今天到了佳乐麝香,打开盖子一看,居然有结晶。而且还很多(天津今天的室外温度18℃)再看标签,赫然写着溶剂是DPG,(之前的溶剂是邻苯二甲酸二乙酯)不知道为何会出现这样的情况,有了解的版友吗,,不同的溶剂,对于佳乐麝香的外观有这么大的影响,?
书上说邻苯二甲酸氢钾溶液是现用现配,我想知道一定要求是当天用当天配吗?如果间隔三四天,这个邻苯二甲酸氢钾溶液的浓度能保持不变吗?
化学需氧量测定中用到邻苯二甲酸氢钾溶液做标准溶液,进行校核实验。这种溶液是不是很容易被氧化,保存不了几天
请问哪里可以买到16种邻苯二甲酸酯混标溶液啊?
土壤中佳乐麝香的检测解决方案人工合成麝香广泛应用于化妆品、个人护理用品等日化产品中。目前,在水体、污泥、大气及人体、鱼、虾、贝类等生物体内均有检测,被认为是环境中新型污染物。由于麝香属挥发性有机物,一般采用GC或者GCMS检测,前处理过程有液液萃取,ASE、SPE、GPC等方法。方法优势迪马科技建立固相萃取-气相色谱串联质谱法检测土壤中佳乐麝香,土壤样品经脱水处理,过20目筛。样品经正己烷提取后,使用ProElut Silica净化,选择合适的净化条件,降低杂质干扰;回收率均在85%以上,保证实验结果的重现性和准确性;方法检出限是5 μg/kg。以下为详细解决方案,敬请参考!1、适用范围本方案适用于土壤中佳乐麝香的检测。方法检出限5 μg/kg。2、样品准备(1) 土壤样品风干,过20目筛;(2) 称取2.0 g样品,加入20 mL正己烷混匀,振荡5 min,超声10 min,6000 rpm离心2 min,收集上清液;(3) 向下层残渣中加入20 mL正己烷,按照步骤(2)重复提取一次,合并上清液;(4) 将上清液在35 ℃水浴下减压蒸干,加入3 mL正己烷,混匀,待净化。3、SPE柱净化——ProElut Silica 500 mg/6 mL(Cat#:63005)(1)活 化: 向柱中加入5 mL二氯甲烷、5 mL正己烷,弃去流出液;(2)上 样: 将待净化液加入柱中,弃去流出液;(3)淋 洗: 依次加入5 mL正己烷、5 mL 30%二氯甲烷正己烷,弃去流出液;(4)洗 脱: 加入10 mL 40% 二氯甲烷正己烷,收集流出液;(5)重新溶解: 将洗脱液在35 ℃水浴下减压蒸干,用甲醇定容至1 mL,供GC-MS分析。4、色谱条件色谱柱:DM-5MS, 30 m × 0.25 mm × 0.25 μm(Cat#:8221)进样口温度:220 ℃升温程序:初始温度60 ℃,保持0.5 min,以20 ℃/min升温至160 ℃,,再以10 ℃/min升温至250 ℃,保持2 min载气:氦气,流速:1 mL/min进样方式:不分流进样进样量:1 μL离子源温度:210 ℃接口温度:260 ℃溶剂延迟:6 min表1选择离子监测组表起始时间/min结束时间/min选择离子816.5243,258,213,157 表2 目标物组分名称、保留时间及特征离子一览表化合物保留时间目标离子参考离子佳乐麝香12.54243258,2135、添加回收结果土壤中佳乐麝香添加回收结果NO.化合物名称添加水平(mg/kg)回收率(%)1佳乐麝香0.5106.4 http://www.dikma.com.cn/u/image/2015/08/11/1439260208724655.jpg相关产品信息http://www.dikma.com.cn/u/image/2015/08/11/1439260234136466.jpg红色产品货号#30039、#30040、#1034、#1035火热促销中
如题,请问校准试验邻苯二甲酸氢钾标准溶液里要加硫酸汞吗?
CODmn,标准溶液cod10g/l或者更低含量,能用邻苯二甲酸氢钾配制吗?
我配制了一桶0.3mol/L氢氧化钠标准溶液,用基准物邻苯二甲酸氢钾标定出来的数据波动很大,根本不平行。可是用硫酸标准溶液标定出来却很平行(己排除了滴定管、基准物、烘箱的准确度因素),而且有个奇怪的现象,就是邻苯二甲酸氢钾标称样量一样数据就能平行,如果称样量不一样数据就不能平行,称样量越大标定出来的数据越高,称样量越小标定出来的数据就越低(比如称1.9g邻苯二甲酸氢钾标定得0.3050mol/L,称1.5g邻苯二甲酸氢钾标定得0.3000mol/L,如果8组都称1.9g邻苯二甲酸氢钾8组都标定得0.3050mol/L,如果8组都称1.5g邻苯二甲酸氢钾8组都标定得0.3000mol/L)这是为什么啊?
各位大神,大家好。我要测试水溶液中的间苯二胺的浓度,这个气相色谱之前没有接触过。问之前的做过的,他直接水稀释进样。但貌似水溶液直接进样对仪器有损伤。请教大神,这个需要设计一个怎样的实验方案。我想萃取的话,萃取出来的间苯二胺就可以用乙醇作溶剂,若果萃取的话,要选择合适的萃取剂。主要是在线监测使用。谢谢大家。
从中国计量院买的正己烷中三种邻苯二甲酸酯类化合物标准溶液,标称体积为两毫升,如何吸取?用移液管感觉不够用,用[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]枪头都是塑料的怕污染不可用,用注射器现在还没有买。小弟头次购买使用。[img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906101642018154_5821_2516124_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2019/06/201906101642019034_3165_2516124_3.png[/img]
用溴麝香草酚蓝溶液测ph的时候是一加到水里摇匀读出来的数值最接近水的ph吗?每次一加,过了几秒颜色就会比刚加低1-2个ph值,所以什么时候读是最准确的呢?
【中文名称】邻苯二甲酸氢钾 【英文名称】potassium acid phthalate 【CAS号:】877-24-7 【结构或分子式】KHC8H4O4 【相对分子量】204.22(按1987年国际原子量) 【密度】1.636 【性状】 无色或白色结晶粉末,能溶于水。 【溶解情况】溶于水,水溶液有酸性反应。 【邻苯二甲酸氢钾(KHC8H4O4)含量测定 】 1 氢氧化钠标准滴定溶液摩尔浓度的标定 称取0.5g于105~110℃干燥至恒重的第一基准试剂(容量)邻苯二甲酸氢钾,称准至0.00001g,置于反应瓶中,加50mL无二氧化碳的水溶解,用231型玻璃电极作指示电极,用232型饱和甘汞电极作参比电极,按GB 10737之规定,用待标定的氢氧化钠标准滴定溶液[b(NaOH)=0.1mol/kg]滴定至终点。 氢氧化钠标准滴定溶液的质量摩尔浓度按式(1)计算: b= m1/(m2×0.20422)…………………………………………(1) 式中:b----氢氧化钠标准滴定溶液的质量摩尔浓度,mol/kg; m1----第一基准试剂(容量)邻苯二甲酸氢钾的质量,g; m2----待标定的氢氧化钠标准滴定溶液的质量,g; 0.20422----与1.0000g氢氧化钠标准滴定溶液[b(NaOH)=1.0000mol/kg]相当的,以克表示的邻苯二甲酸氢钾的质量。 2含量的测定 称取0.5g于105~110℃干燥至的试样,称准至0.00001g,按4.1.1条之规定,用氢氧化钠标准滴定溶液[b(NaOH)=0.1mol/kg]滴定。 含量的测定与滴定分析用标准溶液浓度的标定同时进行。 邻苯二甲酸氢钾(KHC8H4O4)含量按式(2)计算: X= (m3/m4)b×0.20422 ×100 ……………………………(2) 式中:X----邻苯二甲酸氢钾的百分含量,%; m3----氢氧化钠标准滴定溶液的质量,g; b----氢氧化钠标准滴定溶液的浓度,mol/kg; 0.20422----与1.0000g氢氧化钠标准滴定溶液[b(NaOH)=1.0000mol/kg]相当的,以克表示的邻苯二甲酸氢钾的质量; m4----试样的质量,g。 【用途】 滴定分析中的基准物质,用作制备标准碱溶液的基准试剂和测定pH值的缓冲剂。 与氢氧化钠反应生成邻苯二甲酸钾纳。 【制备或来源】 可由邻苯二甲酸酐与氢氧化钾作用而得。 【安全术语:】S24/25 【检验规则】按GB 619之规定进行采样及验收 【包装】按HG 3—119之规定 内包装形式:G-2; 外包装形式:用规格为600g/m的盒板纸制盒,外层裱紫色电光纸; 包装单位:第3类。 【标志】 按HG 3-119之规定。
测自来水的时候刚加入溴麝香草酚蓝溶液是蓝色,可能过了几秒颜色就不蓝,稍微有点绿了,那是不是刚加入那时读出的ph是最接近的呢?还是说要放置一会再读?
原文如下;weigh out 1 part by mass of phenol(AR) and dissolve in 1 part by mass of 1,2-dichlorobenzene (AR),work to an accuracy of 1% or better in the weighings.(称量等质量的苯酚溶解在等质量的邻二氯苯中,称量的精度达到1%或者更佳)疑问一;苯酚是固体,是否能直接溶解在邻二氯苯中,如果在邻二氯苯中溶解较慢,能否加热?疑问二:能否把苯酚直接加热溶解,在热的状态下称量苯酚
三乙胺我们在生产中用作缚酸剂,现在得到一个三乙胺邻二氯苯混合溶液,配置了不同浓度的三乙胺溶液,但是打出来同样溶度的三乙胺峰面积相差挺大的,这个是为什么啊?
配制好的邻苯二甲酸酯标准溶液用什么容器保存??
我配制了一桶20L的0.25mol/L氢氧化钠标注溶液,用邻苯二甲酸氢钾标定数据一点都不稳定,最高的数据0.2495,最低的0.2444,可是用硫酸标准溶液标定却平行得很好,为什么呀?不明白呀?知道的快快指导下吧,我快郁闷死了!
配置了一个二甲苯(分析纯)溶液,溶剂为甲醇(色谱纯),目的是想定性对、间、邻三种组分对应的谱峰,FID,AB-5毛细管柱,30m*0.32mm*0.25[font='Times New Roman']μm,按照出峰顺序是对、间、邻二甲苯呢还是对和间二甲苯分不开,另外一个峰是杂质峰?请高手解答一下,本人[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]小白,谢谢了,图一是色谱图,图二是二甲苯试标签(分析纯)[img=二甲苯谱图,690,517]https://ng1.17img.cn/bbsfiles/images/2021/07/202107131819270806_8009_5017093_3.jpg!w690x517.jpg[/img][img=二甲苯试剂说明(分析纯),690,387]https://ng1.17img.cn/bbsfiles/images/2021/07/202107131819506935_7457_5017093_3.jpg!w690x387.jpg[/img][/font]
[align=right][b]SGLC-GC-040[/b][/align][b]摘要:[/b]本文建立了麝香中麝香酮含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件,采用色谱柱SH-50 分析麝香中麝香酮,麝香酮峰形对称,理论塔板数按麝香酮峰计算远高于1500,满足《中国药典》要求。此方法可为麝香中麝香酮含量测定提供参考。[b]关键词:[/b]麝香 麝香酮 SH-50 GC[b]1. 实验部分1.1 实验仪器及耗材[/b]Shimadzu GC-2030[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url];色谱柱:SH-50(30 m,0.25 mm × 0.25 μm;P/N:227-36162-01;S/N:1553669);SHIMSEN Arc Disc HPTFE针式过滤器(P/N:380-00341-05);[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]认证样品瓶LabTotal Vial(P/N:227-34002-01);SHIMSEN Pipet[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url]:SHIMSEN Pipet PMII-10(P/N:380-00751-02);SHIMSEN Pipet PMII-100(P/N:380-00751-04);SHIMSEN Pipet PMII-1000(P/N:380-00751-06)。[b]1.2 对照品溶液的制备[/b]取麝香酮对照品适量,精密称定,加无水乙醇制成每1 mL含1.5 mg的溶液,即得。[b]1.3 供试品溶液的制备[/b]取(检查)干燥失重项下所得干燥品约0.2 g,精密称定,精密加入无水乙醇2 mL,密塞,振摇,放置1小时,滤过,取续滤液,即得。[b]1.4 分析条件[/b]色谱柱:SH-1 (30 m, 0.25 mm × 0.25 μm P/N:221-75719-30;S/N:1541069 )升温程序:初始温度200 ℃,保持10分钟,以每分钟50 ℃的速率升温至280 ℃,保持2分钟载气:N2进样口温度:280 ℃分流模式:分流(40:1)控制模式:恒线速度(30 cm/s)初始流速:0.93 mL/min检测器:FID,温度:300 ℃进样量:1 μL[b]2. 实验结果[/b]按照上述色谱条件(1.4)进行采集,对照品溶液和供试品溶液色谱图如下:[b]对照品溶液[/b][img=麝香中麝香酮含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-040_1.png[/img][font=arial, &][size=12px][/size][/font][b]供试品溶液[/b][img=麝香中麝香酮含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-040_2.png[/img][font=arial, &][size=12px][/size][/font][b]重现性[/b][img=麝香中麝香酮含量测定]https://img.shimadzumall.com/Storage//userfiles/images/Img_articles/SGLC-GC-040_3.png[/img][font=arial, &][size=12px][/size][/font][b]3. 结论[/b]本文建立了麝香中麝香酮含量测定的GC 方法。结果表明,参照2020版《中国药典》中色谱条件,采用色谱柱SH-50 分析麝香中麝香酮,麝香酮峰形对称,理论塔板数按麝香酮峰计算远高于1500,满足《中国药典》要求。此方法可为麝香中麝香酮含量测定提供参考。
[em63] 请问邻苯二胺水溶液紫外最大吸收波长是多少啊,不甚感激!
[b][font=宋体]问题描述:[back=white]按照[/back][/font][back=white]HJ/T 72-2001[/back][font=宋体][back=white]方法对标准曲线溶液处理后上机后没有出峰,是什么原因?改用[/back][/font][back=white]C[sub]18[/sub][/back][font=宋体][back=white]色谱柱,流动相采用甲醇,甲醇做溶剂,标准曲线线性非常好。明显用反相色谱柱分离效果好,为什么标准选择了正相色谱分离,正相色谱的优势在哪里?[/back][/font][font=宋体]解答:[/font][/b][font=宋体][back=white]([/back][/font][back=white]1[/back][font=宋体][back=white])由于[/back][/font][back=white]HJ/T 72-2001[/back][font=宋体][back=white]《水质[/back][/font][font=宋体][back=white]邻苯二甲酸二甲(二丁、二辛)酯的测定[/back][/font][font=宋体][back=white]液相色谱法》中在配制标准物质工作溶液时,是将甲醇中的标液用正己烷进行萃取,在萃取的过程中可能导致标准物质损失,可以尝试不萃取进样,如果出峰,则基本可以断定是萃取的问题。[/back][/font][font=宋体][back=white]([/back][/font][back=white]2[/back][font=宋体][back=white])实际上检测[/back][/font][font=宋体]水中邻苯二甲酸二甲酯的方法有很多种,包括[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法、液相色谱法、液相色谱串联质谱法等。根据文献记载,液相色谱法[back=white]检测[/back]水中邻苯二甲酸二甲酯,正相色谱法和反相色谱法都可以满足检测要求。[/font][font=宋体][back=white]([/back][/font][back=white]3[/back][font=宋体][back=white])该标准方法测定的是水和废水中邻苯二甲酸酯,包含了[/back][/font][font=宋体]邻苯二甲酸二甲酯等[/font][back=white]4[/back][font=宋体][back=white]个组分,而非[/back][/font][font=宋体]邻苯二甲酸二甲酯单个组分[back=white]。当然,如果在实际检测过程中,使用反相色谱分离[/back]邻苯二甲酸二甲酯[back=white]效果比较好,完全可以采用反相色谱法来检测,但需要对该方法进行评估和验证。[/back][/font][font='微软雅黑','sans-serif'][color=black][back=white]领取更多《实战宝典》请进:[url]http://instrument-vip.mikecrm.com/2bbmrpI[/url][/back][/color][/font][font='微软雅黑','sans-serif'][color=black][back=white] [/back][/color][/font]
请教一下:水溶液中的2,4-二氯苯酚怎么测定,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]还是液相色谱,色谱条件是什么?
请教专家:我在搞毕业论文,急!!!我用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测试邻苯二甲酸酯类时配制标准溶液是用什么溶剂比较好,色谱仪条件应该如何选择,每个样的测试时间如何控制?谢谢
求,分析有机溶液(PTA对苯二甲酸)中对甲基苯甲酸(PT酸)和对羧基苯甲醛(4-CBA)含量的方法,液相也行,最好为[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]方法,当然有滴定的方法,更加,但需测试微量,就是只有150PPM以内的,谢谢了
各位前辈 您们好! 晚辈急需要两种标准溶液,1、TDCPP 磷酸三(2,3-二氯丙基)酯CAS No. 78-43-32、DHNUP 邻苯二甲酸-二(C7-11 支链与直链)烷基酯 CAS :68515-42-4有知道哪里有卖的朋友的 告知一下 或者直接联系我,非常感谢!联系邮箱:2541035023@qq.com QQ:2541035023
我这儿有反应产物,估计里面有铝离子,原液为2,4-二氯苯酚 甲醇溶液(10000mg/L) 用蒸馏水稀释成100mg/L,取50ml溶液经一定的脱氯降解工艺,现在想测定降解工艺的效率,所以要测产物溶液中余下的2,4-二氯苯酚的含量,文献中的检测方法为2,4-DCP的测定采用HPLC(Agilent 1100),色谱柱为4.6 mm×150 mm ZORBAX C18反向柱。流动相组成为甲醇与2%冰乙酸水溶液的体积比为77:23,流速为1 mL/min,检测波长为284 nm。但现在我的问题是,产物溶液里有铝离子,我怕会堵塞柱子,想是不是要对水样进行预处理,比如萃取等等,但同时考虑到50ml水样中2,4-二氯苯酚本来就很少,反应后估计只有1mg左右,担心萃取会不会挥发掉?请各位指教。
心力丸是本所科研成果,药厂注册品种,国家中药保护品种,质量标准收载于卫生部中药成方制剂第十七册(WS3—B—3150—98)。本品具温阳益气、活血化瘀功能,用于心阳不振、气滞血瘀所致胸痹心痛、胸闷气短、心悸怔忡等证及冠心病、心绞痛。该制剂是由麝香、人参、附子、红花、人工牛黄、蟾酥等中药制成的浓缩丸,现国家规定处方中麝香以人工麝香等量替代使用,因此,人工麝香中主要有效成分麝香酮的含量[1],对控制该产品质量和保证临床用药安全有效具有重要意义。已有对六神丸等制剂中麝香酮进行测定的文献[2-5]报道。本文用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定心力丸麝香酮的含量,方法简便、准确,可以有效控制该产品质量。1 仪器和试剂1.1 仪器岛津GC17A[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],FID 检测器,N2000数据工作站(浙江智达) METTLER AE240电子分析天平 AS5150A超声波仪。1.2 试剂、试药麝香酮对照品(中国药品生物制品检定所,批号:071920006,含量测定用,质量分数99.8%) 心力丸(广东省药物研究所制药厂) 无水乙醇、乙醚等试剂均为分析纯 压缩空气、H2、N2均为高纯度的气体(广州气体厂,质量分数达99.9%以上)。2 方法与结果2.1 色谱条件毛细管柱:SPBTM1 FUSED SILICA Capillary Column(30 m×0.32 mm×1.0 μm) 柱温:200 ℃ 进样口温度:210 ℃ 检测器温度:225 ℃ 载气:N2 流速:2 mL/min 柱前压:80 kPa 空气:300 mL 氢气:30 mL 尾吹:30 mL 分流:50∶1 进样量:1 μL。在上述条件下,供试品溶液中麝香酮色谱峰能达到基线分离,供试品与邻峰的分离度大于2.0,峰形对称,人工麝香阴性供试品无干扰,方法具有专属性,按麝香酮计算理论塔板数不低于4 500。色谱图见图1-图3。图1 麝香酮对照品的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(M.麝香酮)(略)Figure 1 Gas chromatograms of muscone图2 心力丸供试品的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(M.麝香酮)(略)Figure 2 Gas chromatograms of Xinli pills图3 人工麝香阴性[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]图(略)Figure 3 Gas chromatograms of negative sample2.2 线性关系考察精密称取麝香酮对照品0.151 94 g,置100 mL量瓶中,加无水乙醇使溶解并稀释至刻度,摇匀 再分别精密量取0.5、1、2、4、5、6、8、10 mL置10 mL量瓶中,加无水乙醇至刻度,摇匀,精密吸取1 μL进样,测定峰面积,以对照品质量浓度(ρ)为横坐标,峰面积(A)为纵坐标,求得回归方程为A=291382. 2ρ+18117.0,r=0.999 6,麝香酮线性范围为0.076~1.5 mgmL-1。2.3 供试品溶液制备取本品,研细,精密称取3.5 g,置具塞锥形瓶中,加入乙醚30 mL,密塞,冷浸12 h,滤过,残渣和具皿用少量乙醚洗涤数次,合并乙醚液,挥去乙醚,残渣加无水乙醇使溶解并定容至5 mL,摇匀,用0. 45 μm滤膜滤过,取续滤液,即得。2.4 测定方法精密吸取供试品溶液和对照品溶液1 μL,分别注入[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]中,记录色谱图,以外标法计算供试品中麝香酮含量。2.5 稳定性试验取同一供试品溶液制备后室温放置,分别在0、1、2、4、6、8、10 h进样测定,每次进样1 μL,测得麝香酮平均含量490.4 μg/g,RSD为0.29%。结果表明供试品在10 h内测定结果稳定。2.6 精密度试验取麝香酮对照品溶液(0.759 7 mg/mL)重复测定6次,测得麝香酮峰面积的RSD值为0.15%。2.7 重现性试验取同一批供试品,平行6次精密称取,按“2.3”项下方法制备供试品溶液,依“2.4”项测定,计算供试品中麝香酮含量,结果其RSD值为1.4%,表明方法重现性好。2.8 回收率试验精密称取已知含量的心力丸(麝香酮含量495. 0 μg/g)共9份,置具塞锥形瓶中 取高(1.5194 mg/mL)、中(0.759 7 mg/mL)、低(0.075 97 mg/mL)3个浓度的对照品溶液各3份,每份2 mL,置蒸发皿中,通风柜中挥干乙醇,残渣用适量乙醚溶解,移入称取有供试品的具塞锥形瓶,用乙醚洗涤具皿数次,将洗涤液并入锥形瓶,乙醚用量为30 mL,其余按“2.3”方法制备,按“2.4”方法测定,结果见表1。2.9 样品测定取本品6批,按“2.3”项下方法制备供试品溶液,按“2.4”法进行测定,结果见表2。表1 麝香酮回收率试验结果(略)Table 1 Recovery test of muscone表2 样品测定结果(略)Table 2 Assay test of samples3 讨论3.1 提取方法的选择分别考察了超声提取法和浸渍法,结果见表3。结果表明:用乙醚作溶剂的2种提取方法有偏差(相对偏差为3.1%),浸渍法提取比超声法提取损失少,或者说提取较完全 在产品含量测定时采用浸渍法提取,提取适宜时间为12 h 在生产过程中的产品质量检测则可采用超声提取方法。3.2 色谱柱的选择曾采用强极性毛细管柱(SHIMADZU CBP20睲25025)进行试验,结果麝香酮很难达基线分离,且被测组分峰有拖尾现象。表3 两种提取方法麝香酮含量测定结果(略)Table 3 Muscone contents from two batches of extractions【参考文献】[1] 唐洪梅,黄樱华,李得堂,等.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定人工麝香中麝香酮的含量[J].中国实验方剂学杂志,2007,13(9):4-6.[2] 林巧玲,卓婷.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定保婴散中麝香酮的含量[J].广东药学院学报, 2006,22(3):257-258.[3] 袁劲松,汤翠娥.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定紫金胶囊中麝香酮含量[J].中国医院药学杂志, 2003,23(2):89-91.[4] 邹巧根,苏建俊.[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法测定麝香保心丸中麝香酮的含量[J].中国中药杂志, 1994,19(7):418-420.[5] 王强,毕开顺,陈金泉,等.六神丸中麝香酮含量的GC法测定[J].中国药科大学学报, 1995,25(6):87-89.
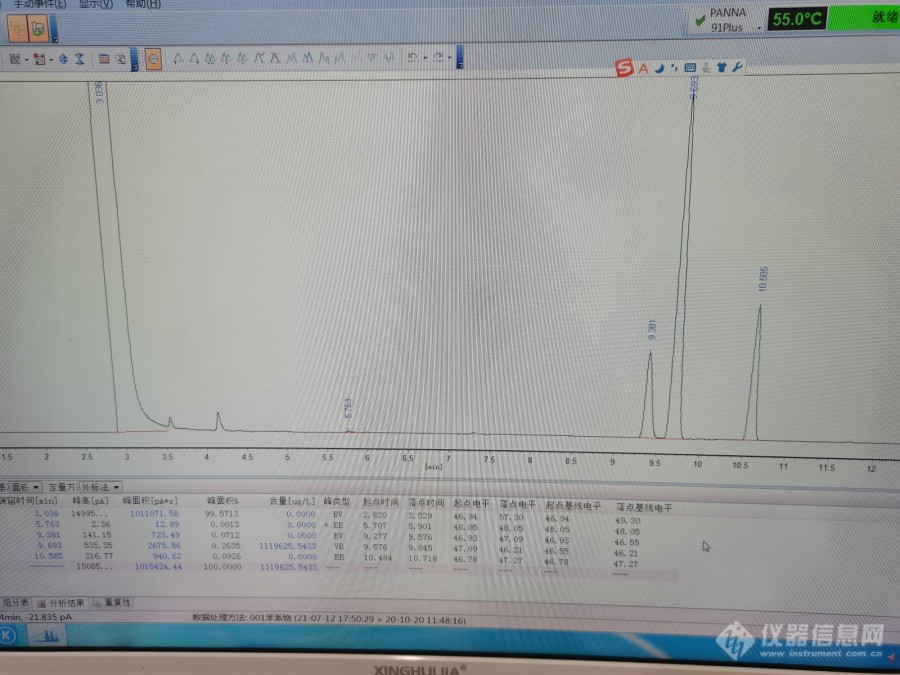
R..T..之前的样品中是有佳乐麝香 但是也不是很多 总和不到10%可,这个样品 在这个位置出佳乐 不太正常吧,而且还把基线抬得老高 难道是柱子里的残留吗??
【摘要】 目的 研究麝香祛痛搽剂中人工麝香质量的控制方法。方法采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法对处方中的人工麝香药材进行定性鉴别 用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法对处方中人工麝香的有效成分麝香酮进行定性测定。结果供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高,同时更能保证检测结果的真实性、准确性、重复性,阴性无干扰,专属性强。结论该方法可用于麝香祛痛搽剂中麝香酮质量的控制。【关键词】 麝香祛痛搽剂 [url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法 麝香酮麝香祛痛搽剂原收载于《中国药典》2005年版Ⅰ部,根据2010年版《中国药典》计划任务表要求,本文将处方中的麝香修订为人工麝香,因为人工麝香为贵重药材,所以应对麝香祛痛搽剂中人工麝香制订质量控制方法,本文采用[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱[/color][/url]法对处方中的人工麝香药材进行定性鉴别,供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高,同时更能保证检测结果的真实性、准确性、重复性,阴性无干扰,专属性强,结果较满意。1 仪器与试药[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]Agilent6890型(FID检测器) Agilent色谱工作站 HP-5弹性石英毛细管柱(柱长30 m,内径0.25 mm,膜厚度0.25 μm) 聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm) 麝香祛痛搽剂样品: 武汉马应龙药业集团股份有限公司产品(批号080402,080403,080413),襄樊隆中药业有限责任公司产品(批号 080101,080201,080401),绍兴华通制药有限公司产品(批号C03A0753,C03A0809),李时珍医药集团有限公司产品(批号 2009020001)。麝香酮对照品,中国药品生物制品研究所产品(批号110719-200512) 无水乙醇为分析醇。2 方法与结果2.1 色谱条件色谱柱为HP-5弹性石英毛细管柱(柱长30 m,内径0.25 mm,膜厚度0.25 μm) 聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm) 柱温:程序升温:初温130℃,保持5 min后,每分钟升高0.8℃至180℃,保持2 min后,再以每分钟升高20℃至220℃保持5 min 检测器为FID检测器 检测器温度:250℃ 进样口温度220℃ 流速1.0 ml/min 理论板数按麝香酮计算应不低于20 000。2.2 色谱条件的确定2.2.1 色谱柱的确定取供试品(绍兴华通制药有限公司,批号:C03A0753)按供试品制备方法制备得供试品溶液,用聚乙二醇(PEG)-20M毛细管柱(柱长 30 m,内径0.32 mm,膜厚度0.5 μm)进样。结果表明,用聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm)能收获较好的分离效果,得到准确高效的分析结果。故选用:聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm)。2.2.2 程序升温条件的确定将“2.3.1”项下制得供试品溶液按程序升温:初温130℃,保持3 min后,每分钟升高1.5℃至180℃,再以每分钟升高20℃至220℃保持5 min 流速1 ml/min。结果表明,在该方案下,供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高。通过试验,确定色谱条件为:色谱柱:聚乙二醇(PEG)-20M毛细管柱(柱长30 m,内径0.32 mm,膜厚度0.5 μm) 检测器为FID检测器 进样器温度:220℃ 检测器温度:250℃ 分流进样(分流比1∶1) 程序升温:初温130℃,保持5 min后,每分钟升高0.8℃至180℃,保持2 min后,再以每分钟升高20℃至220℃保持5 min 流速1 ml/min 进样量:1 μl。2.3 溶液的制备2.3.1 供试品溶液的制备针对麝香酮的脂溶性,采用石油醚(30~60℃)提取的方法,这样组分的转移率较高,同时操作简便。但是因为本品麝香酮的处方量过低 必须进行富集的过程。因为样品为50%的乙醇制剂,与石油醚(30~60℃)会发生混溶,因此加入4倍量的水,再置于分液漏斗中用石油醚(30~60℃)提取2次,100 ml/次,石油醚(30~60℃)液自然挥干,可使麝香酮成分不会损失。取本品50 ml,加水200 ml,摇匀。置于分液漏斗中,用石油醚(30~60℃)提取2次,100 ml/次。合并石油醚,自然挥干。残渣用无水乙醇2 ml溶解,取上清液作为供试品溶液。2.3.2 对照品溶液的制备取麝香酮对照品,加无水乙醇制成每毫升含0.1 mg的溶液,作为对照品溶液。2.3.3 阴性对照溶液的制备按处方量制备不含麝香酮的阴性样品,按供试品溶液制备方法制备阴性对照溶液。2.4 专属性考察取对照品溶液、供试品溶液、阴性对照溶液,分别依法测定。在该色谱条件下,在与麝香酮对照品保留时间相同的位置不出现色谱峰,表明其他成分对麝香酮的测定无干扰。结果见图1~3。2.5 样品测定按正文所述方法,对各厂家各批次样品进行检验,结果均呈正结果。3 讨论因为麝香酮具有脂溶性,采用石油醚(30~60℃)提取的方法,这样组分的转移率较高,同时操作简便。但是因为本品麝香酮的处方量过低,必须进行富集的过程。因为样品为50%的乙醇制剂,与石油醚(30~60℃)会发生混溶,因此加入4倍量的水,再置于分液漏斗中用石油醚(30~60℃)提取,可使麝香酮成分不会损失。但是富集后导致杂质峰较多,用平温或较快的升温速率均达不到较好的分离效果,因此经过摸索将升温速率定为每分钟升高0.8℃,在该方案下,供试品中各组分出峰时间较合适,分离效果较好,而且各组分对应的峰面积和理论踏板数均较高,同时更能保证检测结果的真实性、准确性。【参考文献】[1] 国家药典委员会.中国药典,Ⅰ部[s].北京:化学工业出版社,2005.[/s]
用DR的8P标准品配制标准溶液,,和用Accustandar的8P混标配制出来的标准溶液,对这两套标准溶液分别进样,用DR配制成的标准溶液得到的峰面积要比用8P混标得到的峰面积小的比较多。前面6个的单峰面积要比对应浓度的混标配制得到的峰面积要小三分一左右,后2P(DINP,DIDP)则比对应的峰面积小二分一左右。我是把8P的标准品称量到100毫升的容量瓶里,再用二氯甲烷定容,得到8瓶1000PPM的单标溶液。然后逐级稀释到低浓度点的混标。请问我在用标准品配制混标时哪个步骤出问题的。请问有什么方法可以解决。我怀疑是邻苯标准品没有充分溶解才会导致峰面积比8P混标配制出来的要小。请问你们是怎么样配制邻苯标准溶液的。







 我要推广仪器
我要推广仪器
 下载APP
下载APP