,丁香苷,冬绿苷,豆腐果新苷A,B,C。menisdaurin,3,29-二苯甲酰栝蒌仁三醇,表木栓醇,柴胡皂苷D,毛花洋地黄苷C,黄芪皂苷II,对羟基苯甲酸乙酯,白花前胡丙素,桃叶珊瑚甙,胡黄连苦苷I,和厚朴酚,白花前胡丁素,秦皮乙素,没食子酸,芍药甙,补骨脂素,岑酮,白花前胡素E,胡黄连苦苷II,阿魏酸,龙胆苦苷,丹参素钠,水杨苷,木香烃内酯 Luteolin、穿心莲内酯,右旋比扣扣灵碱,槐果碱,乌头碱,槐胺碱青藤碱、姜黄素系列,靛玉红,豨莶精醇,异古伦宾 银杏系列(白果内酯、银杏内酯A、银杏内酯b)虎杖甙、、芹菜素、茄尼醇、芥子碱硫氰酸盐,常春藤皂苷元,木犀草素, 虫草素、EGCG(姜黄素 Curcumin,去甲氧基姜黄素 Curcumin2,去二甲氧基姜黄素 Curcumin 3、阿魏酸 Ferulic acid、积雪草苷,豨莶精醇 Asiaticoside、柴胡系列柴胡皂甙 A Saikosaponins A、柴胡皂甙 D Saikosaponins D、去氢木香内酯,异土木香内酯,土木香内酯,番泻苷A Sennoside A栀子苷 Caryptoside、山奈酚 Kaempferol、Hyperoside、根皮苷Phloridzin、氢溴酸槟榔碱Arecoline Hydrobromide、2-hydroxyeupatolide、阿卡宁 Alkannin、Salidroside、肉桂醇苷 Rosavin、酪醇 Tyrosol、奇任醇,辣椒素系列、苍术内酯Ⅲ,二氢辣椒素 Dihydrocapsaicin、番泻苷A,二氢辣椒素 Dihydrocapsaicin、乌药醚内酯,吉马酮,雄烯二酮 Androstenedione、10-脱乙酰巴卡丁 III10-DAB 10 III麻醉椒苦素 Methysticin 枸橼酸血根碱,醉椒素Kavain 二氢醉椒素Dihydrokavain吴茱萸碱Evodiamine1-乙酸基-5-去乙酰基-巴卡亭 I,吴茱萸次碱Rutaecarpine 水飞蓟宾 Silybin石衫碱甲 Huperzine-A哈巴饿甙,二氢丹参酮,Harpagoside水杨甙 20-羟基蜕皮甾酮β- 蜕皮甾酮吲哚- Ecdysone甘草酸 Glycyrrhizic acid、异鼠李素Nordihydrocapsaicin、N –Vanillylnonanamide、鸢尾苷,野黄芩苷,乙氧基血根碱,吲哚醇,乙氧基白屈菜红碱N –Vanillyldecanamide、秋水仙碱,白藜芦醇甙Polydatin、、白鲜碱dictamnine、山萘素、Kaempferol、异鼠李素Isorhamnetin、花椒毒酚、淫羊藿苷Icariin、染料木素,芝麻素, 川芎嗪,羟基吴茱萸碱,紫草氰苷,新橙皮甙, 橙皮甙,柚皮甙,橙皮甙二氢查尔酮、柚皮甙二氢查尔酮、东方唐松草苷、苦参碱、茵芋苷,胡黄连苷Ⅰ、氧化苦参碱、贝母甲素,贝母乙素,青蒿素、辛弗林、apiosylskimmin,格列风内酯、高良姜素,芝麻素, 黄芪甲苷、蜕皮激素, 阿马碱, 莽草酸,熊果酸、EGCE。穗花双黄酮,番茄红素(90~95%)5-HTP,大黄素、蜕皮激素(20-β-蜕皮甾酮)阿魏酸,黄芪甙,豆蔻明,甘草酸二铵盐,异甘草素,原花青素B2 Procyanidin B2、丹参酮ⅡA,番茄红素,绿原酸,叶黄素,钩腾碱,水杨甙,灵芝酸, 山奈酚-3-O-芸香糖苷、阔叶冬青苷G、迷迭香酸Rosmarinic, 齐墩果酸Oleanolic acid, 刺芒柄花素Formononetin( 98%美国进口/5mg) , 鞣花酸 Ellagic acid, 熊果酸 Ursolic acid, 连翘苷 phillyrin, 氢溴酸槟榔碱(97%)Arecoline Hydrobromide, 牛蒡子苷Arctiin, 栀子苷 Caryptoside,大黄素甲醚 Physcion, 大黄酚 Chrysophanol, 芦荟大黄素 Aloe_emodin, 金丝桃苷, 薯蓣皂苷元, 甘草酸单胺盐, 熊果苷, 梣酮、人参皂甙系列,ROSAVIN,五味子醇甲,五味子乙素,扁蓄苷、木香烃内酯、五味子甲素,柴胡皂甙A,柴胡皂甙B,银杏内酯A,Ginkgolide A,银杏内酯B,GinkgolideB,去二氢甲氧基姜黄素,去甲氧基姜黄素 ,Curcumin2,, 18-β甘草次酸 18β-Glycyrrhetinic acid ,甘草次酸,五味子酯A GomisinA,羟基吴茱萸碱、 五味子酯N Gomisin N ,白果内酯 Bilobalide,乙酰紫草素Acetylshikonin,苦杏仁苷Amygdalin、牛蒡子苷、苍术内酯Ⅲ、吴茱萸碱、异丁酰紫草素素,甘草酸单胺盐DENG,枸橼酸血根碱、儿茶酸(+)-Catechin,紫草素 Shikonin,咖啡酸、乌药醚内酯、柯里拉京,苦杏仁苷,辣椒素,龙胆苦甙,氯化两面针碱,夏无碱、落叶松树脂醇-吡喃糖苷,马兜铃酸,马钱素,马钱子碱,吲哚醇、拟人参皂苷F11,尿囊素,牛蒡子甙,欧前胡素,七叶甙,秋水仙碱,肉桂酸,异土木香内酯、三尖杉宁碱,山姜素,山奈酚,山奈素,麝香草酚,石杉碱甲,酸枣仁皂苷A,酸枣仁皂苷B,乙氧基白屈菜红碱、阿马碱、天麻素,甜菜碱,土大黄甙,乌索酸,五味子醇甲,西贝碱,延胡索乙素,左旋紫草素,对-香豆酸,枸橼酸血根碱、原人参二醇,南蛇藤素,雷公藤内酯A、雷公藤红素、番泻苷A、槐角苷,岩白菜素,千金子二萜醇,豨莶精醇、靛蓝,槐角苷、斑蝥素,羽扇豆醇,右旋比扣扣灵碱、异土木香内酯、羟基积雪草苷,鸢尾苷,8-Gingerol(8-姜酚),10-Gingerol(10-姜酚)等等。产品在不段更新中。jiehua0501@yahoo.com.cn 提供大部分产品(液相色谱hplc)检测条件(参考)。供应标准品,g级供应可获更大优惠。部分产品可以kg级供货(等)-价格有竞争力,多种规格。以上可以部分产品可以大量生产供货,还有多种的含量与规格。有需要,欢迎你联系。由于种类太多,请您先有邮件和我联系,标明你要的数量等具体要求,我会在最短的时间给你回复,推荐联系方式:jiehua0501@yahoo.com.cn qq: 37144588或MSN:jiehuahua0501@hotmail.com 13482587565







 我要推广仪器
我要推广仪器
 下载APP
下载APP