[font=&]新人小白求助各位前辈,本人第一次使用安捷伦的[/font][url=https://insevent.instrument.com.cn/t/bp][color=#3333ff][url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url][/color][/url][font=&]检测尿液中单羟基多环芳烃,使用混标进行标准曲线的时候发现其中一个单羟基多环芳烃1-羟基芘的线性结果不好,R2只有0.98。有没有可能是1-羟基芘和1-羟基芘-D9没有分开的?内标和物质没分开?1-OHP不稳定分解?还是前处理影响得原因?。[/font]
选择R-(+)-苯乙基异氰酸酯作为手性衍生化试剂,与非索非那定生成氨基甲酸酯衍生物,通过反相高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法实现对映体的分离分析。非索非那定两个对映体衍生物在25~100 ng/ml浓度范围内线性关系良好(R~2=0.9992,0.9989),日内、日间精密度均小于10%。建立的非索非那定对映体柱前手性衍生化反相高效[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析方法灵敏、准确,可用于体外细胞模型中盐酸非索非那定立体选择性分析。 详见姚青青等,浙江大学学报(医学版). 2014,43(02)。
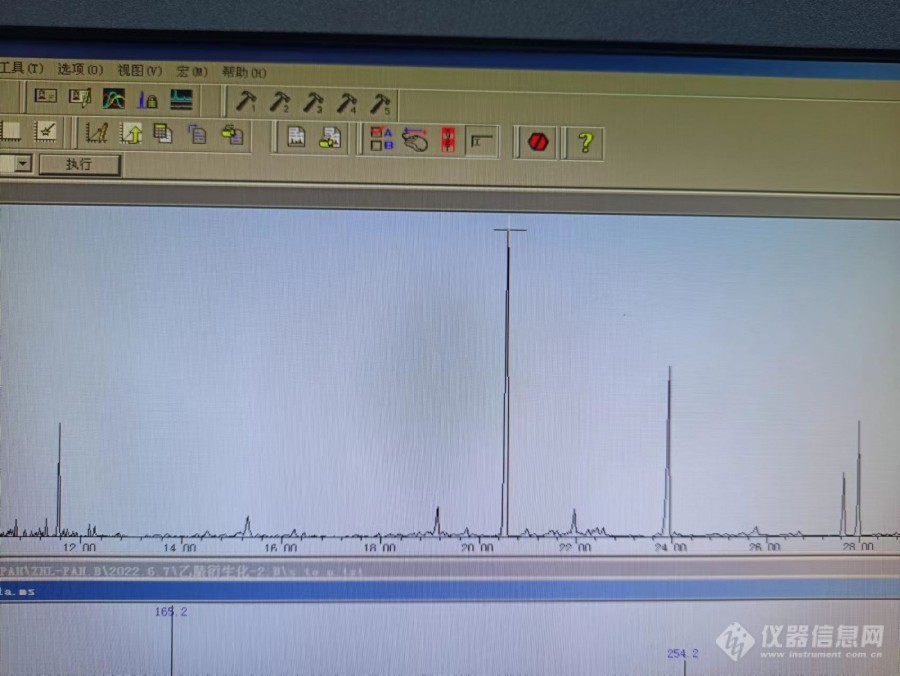
新人小白求助各位前辈,本人第一次使用安捷伦的[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]GC-MS[/color][/url]检测尿液中单羟基多环芳烃,在实际测样过程中,本人发现,样品在进样后在所有待测物质的最后总是出现一个累积的物质峰,这个峰会随着进样过程逐步累积。[img=,690,518]https://ng1.17img.cn/bbsfiles/images/2022/06/202206281637228491_6395_5547635_3.jpg!w690x518.jpg[/img],就像图上所示,我想可能因为是这个峰的原因,导致我检测一段时间后发现我调谐就无法通过,总会显示无法得到恒定峰宽。我在样品前处理时用到了衍生化试剂BSTFA(1%TMCS),所以这个峰是因为前处理过程中样品比较脏造成的,还是因为衍生化试剂对色谱柱损伤造成的呢,我接下来打算老化一下色谱柱再看一下,有没有做过相关实验的前辈指点一下的。
HPLC法在药物对映体的分离和测定中的应用 (转) 摘 要:由于药物对映体之间在药理、毒理及吸收等方面存在较大差异,因此,建立分离和测定对映体化合物的方法十分重要。本文综述HPLC法在分离和测定药物对映体的常用方法,包括手性衍生化试剂、手性流动相和手性固定相在药物对映体分离测定中的应用。对对映体化合物的分析鉴定有指导意义。 手性化合物的拆分是当前分析化学中最为活跃的领域之一,自然界中的许多化合物都是有旋光性的,而合成手性药物中大多(88%)是外消旋体,许多手性药物的对映体在生理过程中显示了不同生理活性。据研究反应停的致畸作用主要是由于其(S)-(-)异构体所致。因此,建立高专属性、高灵敏度、高分离度的对映体拆分和测定方法,对提高药物的活性、减小副作用,深入研究药物的作用机理等具有重要的理论和实际意义。对映体化合物之间除对偏振光的偏转方向不同外,具有完全相同的理化性质,因而其分离比较困难。传统的拆分方法有分步结晶、微生物和酶消化法等,或者用手性衍生化试剂将其转化成非对映体,然后根据其物理性质不同进行分离,但这些方法难于进行微量的分离和测定。80年代以来,随着快速、准确、微量的光学异构体的HPLC拆分及测定方法的建立和发展,使HPLC迅速成为药物对映体分离和测定最为广泛应用的方法。手性HPLC拆分法是以现代HPLC技术为基础,引入手性环境使对映异构体间呈现物理特征的差异而进行分离。通常分间接法和直接法,前者是对映体混合物以手性试剂作柱前衍生,形成一非对映体,然后以常规(偶也见手性)固定相分离。后者是直接以手性流动相(CMP)或手性固定相(CSP)直接进行分离。1 手性衍生化试剂法 手性衍生化试剂(CDR)法是在分子间引入手性中心,其产物为非对映异构体(diastereomer,DSTM),从而进行分离。下列情况通常选用CDR法进行拆分:(1)不宜直接拆分。添加某些基团,以增加色谱系统的选择性。如游离胺类在CSP上往往是颇弱的色谱性质,生成中性化合物后则获显著改善。(2)提高紫外或荧光检测的效果。刘雁鸣等[1]用 NBD-(L)-APY荧光试剂柱前衍生化测定布洛芬对映体,提高了检测灵敏度。对CDR的要求通常为:溶质分子至少有一个(多个时其性质各不相同)功能团供衍生(多为-NH2,-OH或-COOH)。光学活性试剂必需是手性高纯度;反应条件必须温和、简便;宜附有发色或荧光基团。目前,已有许多商品化的CDR可供选用,常见的CDR可分为以下几类:(1)异硫氰酸酯和异氰酸酯类 此类试剂易与大多数醇类及胺类化合物反应进而被分离,如麻黄素类,肾上腺素类,肾上腺素拮抗剂,儿茶酚胺类等。王亚芹等[2]采用S(+)-1-(1-苯基)乙基异氰酸酯为衍生化试剂分析了血浆中普罗帕酮的对应体,并研究了其在健康人体内的药代动力学。邱宗荫等[3]用乙酰葡萄糖异硫氰酸酯(GITC)为柱前CDR,以反相HPLC法测定血浆中地佐西平对映体的血药浓度,线性范围为5~200μg.L-1。陈冰等[4]用GITC为柱前CDR,用反相HPLC法测定血浆中普罗帕酮对映体的血药浓度,适合用于临床药动药效学研究。(2)萘衍生物类 由于此类化合物有利于提高立体选择性和检测灵敏度,因此萘的各种衍生物用作手性试剂十分普遍。Wainer等[5]选用萘甲醛(NDH)为手性试剂,与其缩合成恶唑烷衍生物,成功地分离了麻黄碱、4-甲氧基麻黄碱、伪麻黄碱。Bhatti[6]等用S-(+)-1-(1-萘基)-乙基异氰酸酯为CDR,用HPLC法测定了人血浆中美托洛尔对映体浓度。(3)酰氯与磺酰氯类 此类试剂可与化合物直接缩合,或与样品反应后,再引入其它基团,合成更有利于拆分与检测的衍生物。Sallustio等[7]以SOCl2与芳丙酸类消炎镇痛药如2-苯丙酸、酮洛芬及非诺洛芬的血浆样品提取物反应,然后再与R-2-苯乙胺成酰胺衍生物,产物以NP(Sil,乙腈∶二氯甲烷,5∶95)分离,异构体均可完全拆分。(4)光学活性氨基酸类 为最早采用的色谱手性试剂,为提高反应活性和定量回收率,常将羧基转化成酰氯、酸酐等。此类试剂广泛用于胺、羧酸及醇类药物,尤其是氨基酸类,其衍生化法多基于肽合成原理。本类方法要求手性药物具有活泼反应基团,同时两个对映体的衍生化速度应相同,否则会引起非对映体与原对映体的组成产生差异,另外要求手性衍生化试剂光学纯度高,反应要迅速、彻底,因此应用受到一定限制。
我现在分析一个含多个手性中心的物质,非对映异构体有好几个,其中有几个用反相分不开,用正相分开了,现在要写分析方法,对于这几个杂质的纯度测定应该归属到"相关物质"还是"非对映体纯度"一项不太确定,请大家给点建议!
各位老师: 实验中碰到下面难题:一个含有2个手性中心的化合物,有4个对映异构体;顺利将该化合物四个对映体拆分后,利用在线旋光测出了这四个对映体的旋光性,依次为 (+)/(-)(+)/(-);问题是:这四个对映体的绝对构型(R)/(S)目前条件没有办法得到;在如何描述这个对映体上碰到了麻烦! 对于一个仅含有一个手性中心的化合物A,即便不知道其绝对构型,如果测定出了其对映体的旋光性,可以用 (+)-A 或 (-)-A 来表示该手性化合物的两个对映异构体;但对与含2个手性中心,有4个对映体的化合物,不知道其绝对构型,仅知道旋光性,按照上面的命名显然会造成歧义;目前也没有查到率属于上述情况的能供参考的文献,请各位老师支招,有没有比较好的办法来表示这四个对映体?麻烦了,非常感谢!
最近想要分离一个手性对映体,原料为L-酪氨酸,S构型,其在反应过程中容易生成另外一种构型,看了一些资料文献,说在不用手性柱的情况下也可以尝试分离,请问这个能否用普通C18柱分离?有几个加手性添加剂的条件,使用L-脯氨酸和硫酸铜,普通C18柱能否用这个流动相?(问了工程师,工程师说是不行,但是很多小伙伴貌似都有使用……)如果使用了这个流动相,需要注意什么,仪器如何清洗比较好?是否有做过此类分离的小伙伴,提供一下经验~~~万分感谢~
[size=3][b]请问在对手性化合物(RS)原料药进行光学纯度控制中,用手性柱对其杂质对映异构体(SR)进行控制,而用普通的C18柱对非对应异构体(RR+SS)进行控制,但此RR与SS在C18上是重合的,这样可以吗?前提是该四个异构体无法在手性柱的一个流动相体系中出现,所以才出此下策。[/b][/size]
采用的是CHIRALCEL OJ柱,正相条件下分离多奈哌齐(安理申)。第一个求助问题是:没有左右对映体的对照品,无法确认R或S.目前局限性的条件:1、没有手性制备的条件。2、因为做的是体内药物分析,所以没有办法做旋光度测定。想请问有人做过此法拆分的么?或者类似结构的拆分。结构图见 [img]http://bbs.antpedia.com/attachments/month_1003/20100302_7a36b10b8b06da13ae16yx0neL7fzjbF.jpg[/img] [img]http://bbs.antpedia.com/images/default/attachimg.gif[/img][img]http://bbs.antpedia.com/images/attachicons/image.gif[/img] [url=http://bbs.antpedia.com/attachment.php?aid=3904¬ humb=yes][b]124.jpg[/b][/url] (6.83 KB)2010-3-2 16:21目前有报道采用CHIRALCEL OD柱,正相分离,R先出峰。能以此推断OJ的出峰顺序么?第二个求助问题,由于没有左右旋对照品,那么做血药浓度测定时,左右旋体标准曲线浓度是以消旋体的一半为准么?谢谢!参考:[url]http://bbs.instrument.com.cn/shtml/20100302/2424734/[/url]
各位版友,大家好。 目前手上有1化合物及其对映体,纯度均大于95%,需开发检测方法,其本身无紫外吸收。为提高效率,准备委外与自行开发并行,现寻求各位版友帮助,帮忙提供一些第三方拆分机构信息。PS:已联系大赛璐,YMC,菲罗门(均无对应检测器),研创已送样。望各位版友提供更多的资源,万分感谢。[img]http://simg.instrument.com.cn/bbs/images/default/em09505.gif[/img]
问题是:我要做一个物质非对应异构体(AB两个对映异构体)的液相色谱谱方法学的验证,但是我得不到纯的A或B的对照品。所以在做验证的时候,里面很多项目没办法进行。比如说:检测线、回收率等等。等待大家的支持!
非对映异构体仅仅是构型上的差别,为何理化性质会差别很大呢?而且液相分析时很多非对映异构体用普通的反相就能分开,为什么仅仅一个构型上的差别会导致这么大的理化性质差异呢?求指教
[color=#444444]液相色谱质谱联用时,比如在液相色谱端进一针DL-苯丙氨酸,那么在液相的色谱图上理论上会出现DL-苯丙氨酸对映体的两个峰,那么当样品流到质谱检测器时,质谱的总离子图上是出现一个峰还是两个峰?液相负责分离,质谱负责定性,质谱可以确定化合物的分子量和分子式,但是可以确定化合物的左右旋对映体么?[/color]
各位大侠,有谁能告诉我桧烯有几个对映体呀?在NMR氢谱上可以看出区别吗?最近分挥发油,分到桧烯,可是有对映体,需要测光学纯度,但是条件有限,想知道核磁方法可以解决吗? http://imgsrc.baidu.com/baike/pic/item/814b07d89e5dc77333fa1c8e.jpg
如题,固体样品上的氢都是以羟基的形式存在的。这时候在红外上羟基特征吸收峰的个数和固体核磁氢谱上的峰的个数有对应关系么?简而言之,它俩是相同的么?
请问对映体分离时,两个对映体的有效电泳淌度差和分离度的变化趋势应该是一样的吗,我在考察条件时出现了不一致的现象,不知道是什么原因
使用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]检测尿中多环芳烃代谢物,其中的两个单羟基代谢物为1-羟基芘和1-羟基芘-D9,请问1-羟基芘的性质有哪些?是否会影响实验检测的结果?
一对对映体在正离子模式下两个峰,负离子模式下只有一个峰这种情况可能吗?
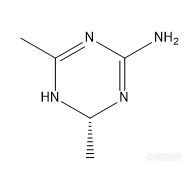
[img=,192,172]https://ng1.17img.cn/bbsfiles/images/2020/08/202008201612416491_7888_3116636_3.jpg!w192x172.jpg[/img]什么衍生化试剂比较合适拆分如图的对映体?
请问外消旋体在碳谱和氢谱上有什么特点,如何利用核磁来区分对映异构体和非对映异构体
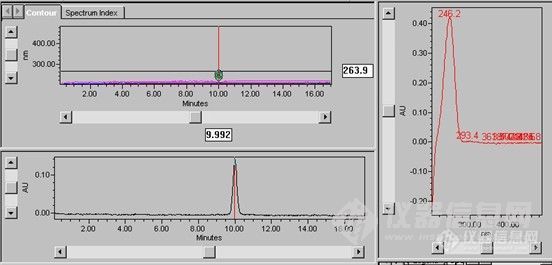
【实验背景】 对于做植物提取分离的人而言,利用各种手段分离得到单体化合物(即通常所说的纯品),然后得用化学法或者NMR(最主要的手段)进行结构鉴定,再进行各种活性筛选及测试,这是一条基本的路子。 这条路子,涉及的一个重要的环节就是化合物比对。分离得到的纯品多了,就要进行化合物比对。比对的目的,是避免将同一单体化合物多次进行NMR测试,这样会造成人力、物力和时间的浪费。 是的,我分到了两个单体化合物,管它们叫A和B吧,因为结构我还没有确定,现在,我要将它们进行比对。以免浪费老板的money。【仪器试剂】水:重蒸水 甲醇:色谱纯,天津大茂 氯仿:分析纯,凯信甲醇:分析纯,凯信 液相:Waters 高效液相,Waters 996 DAD检测器,Waters Model 600 controller液相色谱手机:HTC野火,薄层图谱扫描【薄层色谱条件】硅胶GF254板(青岛海洋化工):2*5cm展开剂:氯仿/甲醇=2/1【液相色谱条件】 色谱柱:Ultimate XB-C18柱(5μm, 4.6x250mm)、某国产品牌色谱柱(5μm, 4.6x250mm)流动相:甲醇/水(V:V),40%(某国产品牌)、45%(Ultimate XB-C18柱)流速:1.0ml/min柱温:柱温箱30度检测波长:全波长扫描进样量:随意【比对过程】1. 正相TLC共薄层,如果Rf值不一样,为不同单体化合物;如果Rf相同,需要用HPLC进一步确认2. 如果HPLC也显示单一峰形,基本可以说明是相同单体化合物;如果是非单一峰形,为不同单体化合物【实验过程】共薄层的意思,说白了就是三个点:A点、B点、AB点。从下图结果来看,正相上色谱行为一致。展开剂:氯仿/甲醇=2:1(右边为刚显色时拍的照,左边为显色30min后拍的照)http://ng1.17img.cn/bbsfiles/images/2011/11/201111142246_330498_1745326_3.jpgTLC显示单一斑点,不能证明他们就属于同一个单体化合物,需要用液相进一步确认。我曾从一个斑点分离得到10多个单体化合物,呵呵,类似的样品很多。最开始的时候用的是某国产品牌色谱柱,流动相为40%甲醇/水。对A、B分别分析(11月5日分析),保留时间相差不大(11.59min、11.24min)。http://ng1.17img.cn/bbsfiles/images/2011/11/201111142255_330501_1745326_3.jpghttp://ng1.17img.cn/bbsfiles/images/2011/11/201111142255_330502_1745326_3.jpg保险起见,对他们进行合并进针,显示同一峰形。http://ng1.17img.cn/bbsfiles/images/2011/11/201111142255_330503_1745326_3.jpg由于此国产品牌色谱柱使用时间较长,柱效已经不能跟新柱子比了。所以,即便显示单峰,我仍真心希望A、B不是同一个单体化合物。于是没办法,再次启用Ultimate XB C18(45%甲醇/水)进行分析。结果,几多欢喜几多愁!!!!!显示单一峰形!!!!!http://ng1.17img.cn/bbsfiles/images/2011/11/201111142258_330504_1745326_3.jpg【结果与讨论】1. 不同时间分析的样品,即使是同样的流动相,同样的色谱柱,同样的温度,由于仪器的不稳定,相同样品的保留时间相差还是挺大的,比如某国产品牌分析的样品,11月5日分析的保留时间是11min多,今天分析的是10min左右,这个不稳定因素,给我增加了很多工作量。2. 如果没有DAD检测器,工作量分增加很多,因为我还得跟其它类似化合物进行比对。有了DAD检测器,能清楚地了解化合物的紫外吸收情况,举个简单的例子,这里提到的A、B就不用跟黄酮类化合物比对了(黄酮一般是3个紫外吸收带)3. 某国产品牌色谱柱对样品的保留比Ultimate XB C18弱,基本上,要高5%。比如,用某国产色谱柱40%分析时的出峰时间,跟Ultimate XB C18用45%分析的出峰时间差不多。4. Ultimate XB C18的柱效,虽然没有具体算出来,但是从图上可以看出,很高。5. 为什么是几多欢喜几多愁?我庆幸的是,A、B是同一个东西,我没有浪费(测NMR很贵的);我也很郁闷,因为A、B是同一个东西,我又少了一个化合物!!!
请问有做1,5-二羟基萘氧化为5-羟基-1,4-萘醌(胡桃醌)的吗?怎么计算反应的转换率啊?用液相还是[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]更好一些呢,我用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]没测出来,柱温为280度。谁有经验哪?麻烦指导一下。拜托了
用普通的反相柱跑对映异构体,为什么会出现双峰?用的是普通反相柱而不是手性柱,对映异构体是纯品,无杂质,双峰峰高基本一致,请教其中的原理。
1、内标的一个选择要求是:与待测物保留时间应接近但不重叠。这里面的“接近”具体怎么理解呀?待测物的保留时间如果是10min,内标的保留时间是6min,这样算是接近吗?2、在测定10几种羟基多环芳烃时,只选了3-羟基菲13C6和6-羟基屈13C6这两种碳标,羟基芘定量的时候用的却是6-羟基屈13C6。按这个道理的话,岂不是这些物质我只选一种内标就可以了?3、针对2,目前看到的做法有两种:一种内标对应几种目标物来定量的;一种同分异构体对应了一种内标来定量的。在经费充足的情况下,一种物质对应一种内标,10种物质对应10种内标,进行定量,是不是更好。实际操作中,内标最终追求的到底是什么?如果我的目的是减少进样量的影响,计算回收率,扣除基质效应,我测十几种物质,选两种内标是不是就可以了,甚至一种。为什么大家有得选的多,有得选的少啊?好奇怪呀,怎么选的呢
为何要在对映体水平研究手性污染物(手性农药)?相比在外消旋体水体的研究,在对映体水平的研究有何不同?
rt核磁能检测并区分手性对映体吗?
求 HG/T 2745-2006(2-羟基-3-萘甲酸)标准一份07年3月才实施的
条件已经建立 这个需要做线性吗 怎么做线性 有必要做线性吗 欢迎讨论药品对映体只有一个
一个药物有多个手性碳,已知一个异构体的结构,能否通过NMR来判断另外几个非对映异构体的结构?希望各位多多帮助,谢谢!
各位版友,小弟我最近在开发一个拥有4个手性中心的化合物的HPLC,目前已经购买了这个化合物的5个杂质对照品了,其中一个杂质是对映异构体,准备买大赛璐的手性柱进行分离,其他4个杂质均为非对映异构体,目前分别采用了0.1%的三氟乙酸作为流动相与乙腈梯度洗脱,5mM磷酸二氢钾(PH7.0)/(PH2.5)与乙腈梯度洗脱,采用前一个流动相峰宽有点宽,后一个流动相峰形很好,但是无法分开,想请教有分离非对映异构体的版友们支招啊!兄弟我不甚感激!







 我要推广仪器
我要推广仪器
 下载APP
下载APP