原料药盐酸米多君标准(国内:YBH09452006),5-氨基乙酰丙酸盐酸盐标准。若有特殊要求也可站内信息联系!有国外标准也行。
[em0815] 哪为老师有盐酸多西环素和多西环素一水物标准BP2008和EP6.0版部分.请分享一下.不胜感激.急需.
烟酸、烟酰胺的标准储备液浓度的校正中,为什么紫外的吸光值测不出来,每次都是0.1A,都是按照国标做的
大家有接触过盐酸联苯胺或其它芳香胺的盐酸盐吗?比如,盐酸联苯胺,4-氨基偶氮苯盐酸盐,等。这些用在纺织品的用途是什么?对人体有什么危害?有标准或技术方法可以检测吗?
产品简介: 盐酸吡哆辛,磷酸吡哆醛(英语:Pyridoxal phosphate;PLP)是维生素B6的活性形式。这种维生素主要包括三种天然有机化合物:吡哆醛、吡哆胺与吡哆醇。磷酸吡哆醛参与催化的几种反应有:转氨基作用、α-脱羧作用、β-脱羧作用、β-消除作用、γ-消除作用、消旋作用以及羟醛反应。作用与用途:1.生化研究。主要用于医药,或进一步合成磷酸吡哆醛和脑复新等药物,也用作饲料添加剂,食品添加剂,作为紫外线吸收剂添加于化妆品中。2.维生素B6在一般动植物性食品中存在,尤其是蛋黄、肉类、鱼类、乳汁及各种谷类粮食中的含量比较丰富,且人体肠道细菌也可以合成,一般不会出现维生素B6的缺乏。我国规定维生素B6可用于强化固体饮料,最大使用量7~10mg/kg;在强化婴幼儿食品中使用量为3~4mg/kg;在强化饮料中最大使用量为1~2mg/kg.3.用作饲料营养强化剂,可促进动物体内氨基酸的代谢,提高饲料的利用率,一般用量为3~6mg/kg。4.大部分用作医药或进一步合成磷酸吡哆醛和脑复新等药物,也用作饲料添加剂,食品添加剂.还作为营养添加剂,紫外线吸收剂用于化妆品中。
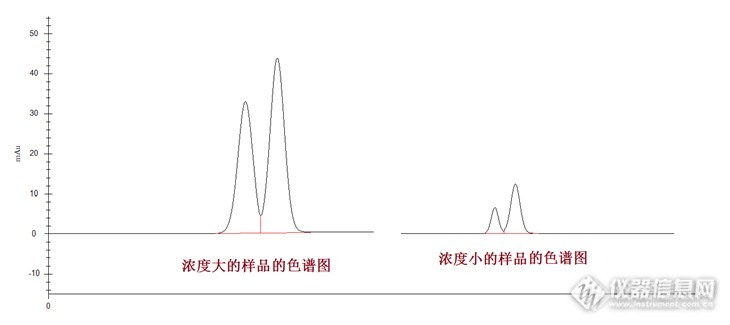
大神们,有做过食品中烟酸和烟酸胺的测定的么,GB5009.89-2016.要参加中检院的能力比对奶粉中烟酸和烟酸胺的测定,第一次接触这个项目,有什么需要注意的地方,包括溶剂哪个厂家好,柱子哪家的出峰效果好,标准溶液买哪里的等等。。。。请做过此项目的同学留下你们宝贵滴经验,先谢谢啦!!
请问大家阿有盐酸付玫瑰苯胺的配制方法啊?我做空气中的二氧化硫,按照标准配出来的颜色是紫红色的也,怎么不褪色?真是急死人

盐酸多西环素注射液(IV)含量的测定实验部分:原理:精密量取处理好的样品及对照品,稀释后注入高效液相色谱系统,C18色谱柱分离,紫外检测器检测,外标法(保留时间定性、峰面积定量)计算,得出该样品中多西环素的含量。仪器及试剂仪器:安捷伦1100高效液相色谱仪,配置紫外检测器+等度泵+柱温箱+在线脱气机等电子天平(万分之一)试剂甲醇:色谱级 已睛:色谱级 草酸铵,二甲基甲酰胺,磷酸氢二铵,氨水,盐酸均为分析纯标准物质土霉素对照品(中国兽医药品监察所生产)土霉素(化学对照品)(中国食品药品检定研究所生产)美他环素(化学对照品)(中国食品药品检定研究所生产)-多西环素(化学对照品)(中国食品药品检定研究所生产)多西环素(化学对照品)(中国食品药品检定研究所生产)色谱条件:流动相:0.05mcl/l草酸铵溶液:二甲基甲酰胺:0.2mcl/l磷酸氢二铵溶液=65:30:5(ph=8.0)柱温:35℃ 检测波长:280nm流速:1.0ml/min进样量:20um对照品及样品的制备系统性试验:称取土霉素、美他霉素、-多西环素、多西环素对照品适量,加0.01mcl/l盐酸溶液溶解并稀释制成每1ml含土霉素、美他环素、多西环素及多西环素均加0.08mg的混合溶液。上机测定:理论塔板数n不低于1500,分离度R大于1.5样品的配制1.1盐酸多西环素注射液(IV)10ml:0.5g精密量取供试品1ml,置于50ml量瓶,加0.01mcl/l盐酸溶液溶解并稀释至刻度,摇匀。精密量取2ml,置于25ml量瓶中,加0.01 mcl/l盐酸溶液溶解并稀释至刻度,摇匀。1.2对照品溶液的制备(多西环素对照品含量为85.2%)精密称取多西环素对照品约25mg,置于25ml量瓶中,加入,加0.01 mcl/l盐酸溶液溶解并稀释至刻度,摇匀。精密量取2ml,置于25ml量瓶中,加0.01 mcl/l盐酸溶液溶解并稀释至刻度,摇匀色谱图系统性试验溶液色谱图 :http://ng1.17img.cn/bbsfiles/images/2015/09/201509281558_568305_2315779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/09/201509281559_568307_2315779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/09/201509281604_568308_2315779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/09/201509281604_568309_2315779_3.png 图2标准品色谱图http://ng1.17img.cn/bbsfiles/images/2015/09/201509281607_568311_2315779_3.pnghttp://ng1.17img.cn/bbsfiles/images/2015/09/201509281607_568312_2315779_3.png 图3 被测样品色谱图计算及结果 样品中被测物含量计算公式:标示量%=http://bbs.instrument.com.cn/xheditor/xheditor_skin/blank.gifA—供试品中被测物峰面积值As—对照品中被测物峰面积值Ms—对照品的重量。单位为g.M—供试品的取样量。单位为mlCs—对照品的含量。85.2%。Ds—对照品的稀释倍数。D—供试品的稀释倍数。B—样品的标示量。经计算该兽药中多西环素含量为99.7%,含量合格,符合药典规定。 结论该方法对药典方法略作修改,得到不错的检测效果。由图1可以看出,该方法的系统实验,理论塔板数为1883,分离度为4.4,满足检测条件。
大家好 请教个问题 我做液相色谱分析标准曲线,用邻氯苯胺盐酸盐做标准曲线,但是样品是邻氯苯胺硫酸盐,怎么计算邻氯苯胺的含量?谢谢
各位老师,关于71-3中0.07mol/L 的盐酸标定,有国标标准时说关于盐酸标定的么?
本人在此急求 中华人民共和国国家药品监督管理局标准(试行)中的关于"盐酸左氧氟沙星注射液"的标准,请大家帮忙!谢谢!
盐酸副玫瑰苯胺测空气中二氧化硫,标准曲线中空白的值低于书中表中所列的值!我的水浴温度是20度,书中列表显示该温度下空白吸光度应该是0.040,但我测得的是0.028,是不是我做的数值一定的跟书上的一样呢!?还是我所得的数据有问题呢?难道某一温度下仅仅对应某一特定的空白值么?有严格的要求么?
请问环保部的氯化物标准液,能否作为测定盐酸的标准物质呢!!!
[align=left] 意想不到盐酸羟胺的不稳定性对实验结果的影响[/align][align=center] [/align][align=left]在前面的二篇中提及了Tris的检测,经过近一个月的努力,顺利完成了Tris的方法开发和验证。今天谈谈另一个化合物的检测验证,盐酸羟胺,在Tris快完成之际,接到另一家客户的要求,检测某一药物中残留的盐酸羟胺。[/align][align=left]天下哪有这么好的事,盐酸羟胺和Tris结构类似,理论上检测方法可以完全套用,除了淋洗液和柱子不一样外,后面的补液模式可以完全照抄!分离方法可以基本按照Tris 的方式(阳离子分离,柱后补碱,安培检测)。[/align][align=left]在开始做之前,我问了对方,盐酸羟胺的稳定性,稳定。同时,我上网简单查了一下,没有提及不稳定的事,这样完全照搬Tris的检测模式,OK。[/align][align=left]在对方厂家测试人员的协助下,按照设计的方案,排好序列,连续进样80针就够了,时间大约14-16小时,为了加快速度,通宵进行,由于担心仪器晚上会突然断开(ICS5000)我通宵查看系统,果然半夜里软件连接突然断了一次,重新连接后就没再断开了,在连续进样的过程中,我感觉有点不对,很低浓度下,怎么没有盐酸羟胺的峰(理论计算是有呀,能看到),峰感觉在变小,似乎不稳定,半夜里只能继续做下去了。[/align][align=left]第二天在分析大批数据后,终于感觉到不对了,盐酸羟胺会分解!所有实验结果泡汤了!赶快上网查资料,的确有明确提及盐酸羟胺不稳定,但如何稳定没说呀!查到相关分析的一篇类似文献,提及用流动相溶解,但其数据和结果似乎表明其检测限比我们高近10倍,他们做的很有问题。怎么办?样品不稳定这样实验做起来就麻烦了,羟胺,盐酸羟胺,我明白了,没有单独的羟胺的实际样品,只有盐酸羟胺,这就意味着只要盐酸的存在,不稳定的羟胺变成了稳定的盐酸羟胺,酸性条件稳定,也理解了论文用流动相溶解的意思。[/align][align=left]重新设计实验,盐酸羟胺稀释不用水稀释而是用跟淋洗液几乎一样的淋洗液浓度进行稀释,在酸性条件下,这样可以避免盐酸羟胺的分解。重新配溶液上机,在仅仅改变样品的溶解稀释方式,整个实验的重复性非常好,盐酸羟胺峰面积稳定回收率合格,而实验仅仅改变了一点!![/align][align=left]在实验中,由于做检测限,盐酸羟胺的浓度非常低,羟胺水解的效应非常明显,当浓度高时,不是很明显,这就是为什么一开始做了半天没发觉的缘故。对比这二个实验,Tris和盐酸羟胺结构类似,测试方法几乎一致,但完全照抄在实际中却遇到问题,说简单很简单,说复杂也不简单。Tris作为缓冲液肯定是稳定的,这是二者的差异。[/align][align=left]这也验证了做方法学的主要关键之一,样品的稳定性必须先验证。[/align]
请教各位一下:在标定盐酸标准溶液时,用无水碳酸钠作基准准确还是用硼砂准确? 假如我要配制0.5mol/L的盐酸,标出的结果是0.4741mol/L,溶液的体积估计为13000mL,请问我应该加几毫升浓盐酸? 谢谢
奶粉中的维生素PP,有烟酸和烟酰胺2个,但是很多奶粉只标记烟酸含量,实际加的确是烟酰胺,而且国家标准也只标记烟酸的指标,我们的检测结果应该如何处理,烟酸和烟酰胺之间是否应该有个换算系数呢?
各位老师们,可以分享下,中国药典第二部2010版 盐酸小檗碱的质量标准不?我找不到这个资源,谢谢大家。
瑞士万通的905自动电位滴定仪,搭配光度电极。用EDTA滴定镍钴锰溶液的总量。如果滴定溶液中不加盐酸羟胺,初始电位很低很低,突越很不明显,加入盐酸羟胺初始电位就有700多mv,正常。但是观察溶液颜色,加入盐酸羟胺前后基本无变化。这是什么原因?滴定用的是可见光波长。

作者:陈云松; (丹东市中心医院;)摘要:目的:建立高效液相法测定多动宁胶囊中盐酸小檗碱含量的方法。方法:采用Diamonsil-C18TM钻石(4.6 mm×250 mm,5μm)色谱柱;流动相:甲醇-水(40∶60);检测波长:345 nm;流速:1.0 ml/min。结果:盐酸小檗碱在0.050 3~0.804 8μg范围内与峰面积呈良好的线性关系(r=0.999 8),平均回收率为98.33%,RSD=0.63%(n=9)。结论:本法简便、准确、重现性好,可用于多动宁胶囊中盐酸小檗碱的质量控制。谱图:http://ng1.17img.cn/bbsfiles/images/2012/08/201208201414_384699_1606903_3.jpg
《环境空气 氮氧化物(一氧化氮和二氧化氮)的测定 盐酸萘乙二胺分光光度法》HJ 479-2009曲线,分享下,吸量管做的标曲,检测条件严格按分析标准执行的(显色忘记放暗处外,是晚上做的一般日光灯亮度下显色;另对氨基苯磺酸高于50度热水溶解,不然溶解不了),空白0.001,室温20度,原始数据无修改,玻璃量具有自校都是A级,试剂和标准溶液都是现配,纵轴A-A0,横轴ug
请问:1,烟酸在什么情况下能转换成烟酰胺?2,我现在用液相法做奶粉中的烟酸烟酰胺,可是每次测都是有烟酰胺,没有烟酸,这是为什么?是不是烟酸都转过去了?谢谢!
瘦肉精莱克多巴、盐酸克伦特罗检测瘦肉精莱克多巴、盐酸克伦特罗检测仪器高效液相色谱仪,紫外检测器超声波振动器溶剂过滤器离心机分析天平恒温摇床固相萃取装置,带萃取小柱匀浆机氮吹仪试剂乙腈:色谱纯磷酸二氢钾:分析纯磷酸甲醇:色谱纯乙醚;分析纯正己烷:分析纯盐酸溶液乙酸缓冲液NaOH溶液超纯水标准品:莱克多巴、盐酸克伦特罗样品制备 看到上面的仪器和试剂,大家可能也会看出前处理的方法了。不管的饲料还是肉制品样品的制备,过程一般都是称取,粉碎(匀浆),溶解,超声提取,离心提取,过滤,固相萃取,吹干,过滤等。只是在提取时样品完全提取可能比较困难,提取条件一定得控制好,比如温度、振动、时间(如果其它前处理设备性能不够理想的话,处理时间尽量长一点)、提取试剂等因数的控制和把握。色谱条件色谱柱:Venusil CN ,4.6mm×250mm,5µm流动相:乙腈:0.01mol/L磷酸二氢钾(用磷酸调节pH值至3.0)=30:70(V:V)检测波长:215nm流速:1.0mL/min柱温:室温进样量:20ul标准品色谱图:http://ng1.17img.cn/bbsfiles/images/2013/12/201312281531_485166_2369266_3.png 分析实验讲的就是准确、可靠,准确可靠的方法就是好方法。实验过程中我们一般没必要把实验做的最好,但我们可以通过优化色谱条件把实验尽量最好。
方法:HPLC基质:标准溶液应用编号:103771化合物:盐酸曲唑酮固定相:Platisil ODS色谱柱/前处理小柱:Platisil ODS 5u 250 x 4.6 mm样品前处理:供试品:0.1g+60mL溶剂,震摇30min,溶剂定容到100mL。对照:供试品用溶剂稀释500倍。 注:溶剂为水:乙腈:二乙胺=650:350:0.4色谱条件:色谱柱: Platisil ODS 250*4.6 mm,5 μm(Cat#:99503) 流动相: 水:乙腈:二乙胺=320:680:0.4 流速: 1.5 mL/min 柱温: 40℃ 检测器: 254nm 进样量: 20μL文章出处:天津应用实验室关键字:盐酸曲唑酮、Platisil ODS、HPLC摘要:Platisil ODS检测盐酸曲唑酮。谱图:http://www.dikma.com.cn/u/image/2016/08/25/1472109147984782.pnghttp://www.dikma.com.cn/u/image/2016/08/25/1472109151321447.png
用盐酸标准溶液滴定氨水,可选择甲基红或酚酞作指示剂?
用盐酸标准溶液滴定氨水,可选择甲基红或酚酞作指示剂?
最近做了1批水样,水中可能有铁离子和铬离子。按照水质监测方法说的,加盐酸羟胺可以有效去除干扰。我想问下 盐酸羟胺是应该时候加入去除干扰,是在消解完水样后加入还是在加完1+9盐酸后加入才对?(PS:能简单解释下盐酸羟胺反应原理吗- - )
我听说现在盐酸不能作为标准溶液了,不知道对不对,请指教下。谢谢
GB/T 601-2002 中氯化锌标准溶液配置是:把氯化锌溶于1L盐酸溶液中(1+2000)。1+2000的盐酸溶液我侧过PH值,大约在3左右。可是,这氯化锌标准溶液是用来做 PAC检测用的(GB 15892-2003 or 2009)。PAC样品最后处理是加入10mL乙酸-乙酸钠缓冲溶液(PH=5.5),用二甲酚橙做指示剂。加入缓冲溶液,样品PH自然到了5.5,可是氯化锌是3,在滴定过程中,随着氯化锌的不断加入,缓冲溶液会不会被破坏掉呢?氯化锌一般是要用40mL的。在配氯化锌的时候能不能少加一点盐酸呢?把PH尽量控制在5.5左右。
我目前急需副产品盐酸标准 HG/T3783,网站搜索不到。请各位帮忙提供下。感激不尽!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
标定盐酸标准滴定溶液的不确定度分析 作者:吴文英 张春雨 唐惠兰 来源:中华医学研究杂志 在理化分析过程中,一切测量结果都不可避免地具有不确定度。盐酸标准溶液是常用化学定量参比物质,其标定值的准确性直接影响常规分析质量。笔者以GB/T601《滴定分析(容量分析)用标准液的制备》为依据配制并标定盐酸根据JJF1059-1999《测定不确定度评定与表示》分析其测量不确定度。简述由标定过程中得到的不确定度。 1 实验部分 1.1 测定方法[1,2] 准确称量270℃~300℃干燥至恒重的基准碳酸钠(99.95%~100.05%)约0.2g左右,电子分析天平(精度为0.1mg),置于三角瓶中,加入50ml水使之溶解,加指示剂,用盐酸标准液滴定至终点同时作试剂空白实验。 1.2 主要计量仪器与试剂 电了分析天平:AG204;酸式滴定管:50ml A级。 1.3 建立数学模型 C=m (V1-V2)×0.05300 式中 C:盐酸标准滴定溶液的浓度(mol/L);m:基准无水碳酸钠的质量(g);V1:盐酸标准滴定溶液用量(ml);V2:试剂空白实验中盐酸标准滴定溶液用量(ml);0.05300:与1.00ml盐酸标准溶液[C(HCl)=1.000mol/L]相当于以克表示的无水碳酸钠的质量。 1.4 盐酸标准滴定溶液的标定结果 为获得标准溶液重复测量的不确定度分量,对同一标准溶液进行8次独立的标定。测定数据见表1。 表1 盐酸标准滴定溶液的标定结果 略 2 测量不确定度来源 从检测过程和数学模型分析,标定盐酸标准溶液的不确定度主要来源,由四个方面所引起。(1)测量的重复性(A类不确定度);(2)基准无水碳酸钠的纯度;(3)测量使用的电子分析天平及量具;(4)其他相关常数。 3 测量不确定度分析 3.1 A类不确定度的分析 利用表1中的测量结果,按照A类评定测量重复性的标准不确定度。具体计算过程:重复测量的平均值计算式:=1 n∑8 i=1xi=0.09951mol/L 单次测量的标准差按贝塞尔公式计算s(x)为 s(x)=∑8 i=1(xi-)2 n-1=0.0001555mol/L 的标准差s()为 s()=s(x) n=0.000155 8=0.0000548mol/L=5.48×10-5mol/L 由测量重复性引起的相对标准不确定度为U(x):0.0000548/0.09951=0.055%。 3.2 B类不确定度分析 3.2.1 基准碳酸钠的纯度 基准碳酸钠的纯度为1.0000±0.0005,视为矩形分布0.00053=0.00029,则标准不确定度为:由基准碳酸钠的纯度引入的相对不确定度u(p)为:0.029%。 3.2.2 天平称量所引入的标准不确定度 干燥器与天平称量仓内均放置同质硅胶,视为相同湿度,称量时无吸潮。电子天平检定证书标出线性为上0.2mg;可视为矩形分布,则标准不确定度为:因为称量采用的是减量法,故称量的标准不确定度为0.2mg /3=0.12mg:因为称量采用的是减量法,故称量的标准不确定度为:2×0.122=0.17mg,则由称量引入的相对标准不确定度u(m)为:0.17mg/0.2018g=0.084%。 3.2.3 标定体积的不确定度 (1)滴定管的校准:滴定使用50ml酸式滴定管(A级),按照检定规程,其最大允许误差为±0.05ml,相对允许误差为±0.1%,按照矩形分布,则滴定体积的相对标准不确定度u(V)为:u(V)=0.1%/3=0.0577%。(2)环境温度:实验环境在空调条件下,室温近似20℃。温度在20℃左右,标准溶液的温度补正值非常小,对实验结果影响可忽略不计,所以在不确定度分析中不把一温度影响引起的不确定度列入考虑范围。(3)滴定终点的判断:终点时的误差±0.05ml(1滴的体积),两点分布,现由终点分布判断引入的标准不确定度为0.05ml:相对标准不确定度为0.05ml/38.32ml=0.13%标定体积的影响引入相对标准不确定度U(V)为0.0572+0.132=0.142%。 3.2.4 其他常数 基准无水碳酸钠摩尔质量引起的标准不确定度很小,可以忽略。 4 合成标准不确定度 测量重复性、基准无水碳酸钠的纯度、天平称量、标定体积等的不确定度相互独立,故将上述数据合成得盐酸的相对合成标准不确定度U(C)为0.0552+0.0292+0.0842+0.1422=0.176%。 5 扩展不确定度 实验测得盐酸标准溶液浓度为0.09951mol/L,则测量结果的合成标准不确定度U(C)=0.09951mol/L×0.176%=0.000175mol/L。若取包含因子K=2,得测量结果的扩展不确定度U=2U(C)=0.00035mol/L。 6 测量结果的表示 盐酸标准滴定溶液的浓度可表示为:(0.09951±0.00035mol/L,K=2)。 【参考文献】 1 姚正堂,将已峰.奶制品中蛋白质测定的不确定度分析.中华医学研究杂志,2005,5(6):6. 2 国家技术监督局.JJF1059-1999测量不确定度与表示.北京:中国计量出版社,1997,81. 作者单位: 214171 江苏无锡,无锡市惠山区疾病预防控制中心







 我要推广仪器
我要推广仪器
 下载APP
下载APP