世界主流药典标准中液相色谱柱应用情况分析
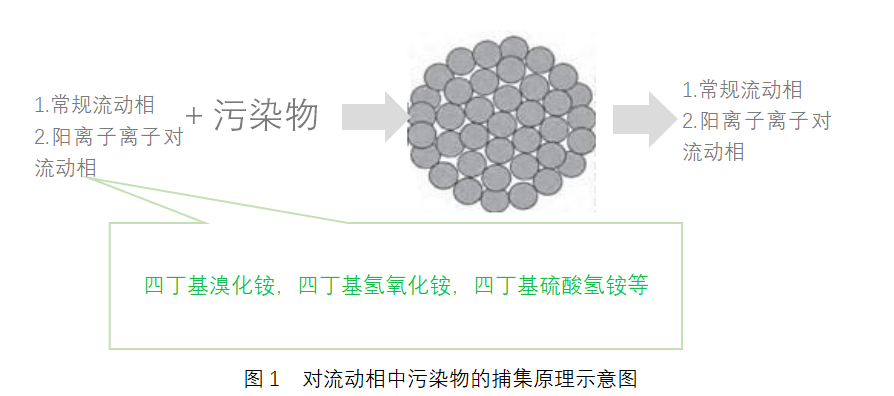
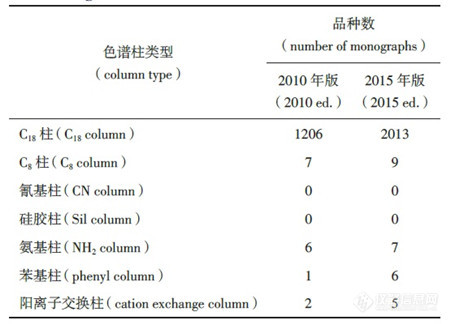
p style="text-align: center " strong液相色谱柱进展及其在药品标准中的应用(三)/strong/pp style="text-align: right "strong ——液相色谱柱在药典标准中的应用情况分析/strong/pp strongspan style="color: rgb(112, 48, 160) "3 液相色谱柱在药典标准中的应用情况分析/span/strong/pp 新颁布的2015 年版《中国药典》自2015 年12月1 日起正式实施。新版药典的最大变化是将原来各部的附录整合成了第四部,形成通则并对通则制定了更为合理的编码,液相色谱法列于2015 年版《中国药典》(四部)中通则0512 中。/pp strongspan style="color: rgb(0, 112, 192) "3.1 《中国药典》中使用的各类色谱柱/span/strong/pp 液相色谱方法在新版《中国药典》中得到了更广泛的应用,使用方法也更加合理。以二部化药为例,在修订的415 个品种中,有的新增了液相色谱检测方法,如本芴醇在有关物质检查项下,采用液相色谱法取代原来的薄层色谱法,规定杂质Ⅰ与主成分的分离度,以及杂质峰面积等要求,并列出了杂质Ⅰ的结构信息,这不仅使杂质的信息更加明确,而且对杂质限量的控制更加准确 有的对流动相进行了修订,如叶酸的含量检测中,通过添加离子对试剂―四丁基氢氧化铵,增加了叶酸的保留,流动相中甲醇的比例也从原来的每升80 mL 增加到270 mL,这样有利于防止色谱柱C18 键合相在高水相比例下产生疏水塌陷。/pp style="text-align: center"img src="http://img1.17img.cn/17img/images/201709/insimg/01c4db8b-eba9-4447-9396-504936620f73.jpg" style="" title="表1_副本.jpg"//pp style="text-align: center " strongspan style="color: rgb(0, 112, 192) "表1 2015 年版和2010 年版中国药典一部中液相色谱柱的使用情况/span/strong/pp style="text-align: center"img src="http://img1.17img.cn/17img/images/201709/insimg/77e42643-539c-48fa-a129-075f52643f0f.jpg" style="" title="表2_副本.jpg"//pp style="text-align: center " strongspan style="color: rgb(0, 112, 192) "表2 2015 年版和2010 年版《中国药典》二部中色谱柱的使用情况/span/strong/pp 但是,与液相色谱柱和填料种类的快速发展相比,在中国药品标准中,包括在《中国药典》中,高效液相色谱柱的应用显得较为单调,缺乏活力。表1、表2 分别列出2015 年版和2010 年版《中国药典》一部和二部使用液相色谱柱的情况。由表2 可以看出,在各类药品分析中,绝大部分方法采用的是反相液相色谱法,色谱柱则是以C18 柱为主 与2010 年版相比,2015 年版《中国药典》中C8 柱的使用数量翻了1 倍 而其他各种类的液相色谱柱使用比例则较少。/pp strongspan style="color: rgb(0, 112, 192) "3.2 各国药典对液相色谱柱规定/span/strong/pp strong3.2.1 关于色谱柱类型描述的差异/strong/pp 美国药典对色谱柱分类则较为详细,收载的各类液相色谱固定相(柱)类型已经超过80 种,除C18 柱、C8 柱、氰基柱、氨基柱、苯基柱外,还有C6 柱、C4 柱、C1 柱、五氟苯基(PFP)柱等。根据是否化学改性,是否封端,是否增加多官能基团以及是核壳结构还是多孔型结构等不同,以C18 为基质的色谱柱分类为L1、L2、L42 和L67等,以C8 为基质的色谱柱分别有L7、L28、L42 和L44 等。L1 柱对应于目前使用的各种C18 分析柱,L2柱常作为保护柱使用。由此可知,美国药典提供的可选择的色谱柱比较丰富。/pp 不过,尽管各厂家或品牌C18 在分离效果上存在一定差异,美国药典却没有对各种商品化C18 再进一步细分。/pp 在英国药典中,当用到特定色谱柱时,色谱柱信息描述会具体到色谱键合相类型、尺寸、键合相官能团描述、是否封端、是否通过碱性脱活处理等。团描述、是否封端、是否通过碱性脱活处理等。/pp 和欧美药典相比,《中国药典》对液相色谱法的色谱柱描述过于简单粗放,色谱柱的种类明显偏少。方法中仅描述色谱柱填料种类的主要大类:如十八烷基硅烷键合硅胶(C18 柱)、辛烷基硅烷键合硅胶(C8柱),氰基硅烷键合硅胶(氰基柱)、氨基硅烷键合硅胶(氨基柱),苯基硅烷键合硅胶(苯基柱)等。使用者无法根据不同性质的化合物选择适合分离的色谱柱。/pp 为解决这一矛盾,满足某些特殊分析目的,或为了简化色谱柱选择的过程,新版药典在某些品种的标准正文中对色谱柱给出了具体描述及品牌的信息。/pp 如在新颁布的2015 年版《中国药典》新增品种拉米夫定及片剂中,含量测定及有关物质测定项对所使用的色谱柱描述为“用十八烷基硅烷键合硅胶为填充剂(Zorbax XDB-C18,4.6 mm× 250 mm,5 μm 或效能相当的色谱柱)”。检测人员可直接选择对应色谱柱进行检测,避免进行盲目的大量色谱柱筛选工作。但详细列明色谱柱信息描述似乎从一个极端走到了另一个极端,从完全的粗放转到特定的选择。在一定程度上,这种具体至色谱柱厂家或品牌仍不是很客观的方法。因为某种色谱柱并不一定仅有1 家公司生产或提供,除非经过同类型不同厂家多根色谱柱的充分研究和实验对比,才能规定具体的色谱柱品牌,否则就意味着可能放弃了使用分离更好的色谱柱。/pp 表3列举了中国药典与英美药典中几个色谱柱使用实例,以便比较各药典对色谱柱分类及应用情况。/pp style="text-align: center"img src="http://img1.17img.cn/17img/images/201709/insimg/b9c5dbd6-b7db-4675-8dc1-e4583a4f4ce2.jpg" title="表3_副本.jpg"//pp style="text-align: center "strongspan style="color: rgb(0, 112, 192) "表3 中国药典与欧美药典中几个色谱柱使用实例的比较/span/strong/pp 由表3可以看出,美国药典列出了色谱柱的尺寸、填料类型编号 而英国药典不仅列出了色谱柱的尺寸和颗粒粒径,还对固定相进行了详细的描述,如封端的十八烷基键合硅胶,适合高比例水为流动相的烷基键合硅胶,碱去活封端十八烷基硅烷硅胶,二异丙基氰基柱等。/pp 另外,以埃索美拉唑(esomeprazole)缓释胶囊为例,表4 列出在美国药典(USP 35-NF 30)官方网站中可以查询到分析用到的色谱柱信息。/pp style="text-align: center"img src="http://img1.17img.cn/17img/images/201709/insimg/4620d675-45bb-4f17-8713-e21264ea69f6.jpg" title="表4_副本.jpg"//pp style="text-align: center " strong表4 美国药典中埃索美拉唑使用的色谱柱信息/strong/pp 从表4 可见,美国药典对方法中用到的色谱柱进行了归类和详细描述,也列出了替代的色谱柱。对于分析人员来说,提供了色谱柱选择方面的便利性。总之,在中国药典中,无论是色谱柱填料种类,还是色谱柱填料粒径和孔径等方面的描述,均显得较为简单、粗放,科学性和严谨度均有待提高。br//pp strong3.2.2 关于使用不同色谱柱时的方法转化/strong/pp 为满足系统适用性的要求,当选择1 根合适的色谱柱时,其尺寸应在一定要求的范围内。根据待分离分析药品的特性和实际分析需要,当使用的色谱柱填料尺寸规格发生变化时,各国药典对色谱柱柱径和填料粒径分别有相应的限定。美国药典( 621 CHROMATOGRAPHY)在色谱适应性要求中对色谱柱长度、粒径、内径等变化范围作了限定。在USP 36及以前的版本中,无论是等度还是梯度条件,色谱柱的粒径可以减小50%,不能增大 柱长有70% 的变化选择余地,流速也可有50% 的变化范围,色谱柱的内径以及进样量可根据情况调整。不过,从USP 37 起,在等度条件下,色谱柱尺寸发生变化的范围采用柱长与粒径的比值(L/dp)或柱效N 来进行限定,要求L/dp 保持恒定,或者N 的值介于-25%~+50% 之间。在梯度条件下,则色谱柱尺寸不宜发生变化,否则需要做方法的验证,见表5。!--621--/pp style="text-align: center"img src="http://img1.17img.cn/17img/images/201709/insimg/51f50c94-cd62-4ae5-a52a-d6da0390e989.jpg" title="表5_副本.jpg"//pp style="text-align: center " strongspan style="color: rgb(0, 112, 192) "表5 美国药典对色谱柱尺寸及条件变化的限定/span/strong/pp 中国药典虽然对色谱柱柱径和填料粒径也有相应规定,但是仅仅区分亚2 微米柱和常规柱(中国药典现在实际上使用的几乎都是常规柱)。某些特殊分析中,如复杂组分、指纹图谱和有关物质的分离,常对色谱柱有更苛刻的要求,即使明确了色谱柱填料具体种类,常规柱的柱内径和填料粒径范围定义太宽,会由于色谱柱的内径和填料粒径的差异,无法实现理想的分离和重现性的效果。/pp 按照仪器公司商业化的概念,采用亚2 微米色谱柱的方法为超高效液相色谱法,采用常规柱的方法为高效液相色谱法。但是,简单地根据粒径的不同将色谱填料分为亚2 微米填料与常规柱填料(3~10 μm)并不是一种科学的分类法,至少未能涵盖粒径为2~3 μm 的色谱填料柱。以美国药典要求的色谱柱粒径变化要求,当选择粒径2.7 μm 的色谱住替代5 μm的色谱柱时,其变化的范围是允许的,只要保持L/dp或N 值在-25%~+50% 范围内。实际上,填料粒径对色谱分离的影响是一个量变过程,粒径在限制性范围内改变不会引起分离机理的改变。但是,量变到一定程度必然引起质变,质变是量变的必然结果,当粒径降低到一定程度时,高效液相色谱仪到超高效液相色谱仪的质变归因于填料粒径大小降低到一定程度引起的压力突变,进而可导致分离机理的改变和各成分峰的保留时间变化。因此,使用常规柱填料或亚2 微米填料的色谱方法转化时,方法验证是必要的,但是,中国药典还没有明确规定应如何验证以及选择何参数进行验证。/pp 尽管中国药典2015 年版没有将超高效液相色谱法作为一个新方法单独收载,并不是否认此技术革新,而是在高效液相色谱法中作了系统的、科学的、实事求是的描述。这样既解决了概念上混乱的问题,也是对这一技术革新在药物分析,特别是在标准中应用的一种认同,对这一技术在药物分析、药品检验中的广泛应用将起着一定的积极推动、引导作用。毫无疑问,亚2 微米填料以及表面多孔型填料技术将是高效液相色谱发展的一个重要方向。/pp strong3.2.3 对药典或药品标准中使用和描述色谱柱的建议/strong/pp 由于商品化的色谱柱填料种类、粒径尺寸、颗粒类型或选择性差异等非常丰富,为了避免方法描述中的不确定性,建议对中国药品标准中包括中国药典使用的色谱柱种类进行归纳总结,国家药典委员会适时对各种可在药品中获得应用的色谱柱进行科学的归类划分,建立相应的色谱柱列表,以便药品标准工作者或检验人员参照使用 各色谱柱生产商或供应经销商应对归类划分工作积极密切配合,提供必要、准确、科学、可靠的相关信息和全面的技术支持。同时,为建立方法提供了更多的选择,应鼓励在建立分析方法时,药物分析工作者应大胆尝试使用各种有利于提高选择性的色谱柱,不要仅限于常规C18 柱等。/pp 从欧美药典对固定相描述或提供的信息来看,细化色谱柱的分类能给色谱分离分析带来积极影响:一方面,由于可从一大类填料中选择到最适合的色谱柱用于分析,从而可获得最佳的分离效果 另一方面,在复杂体系分离时,如中药成分分析或化学药有关物质测定中,如在药品标准中明确规定了色谱填料性质参数的描述信息,有利于克服复杂基质的干扰,提高方法的可靠性,或提高色谱柱的选择性。/pp 在建立相关药品标准时,应适当增加色谱柱尺寸如长度、内径、粒径等的描述 必要时,在充分比对验证的前提下,是否对使用何种色谱柱品牌予以具体规定也是可以探讨的。/pp 为了提高色谱柱的使用寿命,当进行一些具有复杂基质或辅料的原料药或制剂分析时,建议尽可能地使用保护柱,并在方法中说明。在许多品种分离分析中,美国药典都采用了预柱,这对保护色谱柱不受污染,提高色谱柱寿命是极为有利的。/pp 建议中国药典适时在相关的通则中增加对方法转化的描述,提出方法转化的要求,这样有利于分析人员在方法转化时有据可依。/pp strongspan style="color: rgb(112, 48, 160) "4 结语/span/strong/pp 液相色谱柱技术的发展趋势是高效快速分离,亚2 微米填料色谱柱及亚3 μm 的表面多孔型填料在近年来得到了飞速的发展和应用,各种选择性的色谱固定相和多种分离模式解决了许多分离难题。色谱柱填料类型和种类繁多,在制定药典或相关药品标准时,有必要细化色谱柱的分类,从而有利于更科学、更高效地选择和利用恰当的分离技术实现药物中复杂组分的可靠分析。/pp span style="font-family: 微软雅黑, " microsoft=""strong注:近年来,液相色谱柱技术发展的非常迅速,这同时也促进了高效液相色谱法在药物分析中更为广泛的应用。据统计,一个典型的制药企业甚至可能会拥有成百上千支液相色谱柱,在一种药物分析方法的开发过程中,如何选择适当的色谱柱往往会给实验人员带来很多困扰。/strong/span/ppspan style="font-family: 微软雅黑, " microsoft=""strong 本文献原文刊登于《药物分析杂志》2017年37卷第2期,作者为洪小栩、石莹、宋雪洁等八人,分别来自国家药典委员会、扬子江药业、安捷伦科技和江苏省食品药品监督检验研究院等单位。本文为该文献的最后部分,详细介绍了世界主流药典及中国药典中液相色谱柱的使用情况,为广大色谱柱用户以及色谱柱供应商提供了相关参考。/strong/span/ppbr//p


























 我要推广仪器
我要推广仪器
 下载APP
下载APP