我们检测公司做的7种盐酸左旋咪唑的核磁图,提供给初学者解谱练习用。仪器为 布鲁克400,溶剂为重水。1D的有:氢谱、碳谱、dept1352D的有:HMBC HMQC H-HCOSY NOESY
近年来乳制品的质量安全问题时有发生,给消费者的生命安全和财产安全带来巨大损害。默克密理博作为色谱领域的鼻祖,一直在为广大消费者的食品安全分析检测贡献自己的一份力量。默克密理博的应用团队也不断为客户开发出安全可靠的分析检测方法。该贴将给大家分享牛奶中左旋咪唑残留量的测定方法。
大家有知道这些代谢物哪里能提供?CL 288511 /CL 182704是什么编号呀? 咪唑乙烟酸咪唑乙烟酸 的代谢物 CL 288511 咪唑乙烟酸 的代谢物 CL 182704
分享一个检测标准,GB29681-2013 《食品安全国家标准 牛奶中左旋咪唑残留量的测定 高效液相色谱法》 。本标准适用于牛奶中左旋咪唑残留量检测,检测限为2.5μg/kg,定量限为5μg/kg。
2013年10月16日,农业部网站发布消息称,《牛奶中左旋咪唑残留量的测定 高效液相色谱法》等29项标准业经食品安全国家标准审评委员会审定通过。并经农业部、卫生和计划生育委员会审查批准,发布为中华人民共和国食品安全国家标准,自2014年1月1日起实施。 迪马科技在国标基础上对牛奶中左旋咪唑残留量的测定方法进行了优化,取得了优异的回收率结果和重现性,下面为详细解决方案,与大家分享
如题,我按照国标GB/T 22994-2008做左旋咪唑。发现问题如下1、换了两个样品都发现含有少量待测物,含量范围在0.1~2.0ppb。左旋是否普遍存在于奶制品中?那我方法检测限应该怎么做呢?2、样品空白加标回收效果还好,但样品加标回收率很低,是否表示基质对目标物的离子化有影响?
GB 29681-2013 食品安全国家标准 牛奶中左旋咪唑残留量的测定 高效液相色谱法
鄙人最近在做饲料中左旋咪唑的检测,想问各位大侠有没有相关国标或者方法可以参考?我用了GB/T 20766的方法尝试,SCX净化(艾杰尔500mg/3mL),洗脱过程与国标基本一致,用空白试剂溶液配标(由于基质类型比较多,不想做空白基质溶液配标)。样品前加标回收率5%左右。样品后加标回收率10%,说明样品杂质未除净另空白加标也只有10%的回收率,不知道是什么原因?有没有同行也在做相关测试,相互讨论一下。
现本人正在用HP6890N-5973i进行牛奶中左旋咪唑残留[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质[/color][/url]检测,请教哪位做过请提供具体方法,谢谢!包括前处理,试剂,仪器条件,检测标准方法等。请email给我:yzbjm@163.com
如题,求左旋肉碱盐酸盐饲料级标准。请各位老师,哥哥姐姐弟弟妹妹们帮忙。非常感谢!
[size=1][size=3][font=Arial]SPE-HPLC[/font][/size][size=3][font=宋体]法测定土壤中咪唑乙烟酸的残留量[/font][/size][/size][size=1][font=宋体]摘要:建立了[/font][font=Times New Roman]SPE[/font][font=宋体]净化和[/font][font=Times New Roman]HPLC[/font][font=宋体]法测定土壤中常用除草剂咪唑乙烟酸的残留。土壤样品用[/font][font=Times New Roman]0.1 mol/L[/font][font=宋体]的醋酸铵与氨水缓冲液[/font][font=Times New Roman](pH 10)[/font][font=宋体]超声波提取,提取液经酸化后用[/font][font=Times New Roman]200 mg/3mL C18 SPE[/font][font=宋体]柱净化,浓缩,乙腈定容后,供反相高效液相色谱检测,外标法定量。[/font][font=Times New Roman] [/font][font=宋体]添加回收率为[/font][font=Times New Roman]77.2%~94.9%[/font][font=宋体],咪唑乙烟酸的最小检出量为[/font][font=Times New Roman]5.2×10[/font][font=宋体]-[/font][font=Times New Roman]5 mg[/font][font=宋体],经计算可知,咪唑乙烟酸在土壤中的最小检出质量分数为[/font][font=Times New Roman]0.2 mg/kg[/font][font=宋体]。[/font][font=Times New Roman] [/font][font=宋体]该方法大大减少了有机溶剂的使用,方便快速,结果准确可靠,适合一般实验室操作。[/font][/size][size=3][font=宋体][size=1]关键词:咪唑乙烟酸;固相萃取;高效液相色谱;土壤[/size][/font][/size]
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心
下記農薬について、食品中の残留基準を設定・イソキサフルトール(Isoxaflutole,异恶唑草酮,用途:除草剤)・イマザピック(Imazamethapyr,甲基咪草烟; 甲咪唑烟酸,用途:除草剤)※・エタルフルラリン(Ethalfluraline,丁氟消草,用途:除草剤)・フェンブコナゾール(Fenbuconazole,腈苯唑,用途:殺菌剤)・フロニカミド(FLONICAMID,氟啶虫酰胺,用途:殺虫剤)・ぺノキススラム(Penoxsulam,五氟磺草胺,用途:除草剤)・マンジプロパミド(Mandipropamid,双炔酰菌胺,用途:殺菌剤)※今回基準値を設定するイマザピックはイマザピックアンモニウム塩として暫定基準が設定されていたため、イマザピックアンモニウム塩として経過措置を設定しているが、各種試験はイマザピックを用いて実施されていること、海外における基準値はイマザピックの残留量を考慮して設定されていることから、今後は告示においては、イマザピックアンモニウム塩は「イマザピック」とする。・フェンブコナゾール:かき等6食品・フロニカミド:小豆等27食品・ぺノキススラム:ぶどう等5食品・マンジプロパミド:だいこん類(ラディッシュを含む。)の葉等7食品・イソキサフルトール:米(玄米をいう。)等7食品・イマザピック:豚の筋肉等17食品・エタルフルラリン:きゅうり(ガーキンを含む。)等9食品・フロニカミド:羊の筋肉等15食品・イソキサフルトール:とうもろこし等19食品・イマザピック:牛の脂肪等9食品・フェンブコナゾール:みかん等10食品・フロニカミド:クレソン等32食品・マンジプロパミド:はくさい等20食品≪施行・適用期日≫ 平成24年6月14日 ※ただし、下記の農薬等ごとに掲げる食品に係る残留基準値については、 平成24年12月14日から適用。 ◆イソキサフルトール 米、小麦、大麦、ライ麦、とうもろこし、そば、その他の穀類、 その他のスパイス、豚の肝臓、その他の陸棲哺乳類に属する動物の肝臓、 乳、鶏の卵及びその他の家きんの卵 ◆イマザピック 豚の筋肉、豚の脂肪、豚の肝臓、豚の腎臓、豚の食用部分及び乳 ◆エタルフルラリン きゅうり、かぼちゃ、しろうり、すいか、メロン類果実、まくわうり、 その他のうり科野菜、えだまめ及びべにばなの種子
1.国产咪唑苯脲二丙酸盐在牛体内的药代动力学及组织残留沈春岚 吴弋麃 宋鲁敏 张金子 戴国华 【摘要】:给牛单剂量肌注咪唑苯脲二丙酸盐(2mg/kg)。用紫外分光光度计测出不同时间的血药浓度,并按有吸收一室模型=M(e~(-ket)—e~(-kat))公式,计算出咪唑苯脲的主要药代动力学参数:吸收速率常数(k_a)为2.027h~(-1) 清除速率常数(k_e)为0.419h~(-1),峰时间(T~(max))为1.18h 峰浓度(C~(max))为1.746μg/ml 吸收相半衰期(t1/2k_a)为0.342h,消除相半衰期(t1/2k_e)为1.165h 表观分布容积(Vd)为0.88L/kg 体清除率(BIC)为0.25L/kg/h。咪唑苯脲在牛的肝、肾、心,胆汁、脑、肌肉、脂肪中的残留【作者单位】: 兽医大学药理教研室 兽医大学药理教研室 兽医大学药理教研室 兽医大学药理教研室 兽医大学中心实验室 【关键词】: 咪唑苯脲 药代动力学 组织残留 牛 【DOI】:CNKI:SUN:ZSYX.0.1987-02-001【正文快照】: 咪哩苯脉(Imidocarb)具有抗巴贝斯梨形虫的作用,最早是schmidt等①在应用鼠骆氏巴贝西虫筛选一组均二苯脉类化合物时发现的,同年Beveridge②进一步证实了该药的LD:。低于其它通用的抗巴贝斯梨形虫药。随后,该药广泛用于世界各国,并证实其对各种巴贝斯梨形虫和无定形体(边虫)http://www.cnki.com.cn/Article/CJFDTOTAL-ZSYX198702001.htm2.国产咪唑苯脲对驽巴贝西虫病治疗试验李德昌 胡力生 阎仲堂 赵权 【摘要】:应用国产抗巴贝西虫新药咪唑苯脲(Imidocarb)二盐酸盐。对3匹马分别按每kg体重2、4、8mg剂量进行了安全性试验。结果,2、4mg剂量的马,间隔24h肌肉注射2次,临床、血液、肝功、肾功均未见明显变化 8mg剂量的马,仅注射1次就出现呼吸困难、流涎、腹痛和排稀粪等反应,30min后消失。对14例自然发病的驽巴贝西虫病马,按2mg/kg的体重剂量进行了试治,其中8例间隔24h共用药2次,6例仅用药1次,结果均获痊愈,且无任何副作用。【作者单位】: 兽医大学寄生虫病教研室 兽医大学寄生虫病教研室 兽医大学寄生虫病教研室 吉林农业大学兽医系 【关键词】: 咪唑苯脲 安全试验 驽巴贝西虫病 【DOI】:CNKI:SUN:ZSYX.0.1987-02-003【正文快照】: 3.咪哩苯脉(I midocarb)的抗巴贝西虫作用,最早为schmidt等(1969)①在应用鼠骆氏巴贝西虫筛选一组均二苯脉类化合物时发现。同年,Beveridge②进一步证实了该药的半数有效量(EDS。)低于其它通用的抗巴贝西虫药。随后,该药被广泛应用于非洲、拉丁美洲、北美洲、澳大利亚、爱尔兰咪唑苯脲——一种抗巴贝西虫新药李德昌 【摘要】:正 咪唑苯脲(Imidocarb)为均二苯脲(Carbanilide)类中的联脒(diamidine)的衍生物。商品名为 Imizol。化学名称为#结构式为#本药有两种盐类,即二盐酸盐和二丙酸盐,在10%(w/v)溶液时,后者 pH为6.5,前者 pH 为3.1,并且后者具有较前者易溶于水的优点。咪唑苯脲的抗巴贝西虫的作用最早为Schmidt 等(1969)在应用鼠骆氏巴贝西虫(Babesia rodhaini)筛选一组均二苯脲类化合物时发现,同年 Beveridge(1969)3,3′—双(2-咪唑啉)均二苯脲二盐酸[3,3′-his(2—imidozoline—2—yl)—Carbanil【作者单位】: 【关键词】: 巴贝西虫病 肌肉注射 咪唑苯脲 剂量 丙酸盐 二苯脲类 皮下注射 预防作用 静脉注射 衍生物 【DOI】:CNKI:SUN:JLXS.0.1986-03-031【正文快照】: 咪哩苯脉(Imidocarb)为均二苯脉(Carbanilide)类中的联眯(diamidine)的衍生物。商品名为I,nizol。化学名称为 #结构式为#3,3‘一双(2一咪哇琳)均二苯脉二盐酸 〔3,3/一1)15(2一imidozoline一2一yl)一Carbanilide dihydroehloride〕。 /‘一\一,,,。~、… \/一二、11—七L月—4.一种抗梨形虫药物咪唑苯脲及其盐的合成研究李光壁 【摘要】:抗梨形虫药物咪唑苯脲为均二苯脲类联脒衍生物,是一种重要的具有生物活性的化合物,一般以二盐酸盐和二丙酸盐最为常见。它们具有广谱、低毒、应用范围广、作用时间长、用药剂量小等优点,对家畜梨形虫病、无浆体病及猪犬等的附红细胞体病不仅有很好的治疗作用,也具有良好的预防效果,为新一代最佳的抗梨形虫药物,并且该药也是美国药典唯一收录的允许应用于梨形虫病治疗的药物。随着梨形虫病在世界各地的广泛传播,对该类药物的需求量越来越大。面对国内外的迫切需求,探求一条新的适宜工业化生产的路线,促进兽药行业的快速发展以及满足国内外的需求都具有重要的经济效益和社会效益。 本文合成了咪唑苯脲及其二盐酸盐和二丙酸盐。咪唑苯脲又称N,N’-双[3-(4,5,-2H-1H-咪唑啉-2-基)苯基)]脲。目前,据文献报道,有五种方法可以合成咪唑苯脲,如下所述: (1)3,3’-二氰二苯脲在乙醚-氢硫酸或乙醇-盐酸体系中与乙二胺反应: (2)3,3’-甲酸酯二苯脲与乙二胺在氯化铵溶液中反应(R与R’为含碳原子较少的烷烃基团): (3)将间硝基苯甲酸乙酯在三甲基铝存在下,与乙二胺反应得到2-(3-硝基苯基)咪唑啉,然后还原制得2-(3-氨基苯基)咪唑啉,最后在醋酸钠-水溶液中与光气发生缩合反应:【关键词】:咪唑苯脲 二盐酸盐 二丙酸盐 缩合 合成 【学位授予单位】:山东大学【学位级别】:硕士【学位授予年份】:2006【分类号】:TQ463.53【DOI】:CNKI:CDMD:2.2006.164426【目录】: 摘要6-9ABSTRACT9-12符号说明12-13第一章 前言13-351.1 梨形虫病及其治疗药物概述13-221.1.1 梨形虫病的种类14-171.1.2 抗梨形虫病药物国内外研究进展17-221.2 均二苯脲类联脒衍生物的生物活性及国内外研究进展22-281.2.1 均二苯脲类联脒衍生物的生物活性及国外研究进展22-271.2.2 国内研究进展27-281.3 课题的确立及应用价值28-291.4 咪唑苯脲的合成方法29-331.5 本文研究的主要内容33-351.5.1 2-(3-氨基苯基)咪唑啉二盐酸盐及其中间体的合成33-341.5.2 咪唑苯脲及其盐的合成34-35第二章 实验部分35-392.1 实验仪器与原料35-362.1.1 基本仪器352.1.2 基本原料35-362.2 合成部分36-392.2.1 间硝基苯甲腈的合成362.2.2 2-(3-硝基苯基)咪唑啉的合成36-372.2.3 2-(3-氨基苯基)咪唑啉二盐酸盐的合成372.2.4 N,N’-二-(3-(4,5-2H-1H-咪唑-2-基)苯基)脲的合成37-382.2.5 N,N’-二-(3-(4,5-2H-1H-咪唑-2-基)苯基)脲二盐酸盐的合成382.2.6 N,N’-二-(3-(4,5-2H-1H-咪唑-2-基)苯基)脲二丙酸盐的合成38-39第三章 结果与讨论39-683.1 间硝基苯甲腈的合成39-433.1.1 原料及工艺路线的选择393.1.2 反应条件的选择39-413.1.3 重结晶溶剂的选择413.1.4 结构分析与确定41-433.2 2-(3-硝基苯基)咪唑啉的合成43-483.2.1 结果433.2.2 讨论43-453.2.3 结构分析与确定45-483.3 2-(3-氨基苯基)咪唑啉二盐酸盐的合成48-523.3.1 结果483.3.2 讨论48-503.3.3 结构分析及确认50-523.4 N,N’-二-(3-(4,5-2H-1H-咪唑-2-基)苯基)脲的合成52-593.4.1 结果52-533.4.2 讨论53-543.4.3 结构分析及确认54-593.5 N,N’-二-(3-(4,5-2H-1H-咪唑-2-基)苯基)脲二盐酸盐的合成59-623.5.1 结果593.5.2 讨论59-603.5.3 结构分析及确认60-623.6 N,N’-二-(3-(4,5-2H-1H-咪唑-2-基)苯基)脲二丙酸盐的合成62-683.6.1 结果623.6.2 讨论62-643.6.2 结构分析及确认64-68第四章 实验结论68-69参考文献69-74致谢74-75[em09502]
原料药盐酸米多君标准(国内:YBH09452006),5-氨基乙酰丙酸盐酸盐标准。若有特殊要求也可站内信息联系!有国外标准也行。
各位老师,现在实验室在做氯丙醇脂肪酸酯,参照标准[font=Verdana, Arial][color=#333333][back=#f4f1e2]GB 5009.191-2016 食品安全国家标准 食品中氯丙醇及其脂肪酸酯含量的测定。其中衍生剂七氯丁酰基咪唑需要用气密针吸取,但操作过程中衍生剂容易变成白色固体,用气密针吸不起来,所以改成[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]吸,但好像衍生效果不好。请教各位是不能用[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]吸吗?为什么标准中规定要用气密针?万分感谢[img]https://simg.instrument.com.cn/bbs/images/default/em09512.gif[/img]。[/back][/color][/font]
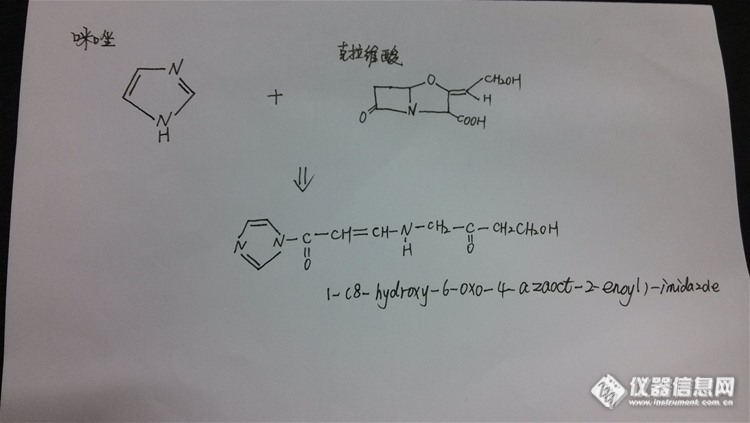
根据国外文献报道,血浆中克拉维酸的高效液相检测方法中常用咪唑衍生化试剂进行衍生以增强其紫外吸收。现在有个问题:衍生化后的咪唑色谱峰对咪唑衍生产物色谱峰有干扰,调节pH值及流动相比例未见效。我将反应式和液相条件列出来,请大家帮忙看看,如何改变液相条件以实现两者分离,感激不尽。 液相检测条件:C18(250mm长柱); 流动相:0.05M硫酸二氢钠溶液(pH调至2.2):乙腈=98:2; 柱温:40℃; 流速:0.6ml/min 克拉维酸衍生反应(文献报道4体积的克拉维酸加1体积的咪唑,具体摩尔比不明):http://ng1.17img.cn/bbsfiles/images/2014/05/201405101115_499105_1625629_3.jpg
哪位大侠,有优级纯(GR,Guaranteed reagent)、分析纯(AR,Analytical reagent、化学纯(CP,Chemical pure)盐酸的杂质含量标准,小弟十分感谢![em09511]
有人做盐酸苯肼的吗,我想找他的分析方法,买来的东西厂家直说是滴定法座含量,有标准或分析方法吗?谢谢了
[font=宋体][size=14px][/size][/font][font='微软雅黑','sans-serif']盐酸中甲苯测定,可以直接萃取后进样分析吗?标准样品配制需要在盐酸中加入不同量的甲苯配制吗?[/font][font='微软雅黑','sans-serif']求助问题来自微信群。[/font][font=宋体][size=14px][/size][/font]
大家好: 我现在有个离子液体样品,里面含有大概1%的N-甲基咪唑量,因为用气相色谱做这个样品对柱子有损坏作用,现在想通过液相色谱法对N甲基咪唑做定量分析。我公司有安捷伦的液相色谱,柱子是C18的,检测器有紫外和视差检测器。我们自己通过液相实验后分析不出样品中的N甲基咪唑,但进纯样N甲基咪唑后有信号峰出现。想求教下大家有什么好方法能分析出离子液体样品中的N甲基咪唑。
[center][url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]法检测动物肝脏中的左旋咪唑(EEC,Sg4.5,本法可扩展为确正方法)[/center]the Determination Method of Residues of Levamisolein Anamal Livers by [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-MS安全操作注意事项 实验室操作人员在处理动物组织时,要戴一次性手套,所有操作包括溶解过程都必须在通风橱中进行。 引言 左旋咪唑是一种广谱杀虫剂,主要用于控制反刍类动物、马及猪的胃肠道、肺中的线虫。 1. 主题内容 本法可用于检测肝脏组织中的左旋咪唑残留量,也可做为常规的筛选法。同时本方法也易扩展为确正方法。 2. 应用范围 本法适用于肝脏组织中左旋咪唑残留量的检测,其检测低限为5μg/kg肝脏。 3. 参考文献 —— 欧盟委员会93/256/EEC指令,制定检测内分泌器官的药残方法。[OJ.NO.L.118,14.4.93 PP64-74]。—— ISO78/2-1982标准集——第2部分:化学分析标准。—— [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]法检测肝脏中的左旋咪唑 Neats,S.(1993)标准操作方法 ACU 0267A,CVL.,UK—— [url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]法检测肝脏中的左旋咪唑 Porter,S.,Patel,ER.Neate,S.和Osso,P.(1993).食品中的农药残留Ed.Haagsma,N,Ruoter,A & Czediik-Eysenberg,P.B., Veldhoven,NL.PP548-552 .4. 定义 本检测方法所测得的左旋咪唑含量是指被测物质中左旋咪唑的残留量,不论其以何种化学形式存在,检测结果表示为μg/kg 。 5. 方法提要 本方法包括4个步骤:(1)将样品中加入碱和乙酸乙酯均质。(2)用液-液分配净化。(3)[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]电子轰击模式测定。(4)通过标准曲线采用内插法计算出左旋咪唑含量,并用左旋咪唑的回收率加以校正。6. 化学试剂 6.1.1 三氯甲烷。 6.1.2 乙酸乙酯。 6.1.3 氢氧化钾。 6.1.4 甲醇:色谱纯。 6.1.5 浓盐酸 。6.1.6 无水颗粒状硫酸钠。 6.1.7 乙酸乙酯 。6.1.8 水:去离子并经0.2μm滤膜过滤。6.2 溶剂6.2.1 50%氢氧化钾溶液:将250克氢氧化钾溶解于400mL水中,冷却后定容至500mL。6.2.2 0.5M盐酸溶液:将22mL浓盐酸加入400mL水中,冷却后定容至500mL。6.3 标准品6.3.1 左旋咪唑盐酸盐()6.3.2 标准贮备液:将50mg左旋咪唑溶解于甲醇中,并稀释至50mL,当月配制。6.3.3 标准工作液6.3.3.1 将1ml贮备液加入到100mL三氯甲烷中,最终浓度为10μg/mL,当天配制。 6.3.3.2 分别取6.3.3.1溶液1mL,2mL,5mL加入到10mL三氯甲烷中,最终浓度分别为1,2和5μg/mL,其分别相当于农药残留限量的1、2和5倍,当天配制。6.4 加样方法取上述标准工作液(6.3.3.1和6.3.3.2)各1mL加入到5g样品中,静置15分钟。 7. 仪器和设备 7.1 离心管:50mL 材质:Sarsted聚丙烯,螺旋盖。7.2 锥形离心管:12mL 材质:聚丙烯,带帽。7.3 锥形离心管:10mL 材质:玻璃,塑料帽。7.4 量筒:25mL,50mL,1L。7.5 容量瓶:10mL,25mL,50mL,100mL,500mL。7.6 移液管:1mL,2mL,5mL。7.7 烧杯:100mL,250mL,500mL,1L。7.8 乙酸纤维素滤膜:0.2 μm。7.9 Pasteur吸管。7.10 微量注射器:100μL 。7.11 7.11 混合器(涡旋型)。7.12 离心机—MSE Mistral 2L或相当者。7.13 均质器。7.14 振荡器:机械平板摇床(Denley)。7.15 微量管:1.5mL有卷曲金属帽和塞。7.16 Falcon 710S瓶盖(Becton Dickinson)7.17 200μL微量管(Chromacol 型号02-CVTG)。7.18 天平。7.19 旋转蒸发器(氮)。7.20 可调节型移液管:200μL,1000μL,5000μL。7.21 [url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-MS气-质谱联用仪。质量选择检测器(质谱仪)与HP-5890[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]、HPVectra486/33uPC机联用。[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]-MC装有自动进样器(Henlett Packard 7673A)。色谱柱:15m×0.25mm(内径)×0.25μm(膜厚),DB5毛细管柱。
有谁做过1-丁基-3-甲基咪唑氯盐的分析?(1)色谱柱为 ODS 型C18色谱柱;流动相为水和甲醇,流动相比为水∶甲醇 = 1∶9,流速为1.0 mL/min;室温条件下检测,紫外检测波长为215 nm。(2) BDS型C18色谱柱;流动相为水(pH = 1.63)∶甲醇 = 85∶15,流速为1.0 mL/min;室温下检测,紫外检测波长为215 nm。我用第一种方法做过,在2.5分钟有一个很大的峰出来,但是接着就有一个很小的峰出来.两个峰分不开.
本人在此急求 中华人民共和国国家药品监督管理局标准(试行)中的关于"盐酸左氧氟沙星注射液"的标准,请大家帮忙!谢谢!
默克密理博应用实验室 2013-07-15近日,百事可乐的产品在美国10个州中被爆出4-甲基咪唑(4-Methylimidazole)严重超标。4-甲基咪唑是一种有机中间体,主要用于合成大宗胃药西咪替丁,也可用作环氧树脂固化剂和金属表面防护剂等。可乐中的4-甲基咪唑是在以亚硫酸铵为原料生产焦糖色素时产生的。 4-甲基咪唑白色至类白色结晶粉末,易溶于水和乙醇,有腐蚀性,是一种能诱发肿瘤的化学物质。http://blog.merckmilliporechina.com/editor/upload/image/4C619C0F_7B615A74.PNG默克密理博致力于分析方法的开发,为客户提供简便、快速的解决方案。4-甲基咪唑及其异构体2-甲基咪唑均有较强极性,适合使用默克密理博的两性离子型亲水作用色谱柱(ZIC®-HILIC)分离。本实验采用默克密理博两性离子型(ZIC®-HILIC)色谱柱直接分析甲基咪唑的液相色谱方法。该方法前处理简单,不需要衍生化,也不需要添加离子对试剂。1 材料试剂1.1 对照品:4-甲基咪唑,2-甲基咪唑。1.2 色谱柱:ZIC®-HILIC 250-4.6mm 5um 200Å(默克密理博,货号:1.50458.0001)1.3 乙腈(默克密理博,货号:1.00030.4008)1.4 甲醇(默克密理博,货号:1.06007.4008)1.5 磷酸二氢钾(默克密理博,货号:1.04873.1000)1.6 可口可乐及百事可乐样品1.7 实验用为为超纯水(默克密理博Milli-Q Advantage)1.8 PVDF0.22um针头过滤器(默克密理博,货号:SLGV033NB)1.9 标准溶液配制:使用70%乙腈溶液,分别配制1mg/ml的4-甲基咪唑,2-甲基咪唑对照品原液。取两个对照品原液,1:1混合、稀释、定容,成,得100ug/ml的混合对照品母液。混合母液用70%乙腈溶液配制浓度为0.
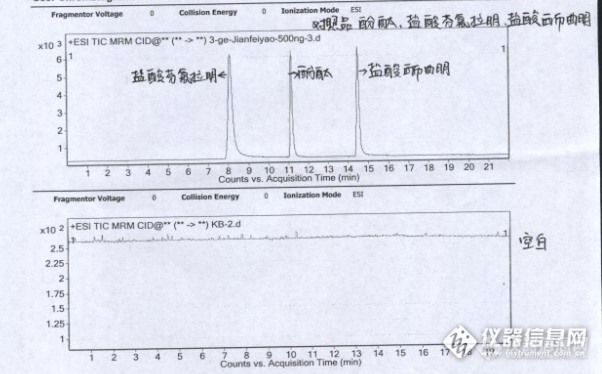
减肥产品中酚酞、盐酸西布曲明及盐酸芬氟拉明的检测盐酸西布曲明,盐酸芬氟拉明,酚酞已被国家食品药品监督管理总局明令禁止用于食品(含保健品)了,目前还有部分违规企业在减肥产品中违法添加。盐酸西布曲明曾为处方药,但目前已在全球大多数国家停止使用。盐酸西布曲明(Sibutramine Hydrochloride)是西布曲明(Sibutramine)的氯化物,是一种中枢神经抑制药物,曾用于肥胖症的治疗。酚酞是化学品和临床处方药,有严格的适应症,需在医生指导下应用,若长期过量服用可能引发严重的副作用。在制药上作为医药原料,其药品名称为酚酞片(Phenolphthalein Tablets),主要用于治疗习惯性、顽固性便秘。过量或长期滥用,可造成人体电解质代谢紊乱,严重时甚至可诱发心律失常。婴儿和哺乳期妇女禁用,幼儿和孕妇慎用。市场上抽检的三批次的减肥产品违法添加盐酸西布曲明,酚酞的检测:仪器型号及编号 Agilent 1200 LC/MS 6410B 天平型号及编号 BP211D,LD310-2 色谱条件:色谱柱:phenomenex C18柱(100x3.0 mm,2.6 μm) 预柱 Agilent 预柱流速(mL/min) 0.2 进样量(uL) 5 柱温(℃) 25℃ http://ng1.17img.cn/bbsfiles/images/2017/01/201701191701_668681_2166779_3.png质谱条件:电喷雾离子化源(ESI) 碰撞气压力(Mpa) 0.15 Nebulizerpressure(Psi) 15 drying GasFlow(L/min) 6 Dry Temp(℃) 350 电离源 ESI ,正离子模式 http://ng1.17img.cn/bbsfiles/images/2017/10/2016071708440193_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071708440482_01_2166779_3.png将标准品分别配制成1mg/mL的酚酞,盐酸芬氟拉明,盐酸西布曲明标准储备液,分别吸取标准储备溶液进行稀释,得到100ng/mL,80ng/mL,50ng/mL,20ng/mL,10ng/mL,5ng/mL的标准工作溶液。2.标准曲线的制作取各标准工作溶液5 uL注入液质仪,采集数据。以峰面积为纵坐标(Y),以标准工作溶液浓度(X)为横坐标绘制标准曲线。酚酞,盐酸芬氟拉明,盐酸西布曲明标准品及空白的色谱图、质谱图及工作曲线:http://ng1.17img.cn/bbsfiles/images/2017/10/2016071717420205_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717421213_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717422190_01_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717433884_01_0_3.pnghttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071717434225_01_2166779_3.png3.试样提取取各试样适量(约相当于一次用量),置50mL离心管中,精密加入甲醇20mL,超声处理15min,放冷至室温,10000r/min离心5min,取上清液用50%甲醇稀释。稀释过程:①0.2→2.0ml (稀释10倍);②0.1→2.0ml(共稀释200倍); ③0.1→2.0ml(共稀释4000倍)③0.1→2.0ml(共稀释80000倍)样品1、2、3号的酚酞,盐酸西布曲明,盐酸芬氟拉明色谱图http://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600844_2166779_3.png样品1号酚酞,盐酸西布曲明,盐酸芬氟拉明的质谱图http://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600845_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607171745_600846_2166779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607171746_600847_2166779_3.png从图中可见1号未检出盐酸芬氟拉明;2、3号样品的质谱图略。定量分析 酚酞的含量(mg/粒)= C样×V样 ×样品稀释倍数×W平 /W样×10-6 供试品编号 10粒内容物装量(g) 平均装量(g) 取样量 (g) 检测结果(ng/mL) 含量 (mg/粒) 平均含量 (mg/粒) 1号 2.6327 0.2633 0.2692 16.5391 25.88 27.8 0.2623 18.5693 29.82 2号 2.4988 0.2499 0.2691 15.0804 22.41 [align=c
食品安全国家标准 食品营养强化剂 L-盐酸赖氨酸
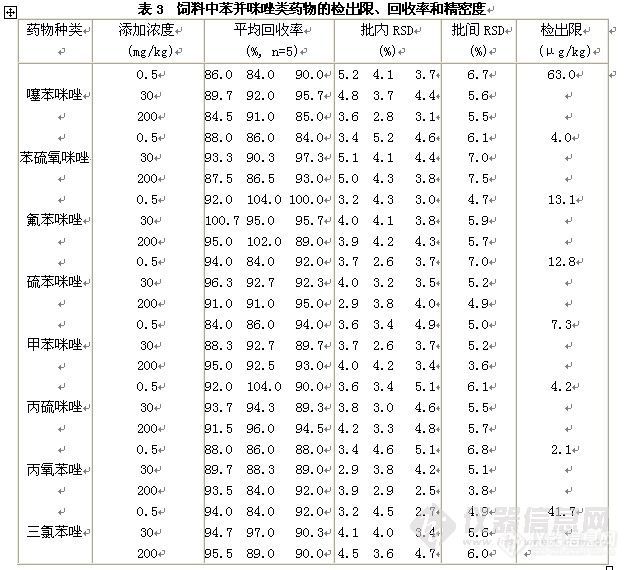
液相色谱串联质谱法测定饲料中8种苯并咪唑类药物摘 要 建立了同时测定饲料中8种苯并咪唑类药物(噻苯咪唑、丙硫咪唑、硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑)的液相色谱串联质谱分析方法。饲料样品直接用酸化乙腈提取,提取液用甲酸溶液稀释后直接进行分析。分析时采用XBridgeTM C18色谱柱,以甲酸溶液-乙腈体系进行梯度洗脱,MRM方式测定,基质外标法定量。苯并咪唑类药物在0.02~10 mg L-1浓度范围内呈良好的线性,线性相关系数均大于0.990,苯并咪唑类药物在饲料样品中最低检测限为2.1~63.0μg/kg。饲料中苯并咪唑类药物在0.50~200 mg/L范围内的回收率为84.0%~104%之间,相对标准偏差(RSD)均小于10%。 关键词 苯并咪唑类药物;液相色谱串联质谱法;饲料 苯并咪唑类药物(benzimidazoles, BMZs)属于广谱、高效、低毒抗蠕虫药,由于对胃肠线虫具有很强的驱杀作用,至今仍在广泛使用。但由于BMZs在实验动物和靶动物显示致畸和致突变作用,目前使用的BMZs多数是食品残留中重要的监控对象,且BMZs在体内转化的代谢产物仍具有毒理作用,所以我国以及联合国粮农组织、欧盟、美国、日本等国家和组织都将苯并咪唑类药物列入限制使用的兽药药物,并制订出各种苯并咪唑类药物在不同动物体内(肌肉、组织、奶等)的最高残留限量。饲料安全直接关系到动物性食品的安全,考虑到苯并咪唑类药物经常被添加到饲料中使用,故很有必要进行饲料中苯并咪唑类药物的分析研究。 目前对于动物组织中苯并咪唑类药物的分析方法较多,而饲料中苯并咪唑类药物分析方法国内未见发表,国外也较少,涉及的种类也较少,最多的仅有5种药物。动物组织和饲料中BMZs分析涉及的主要分析手段有:酶联免疫吸附法( ELISA) 、气相色谱-质谱法(GC-MS)、高效液相色谱法(HPLC)及高效液相色谱串联质谱法(HPLC-MS/MS),高效毛细管电泳法(HPCE)。考虑到苯并咪唑类药物在我国使用情况,本研究选择了8种常用苯并咪唑类药物,考虑到LC-MS/MS法灵敏度高的特点,样品酸化乙腈提取后直接稀释后进行液相色谱串联质谱分析。1 材料与方法1.1 仪器与试剂 Waters 2695 Quattro MicroTM API高效液相色谱串联质谱仪(美国Waters公司),配置电喷雾离子源;固相萃取仪(美国Supelco 公司);Sigma离心机。噻苯咪唑和丙硫咪唑标准品(Accustandard 公司);硫苯咪唑、苯硫氧咪唑、氟苯咪唑、甲苯咪唑、丙氧苯唑和三氯苯唑标准品(Dr. Ehrenstorfer)。乙腈、二甲亚砜和甲酸为色谱纯(Fisher公司)。1.2 仪器条件 XBridgeTM C18色谱柱(150 mm×2.1 mm,内径3.5 μm);流动相A为0.1%甲酸溶液,B相为乙腈,梯度洗脱条件:B相在1.0 min内从15%线性增加到25%,再在2.5 min内线性增加到95%,保持3.5 min,然后在0.1 min内降至15%,保持4.9 min;流速:0.3 mL/min;进样量:10 µL;柱温:30℃。 质谱条件:ESI源正离子模式电离;多反应监测(MRM);毛细管电压:3.0 KV;萃取锥孔电压:20 V;RF透镜电压:0.5 V;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔气流速:50 L/h;脱溶剂气流速:600 L/h;倍增器电压:650 V;二级碰撞气:氩气;其它条件详见表1。http://ng1.17img.cn/bbsfiles/images/2010/11/201011301506_262957_1759541_3.jpg1.3 样品处理 称取2g试样(精确到0.01g)于50 mL离心管中,加入20 mL0.5 %甲酸乙腈,涡旋1 min,然后超声提取10 min,以5000 r/min的速度离心5 min后吸取1.0 mL上清液于5 mL刻度试管中,加入3 mL0.1 %甲酸溶液于试管中,混匀后过0.22 μm滤膜,进行液相色谱串联质谱分析。1.4 线性实验 准确称取各10.0 mg BMZs标准品于相应的10mL容量瓶中,噻苯咪唑、甲苯咪唑、丙氧苯唑和丙硫咪唑用二甲亚砜溶解并定容至刻度,其余4种BMZs用甲醇:二甲亚砜(2:3 v/v)溶解并定容至刻度,即得均为1000 mg/L标准储备液。分别吸取1.0 mL各标准储备液于同一10mL容量瓶中,用甲醇稀释至刻度,即得100 mg/L的混合标准工作液。分别准确移取苯并咪唑类药物混合标准中间液适量,配制浓度为0.2.、0.8、2.0、10.0、40.0和100.0 mg/L的系列标准溶液,吸取0.1 mL于5 mL刻度试管中,再吸取空白试料提取液0.9 mL于该5 mL刻度试管中,加入3 mL0.1%甲酸溶液后混匀过膜,进行上机测定,以定量离子对峰面积为纵坐标,标准溶液浓度为横坐标,绘制基质校准标准曲线。2 结果与分析2.1 液相色谱质谱分析 苯并咪唑类药物色谱分析时,通常采用反相分离体系,主要有三类流动相体系:离子增强体系,pH2~3,一般使用乙腈-磷酸或磷酸盐体系;离子抑制流动相体系,pH5~7;离子对流动相体系,离子增强流动相中加入阴离子对试剂。对于多组分苯并咪唑类药物液相色谱质谱分析时,通常采用离子增强体系进行梯度洗脱,如0.1%甲酸溶液-乙腈体系,因为该体系和纯水-乙腈体系相比色谱峰的拖尾现象得到了明显改善。 苯并咪唑类药物属弱碱性物质,中等极性,在酸性条件下很容易质子化,于是本方法选择ESI+进行分析。以乙腈/0.1%甲酸溶液(3:7,v/v)为溶解液,用蠕动泵(20μL/min)对苯并咪唑类药物的质谱条件进行优化。经过优化的条件为:毛细管电压:3.0KV;离子源温度:110℃;脱溶剂气温度:350℃;锥孔气流速:50L/h;脱溶剂气流速:600L/h。其它条件详见表1。2.2 提取净化方法的选择和优化 [font=宋体
谁能告诉我4,5-咪唑二甲酸分析的方法?
谁有YS/T 239.5-1994 三硫化二锑化学分析方法 重量法测定盐酸不溶物标准,请上传一下进行分享。







 我要推广仪器
我要推广仪器
 下载APP
下载APP