大家知道脱水四环素、差向脱水四环素的cas号,结构式?谢谢
[color=#444444]大家好,我最近做盐酸四环素的标准曲线,标品溶于甲醇中,用液相色谱分析,流动相为甲醇-乙腈-0.01mol/L草酸水溶液,配比有待优化。我参考相关文献对配比调试了多次,但是10mg/L浓度的盐酸四环素甲醇溶液的响应值最高峰高才11.5!很多配比下都是4或者5的响应值。按道理响应值应该是几十几百才对!根本问题出在哪?急!!!![/color]
我现在要配制盐酸土霉素,盐酸金霉素,盐酸四环素,盐酸多西环素,这四种物质除了盐酸金霉素是微溶于水的,其他三种都是溶解于水,可是我配制1000mg/L,发现还是有没有溶解的,在配制的过程中感觉盐酸多西环素还是比较容易溶解于水的,但那三种配的过程中就有悬浮的放置一个晚上后好多沉淀有没有哪位大侠做过这方面研究啊,指点一下,十分感谢,十分感谢我用的液相色谱测峰,之前配过盐酸四环素,盐酸金霉素,土霉素,多西环素,也是溶解的不好,所以才改全是盐酸盐,但溶解的还是不好做标线用甲醇配的,溶解的还可以,出峰也不错,但在水里我是怎么也溶解不了
借用xuanleer的帖子提一下几个疑问:不知道从何说起,我们在做某些碱性化合物检测时,购买的标准品通常是盐酸盐、硫酸盐、草酸盐之类的,如下面的糠氨酸(二盐酸盐)以及莱克多巴胺盐酸盐、四环素盐酸盐等等,疑问:(1)想问下大家这类目标化合物在进(HPLC、GC)色谱分析时,(色谱峰)是以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在还是以游离碱盐酸盐的形式存在?(2)这类化合物不少选用酸性环境下进行HPLC/LC-MS分析,其原因是否是让目标化合物以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在? (3) 如果第(2)个问题是对的,哪进GC分析时得到色谱峰是以何种形式存在的?也是游离碱吗?欢迎各位老师专家解答啊!顺祝大家节日快乐!参考资料如下:糠氨酸的鉴定适用于《NYT 939-2005 复原乳的鉴定》,具体见附件。色谱柱:LAEQ-462572 CNW Athena C18-WP 液相色谱柱,4.6*250mm,5um流动相:A=0.1%三氟乙酸水溶液;B=0.1%三氟乙酸乙腈平衡:A:B=99:1梯度:0min:99%A/1%B,25min:79%A/21%B检测波长:280nm流速:1ml/min进样浓度:2ppm柱温:室温标准品:CDDD-SC494-10MG,糠氨酸(二盐酸盐),品牌 NeoMPS,现货供应。参考:http://bbs.instrument.com.cn/shtml/20120903/4223471/
借用xuanleer的帖子提一下几个疑问:不知道从何说起,我们在做某些碱性化合物检测时,购买的标准品通常是盐酸盐、硫酸盐、草酸盐之类的,如下面的糠氨酸(二盐酸盐)以及莱克多巴胺盐酸盐、四环素盐酸盐等等,疑问:(1)想问下大家这类目标化合物在进(HPLC、GC)色谱分析时,(色谱峰)是以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在还是以游离碱盐酸盐的形式存在?(2)这类化合物不少选用选型环境下进行HPLC/LC-MS分析,其原因是否是让目标化合物以游离碱的形式(糠氨酸、莱克多巴胺、四环素)存在? (3) 如果第(2)个问题是对的,哪进GC分析时得到色谱峰是以何种形式存在的?也是游离碱吗?欢迎各位老师专家解答啊!顺祝大家节日快乐!参考资料如下:糠氨酸的鉴定适用于《NYT 939-2005 复原乳的鉴定》,具体见附件。色谱柱:LAEQ-462572 CNW Athena C18-WP 液相色谱柱,4.6*250mm,5um流动相:A=0.1%三氟乙酸水溶液;B=0.1%三氟乙酸乙腈平衡:A:B=99:1梯度:0min:99%A/1%B,25min:79%A/21%B检测波长:280nm流速:1ml/min进样浓度:2ppm柱温:室温标准品:CDDD-SC494-10MG,糠氨酸(二盐酸盐),品牌 NeoMPS,现货供应。参考:http://bbs.instrument.com.cn/shtml/20120903/4223471/
用液相色谱做四环素的标液时,出现了土霉素的峰。本人同时做土霉素、四环素、金霉素和强力霉素。单标进样只有四环素中有土霉素的峰,而且土霉素峰还比四环素的高,其他标物单标均十分干净,包括土霉素单标,也未见杂峰。不是进样针头污染问题,也不是进样瓶污染的问题。难道是标物的问题?我同时做了盐酸四环素和液体四环素标物两种,均有土霉素的峰。请问到底是怎么回事?请大家给个意见。
各位大侠,国标GB/T 5009.116-2003畜、禽肉中土霉素、四环素、金霉素残留量的测定中所用的标品是:土霉素CAS:79-57-2; 四环素CAS:60-54-8; 金霉素CAS:57-62-5还是盐酸土霉素CAS:2058-46-0;盐酸四环素CAS:64-75-5;盐酸金霉素:64-72-2?
各位大侠,国标GB/T 5009.116-2003畜、禽肉中土霉素、四环素、金霉素残留量的测定中所用的标品是:土霉素CAS:79-57-2; 四环素CAS:60-54-8; 金霉素CAS:57-62-5还是盐酸土霉素CAS:2058-46-0;盐酸四环素CAS:64-75-5;盐酸金霉素:64-72-2?
[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]检测四环素,峰拖尾严重,峰形也不好,请问用什么色谱柱比较适合做四环素?我用的流动相是甲醇和0.1%甲酸水。现在用的是安捷伦的300SB-C18还有就是在前处理时应注意些什么?
[color=#444444]想测量一下盐酸四环素催化臭氧化的反应机理,只有反应之后的溶液,怎么知道反应生成了什么物质。我送去检测[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url],人家和我说要流动相比例,停留时间什么什么类的还不能有无机盐。求各位大神帮忙指点应该怎么测定啊?我听说可以用[url=https://insevent.instrument.com.cn/t/bp][color=#3333ff]气质联用[/color][/url]或者是紫外可见光度计全扫,这都是怎么回事啊?[/color]
本人是质谱新手。仪器 AB 5500。最近在优化四环素类质谱参数,在第一步找母离子的时候目标质量数的响应值只有3e5,而且还不是响应值最高的,有其他的质量数跟目标质量数的响应值一样。按照工程师的培训的要求母离子响应值应该在1-3e6。 最近一次调谐是在两个星期前。各位老师帮忙分析一下是哪的问题?浓度是100ng/ML,溶剂是1:1甲醇水 标注品是固体 盐酸金霉素、盐酸土霉素、盐酸强力霉素、盐酸四环素。
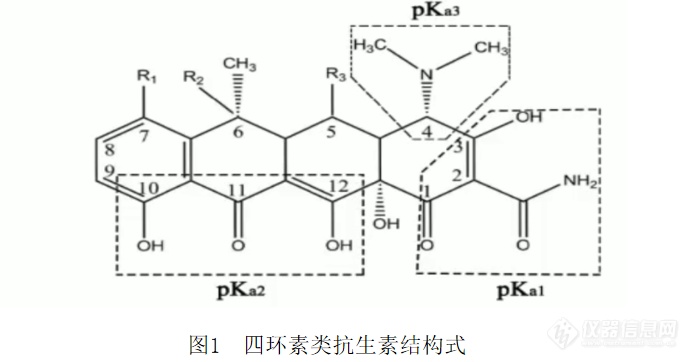
四环素类兽药残留检测重点、难点解读摘要:本文从四环素类化合物的结构式讲述了其在pH<2时,以脱水四环素类的形式存在于溶液中,在2<pH<6时,以差向四环素类的形式存在于溶液中,在pH>9时,四环素类化合物会发生内酯化的现象,因此检测时要注意控制溶液的pH值;并以几个具体的标准方法标准(GB/T 21317-2007、GB 31658.6-2021、GB/T 18932.23-2003、GB/T 22961-2008、SC/T 3015-2002、GB/T 31656.11-2021)为例分别从适用范围、提取、净化、仪器设备、定量方式、检测限与定量限进行讲述;总结了检测四环素类化合物时须注意的事项。关键词:[font=calibri]四环素类;[/font]相关标准;提取溶剂;净化方法1四环素类药物结构解析[img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709317213_9799_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709320700_4239_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709322018_9804_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709327335_7158_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709331085_5576_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709332521_7603_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709333957_9631_2166779_3.png[/img] [font=宋体][size=16px]以上从pH值的变化对四环素类化合物的存在形式影响,与金属离子的螯合方面进行了结构的解析。[/size][/font]1四环素类化合物检测相关标准解读[img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709335314_8942_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709334069_4747_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709335035_1376_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709336012_5471_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709337037_331_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709338043_5929_2166779_3.png[/img]备注: 艾杰尔的pep小柱等效于沃特世的HLB小柱。[img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709339283_4003_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709342824_553_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709344299_6326_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709342701_7969_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709343971_2771_2166779_3.png[/img][img]https://ng1.17img.cn/bbsfiles/images/2022/10/202210011709345221_3087_2166779_3.png[/img]1四环素类化合物检测注意事项提取:四环素类抗生素在酸性和碱性条件下均不稳定,其含有许多羟基、烯醇羟基及羰基,在中性条件下能与多种金属离子形成不溶性螯合物,由于生物基质中富含有钙元素、镁元素等,所以在检测四环素类药物时,通常会用含有EDTA的提取溶剂,用来克服因痕量金属离子存在而引起的重现性差的问题。因此提取剂要采用含有EDTA络合剂的柠檬酸-磷酸盐组成的缓冲液体系(pH=4.0)。样品前处理:四环素类化合物易降解,做实验前要合理安排实验的流程及准备好实验的材料,检测的时间要控制再2个小时内完成。净化:建议使用常规的HLB、C18柱,使用阴离子交换柱及阳离子交换柱要特别注意,不建议使用。四环素类化合物:母液浓度建议配成10ug/mL,避光,高浓度的母液在保存过程中会出现结晶析出的现象。堵柱子的问题:建议大家再前处理时多下功夫,低温高速离心,多离心几次。总结:本文从四环素类化合物的结构式讲述了其在pH<2时,以脱水四环素类的形式存在于溶液中,在2<pH<6时,以差向四环素类的形式存在于溶液中,在pH>9时,四环素类化合物会发生内酯化的现象,因此检测时要注意控制溶液的pH值;并以几个具体的标准方法标准为例分别从适用范围、提取、净化、仪器设备、定量方式、检测限与定量限进行讲述;总结了检测四环素类化合物时须注意的事项。
如题,对于DMF我在百度上查了它的基本知识,但没有用过,没有了解我想配置四环素类抗生素,盐酸土霉素、盐酸金霉素、盐酸四环素和盐酸多西环素,想模拟制药废水,浓度比较高,但溶解不了,不知道加入DMF效果如何?不知道应用起来有什么注意的吗用过的朋友我想问下,加入这个溶剂是否可以加大溶解呢?急迫得到您的意见与指导谢谢啦。十分感谢
[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]法测四环素类,盐酸金霉素和金霉素、盐酸土霉素和土霉素,有什么区别呢?保留时间一样吗?
最近在做四环素类药物,开始液相做的,磷酸盐缓冲液流动相不行,某一个国标的,后来换草酸,出峰和响应均能达到要求,不过实际样品处理的时候回收率不好,前处理是1%高氯酸提取,重复,过C18小柱,也是国标处理方法,纯标准品过柱回收率还可以。液质目前问题是响应不好,仪器是安的6410,只能到50ppb,流动相是0.3%甲酸水和0.3%甲酸甲醇。做的好的达人请指点,多谢
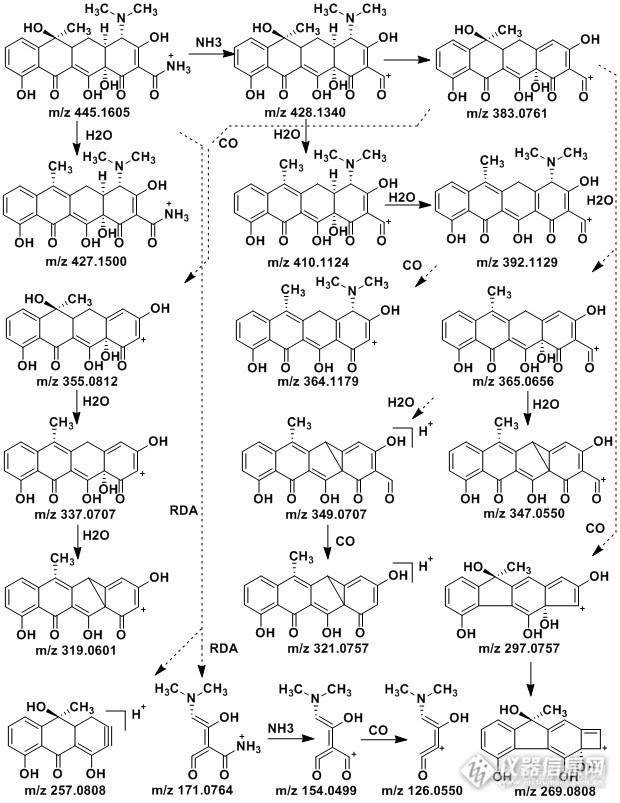
基于密度泛函理论研究四环素的电喷雾质谱裂解机理摘要: 基于密度泛函理论(Densityfunctional theory,DFT)方法,考察四环素的优势构像极其在电喷雾正离子模式下准分子离子峰处于基态的最优构型,结合构形参数及质谱测定对准分子离子的最优构型进行了确认,并通过全几何结构优化,对四环素的优势构像及其在电喷雾质谱(LC-ESI-Q-Orbitrap-MS)正离子模式下准分子离子的二级谱中碎片离子的最优构型进行研究。结合高分辨率质谱数据对其质谱裂解机理进行解释。该研究可以为进一步探索四环素类化合物及其衍生物ESI-MS正离子模式下的质谱裂解规律提供参考和理论指导依据。关键词:密度泛函理论(DFT);静电轨道离子阱(Orbitrap);四环素(Tetracycline)1 实验部分1.1 仪器与试剂Thermo Scientific:Q Exactive Orbitrap ,Merck:CH3OH,Standard: Tetracycline(上海士锋生物科技有限公司)1.2 分析条件质谱(Mass Spectrometry):Ion Source:ESI, MS Type:MS2,Ion Mode:Positive(+),Fragmentation Mode:HCD,Collsion Energy:30ev色谱(Chromatography):Column Name:WatersXBridge TM(Waters,C18)3.5um,2.1*50mmFlow Gradient:90A(0min)-50A(5min)-5A(25min)-90A(30min),FlowRate:200ul/minSolvent A:H2O+0.1%Acid,Solvent B:CH3OH+0.1%Acid1.3 量子化学计算 使用密度泛函的B3LYP方法,以6-311+G*为基组,对反应势能面上的各驻点的构型进行了全几何参数优化,并由频率分析确认了稳定点的正确性,为了得到更精确的能量信息,又在B3LYP//6-311++G(3df,3pd)水平上计算了各驻点的单点能,所有计算采用Gaussian 03程序包完成。前言 四环素类(Tetracyclines,TCs)是由链霉菌产生的一类广谱抗生素(1),在化学结构上都属于多环并四苯羧基酰胺母核的衍生物。四环素类可分为天然品和半合成品两大类。天然品为从放线菌金色链丛菌的培养液等分离出来的抗菌物质,四环素类药物为广谱抗生素,广泛用于临床治疗,并常被用做动物促生长剂,但耐药性的出现限制了该类药物的使用。目前关于四环素类抗生素的分析大多采用液相色谱质谱联用技术分析(2-9),并多数是采用电喷雾离子源。随着串联质谱技术的不断发展,采用量子化学方法及理论计算从分子水平研究化合物的质谱裂解规律及机理受到广泛而长期的关注。采用量子化学理论在质谱的裂解机理计算中,准分子离子几何构型的可靠性直接影响后续更加深层次的分析,而确定准分子离子最可能的最优构型是解析谱裂解机理的首要解决问题,本研究采用量子化学计算方法,依据密度泛函理论,并借助高斯软件Gaussian 03计算分析,计算了四环素正离子模式下准分子离子的最优构型,并且结合高分辨率质谱静电轨道离子阱质谱(Q-Orbitrap-MS)给出的可靠数据,对特征离子的裂解做以归属,为此类化合的鉴定解析提供理论依据。四环素的结构及其空间三维立体模型见图1http://ng1.17img.cn/bbsfiles/images/2015/09/201509221738_567179_2359621_3.bmp图1 Tetracycline结构及其空间立体构型2 结果分析2.1 量子化学计算各质子化位点的质子亲和势能 由于化合物结构有多个质子化位点,所以需通过计算确定其最稳定构型及最大可能质子化位点,质子化反应方程为:RX+H+→RH+分子的气相碱性由其质子化方程的焓变ΔrH来确定,即质子亲和能EPA=-ΔrH,质子亲和能较大的化合物,其气相碱性较强,按照分子轨道理论,质子化方程的气相质子亲和能WPA与分子RX的最高占据道HOMO和质子H+的最低未占据轨道LUMO的差值有关,由于H+的LUMO是一个定值,所以可以认定WPA只与RX的HOMO相关并呈线性关系,原则上RX分子的HOMO能级值可以由量子计算得到。在B3LYP/6-311++G(d,p)//B3LYP/6-311++G(d,p),B3LYP/6-311++G(3df,2p)//B3LYP/6-311++G(3df,2p)和B3P86/6-311++G(3df,2p)//B3P86/6-311++G(3df,2p)基础下,计算了各质子化位点的平衡几何构型,优化得到的分子平衡几何构型都经频率计算证明是势能面上的极小点(无虚频),获得各质子化位点的质子亲和能(E),各质子结合位点的质子亲和能计算结果见表1http://ng1.17img.cn/bbsfiles/images/2015/09/201509221752_567196_2359621_3.bmp表1 四环素各质子结合位点的质子亲和能EPATabel 1 Protonaffinity for proton binding sites of Tetracycline(EPA)通过表1可以看出质子结合位点位于氨基上具有较高的质子亲和能,表明N上孤对电子可能占据HOMO轨道,所以质子化位点极可能位于氨基上。2.2 四环素在LC-ESI-Q-Orbitrap-MS下的质谱裂解途分析通过以上计算,以质子化位点位于氨基上为起点,并结合高分辨率质谱数据对其质谱裂解途径和机理进行分析,使用(LC-Q-Orbitrap-MS)获得准分子离子峰m/z 445.1594的二级谱,质谱碎片离子及相对丰度见表2http://ng1.17img.cn/bbsfiles/images/2015/09/201509221750_567191_2359621_3.bmp表2 四环素电喷雾离子源下准分子离子(MS2)的碎片离子及其相对丰度Tabel 2 Relative abundancesof characteristic ions in the ESI(MS2) mass spectra of Tetracycline依据表1计算结果,对比质子亲和能,质子最可能的结合位点为氨基上氮原子,氮原子的一对未成键电子最可能占据HOMO轨道,所以以质子结合到氨基上所形成的准分子离子峰为起始点(备注:只是最可能概率最大的,但是不排除其他小概率的质子结合位点所引发的裂解),对其可能的质谱裂解途径做以下分析。准分子离子峰失去H2O中性分子后得到碎片离子m/z427.1500,与理论误差为-2.61ppm。而失去H2O中性分子可能有多个不同位点,1.2-消除脱水和-2.4消除脱水,从空间立体构型中可以看到氢和羟基均位于一侧,所以有利于发生1.2-消除和2.4-消除,如此就有了三种可能的脱水方式,所以通过计算得到不同三种方式下脱水后生成离子的稳定构型及其能量,见表3。由表3可以看出第一种模式下生成的离子能量最低,表明此方式为主要途径,更容易进行。准分子离子通过正电荷转移失去NH3可以生成离子m/z 428.1340,与理论误差为0.02ppm,β为的氢重排到侧链氮原子上可以脱去侧链CH3NHCH3得到碎片离子m/z 383.0761,与理论值误差为1.01ppm。该离子进一步通过1.2-消除脱H2O后生成离子m/z 365.0656,与理论值误差为0.19ppm。后通过2.4-消除脱水生成离子m/z 347.0550,与理论值误差为-1.91ppm。,由于2.4-消除相比1.2-消除难所以生成的离子丰度相对较低,离子m/z
[color=#444444]做了三个月的光催化实验,目标污染物是盐酸四环素,休假后回实验室用高效液相色谱测四环素测的时候出很小的峰,倒是清洗的时候会出现分裂的峰。[/color][color=#444444]我是做光催化的,所以只是等梯度洗脱四环素一种物质,条件如下:[/color][color=#444444]1 流动相:甲醇/水=70:30,不调pH.样品的pH在5.0左右[/color][color=#444444]2 普通的C18柱,安捷伦的[/color][color=#444444]3 测试波长是356nm[/color][color=#444444]出现的情况是:前段时间测还是正常的,结果换了流动相之后就有问题了。先是不出峰,超出检测时间(5min)后才出现很宽的峰(大概2.5min) 后来是所幸不出峰了,但是用纯甲醇(也是过滤超声过的)清洗3min左右的时候会出峰,两天前是很对称的峰,今天就是分裂的峰了。[/color][color=#444444]在此期间我重新配了两次流动相,情况并没有好转(因为之前也没有调过pH,加上和其他人共用,清洗会比较麻烦所以就没调,但是前后pH值是一致的);[/color][color=#444444]样品也确定没有问题;[/color][color=#444444]样品池参比池的能量也和以前一样;[/color][color=#444444]期间柱压正常,自检正常,基线也挺稳的;[/color][color=#444444]别人用这个色谱柱测其他样都挺好的,所以色谱柱应该没什么问题。[/color][color=#444444]还请各位大神不吝相助。[/color]
小弟用三重四级杆液质联用检测四环素类抗生素,结果发现TC、CTC、OTC的峰型都很差,就是峰太宽,峰高低,而且峰呈锯齿形,导致信噪比小,难以检测。同步检测的其他抗生素如磺胺类和大环内酯类是没有问题的。样品用磷酸盐乙腈混合溶液提取,色谱条件是C18柱,A:0.1%甲酸水溶液,B:甲醇,梯度洗脱。请教做好这方面实验的兄弟姐妹们LC-MS检测四环素要注意什么?是否出现过上述情况?大家用的色谱质谱条件怎样的?多谢。
如题,对于DMF我在百度上查了它的基本知识,但没有用过,没有了解我想配置四环素类抗生素,盐酸土霉素、盐酸金霉素、盐酸四环素和盐酸多西环素,想模拟制药废水,浓度比较高,但溶解不了,不知道加入DMF效果如何?不知道应用起来有什么注意的吗用过的朋友我想问下,加入这个溶剂是否可以加大溶解呢?急迫得到您的意见与指导谢谢啦。十分感谢
基于浊点提取和[url=https://insevent.instrument.com.cn/t/5p][color=#3333ff]液相色谱[/color][/url]分析,建立了食品样品中四环素残留量的灵敏预浓缩和测定方法。在 pH 5.0 缓冲液和高电解质浓度存在下四环素 (TC) 分子被萃取到聚乙二醇相中(PEG-6000) 。将样品离心以增加相分离,用甲醇:水 (1:1) 的混合物稀释样品后通过 0.45 μm 膜后 HPLC 测定。流动相:pH 4.0草酸盐缓冲液:甲醇:乙腈(70:10:20)等度洗脱,色谱柱 Luna Omega C18,在整个研究过程中,所有分析均使用 276 nm 和 358 nm 的 DAD 检测器进行。通过优化所有实验参数,两个波长的线性范围计算为 30–500 ng mL-1和 65–800 ng mL - 1。两个波长的检测限分别为 9.56 和 21.11 ng mL - 1 。该研究新颖之处在于使用聚乙二醇6000的浊点提取。详见[url]https://doi.org/10.1016/j.microc.2019.104170[/url]
想用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]测四环素,但是手头没有EDTA,无法按照国标做前处理。请问各位小伙伴有没有其他的前处理方法,现在实验室有盐酸,硫酸,偏磷酸,乙酸,还有HLB的小柱。
最近在做兽药残留的液质方法,四环素类的做了四环素,土霉素,金霉素和强力霉素,四环素经基质加标后处理上机,四环素有40%左右已转化为差向四环素,不知大家有没遇到此类情况?我在提取时有超声10分钟/次×3次,感觉是因为超声的关系。有过相关经验的请麻烦指教
[color=#444444]三重四级杆[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]检测四环素类抗生素,结果发现TC、CTC、OTC的峰型都很差,就是峰太宽,峰高低,而且峰呈锯齿形,导致信噪比小,难以检测。同时检测的其他抗生素如磺胺类和大环内酯类是没有问题的。[/color][color=#444444]样品用磷酸盐乙腈混合溶液提取,色谱条件是C18柱,A:0.1%甲酸水溶液,B:甲醇,梯度洗脱。请教做好这方面实验的兄弟姐妹们[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]检测四环素要注意什么?是否出现过上述情况?大家用的色谱条件怎样的?[/color]
1.实验目的 据标准GB/T 14931.1-94测定畜禽肉中土霉素、四环素、金霉素残留量。最低检出浓度为:0.15,0.20,0.65mg/kg。 2.试验原理 样品经提取,微孔滤膜过滤后直接进样,用反相色谱分离,紫外检测器检测,与标准比较定量,出峰顺序为土霉素、四环素、金霉素。标准加法定量。 3.试剂 3.1乙腈(分析纯)。 3.20.01 mol/L磷酸二氢钠溶液:称取1.56 g(±0.01g)磷酸二氢钠(NaH2PH4.2H2O)溶于蒸馏水中,定容到100 mL,经过滤器过滤(选用微孔滤膜为0.45 μm),备用。 3.3土霉素(OTC)标准溶液:称取土霉素0.0100g(±0.0001g),用0.1mol/L盐酸溶液每毫升含土霉素1mg。 3.4四环素(TC)标准溶液:称取四环素0.0100g(±0.001g),用0.01 mol/L盐酸溶液溶解并定容10.00 mL,此溶液每毫升含四环素1mg。 3.5金霉素(CTC)标准溶液:称取金霉素0.0100g(±0.0001g),溶于蒸馏水并定容成10.00mL,此溶液每毫升含金霉素1mg。以上标准品均按1000单位/mg折算。3.3~3.5溶液应于4℃以下保存,可使用1周。 3.6混合标准溶液:取3.3、3.4标准溶液各1.00 mL,取3.5标准溶液2.00 mL,置于10mL容量瓶中,加蒸馏水至刻度。此溶液每毫升含土霉素、四环素各0.1mg,金霉素0.2mg,临时现配。 3.75%高氯酸溶液。 4.仪器 4.1 高效液相色谱仪(HPLC):具紫外检测器。 4.2 振荡器:郑州中谱仪器设备有限公司 4.3 过滤器:郑州中谱仪器设备有限公司 4.4 超声波:郑州中谱仪器设备有限公司 5.色谱条件 5.1柱:ODS-C18 (5 μm) 6.2mm×15 cm。 5.2检测波长:355 nm。 5.3灵敏度:0.002 AUFS。 5.4柱温:室温。 5.5流速:1.0mL/min。 5.6进样量:10 μL。 5.7流动相:乙腈/0.01 mol/L磷酸二氢钠溶液(用30%(V/V)硝酸溶液调节pH2.5),35:65(V/V),使用前用超声波脱气10 min。 6.操作方法 6.1样品测定:称取5.00 g(±0.01 g)切碎的肉样(<5 mm),置于50 mL锥形烧瓶中,加入5%高氯酸25.0mL,于振荡器上振荡提取10 min,移入到离心管中,以2000 r/min离心3min,取上清液经0.45μm滤膜过滤,取溶液10μL进样,记录峰高,从工作曲线上查得含量。 6.2工作曲线:分别称取7份切碎的肉样,每份5.00 g(±0.01 g),分别加入混合标准溶液0、25、50、100、150、200、250 μL(含土霉素、四环素各为0、2.5、5.0、10.0、15.0、20.0、25.0 μg, 含金霉素0、5.0、10.0、20.0、30.0、40.0、50.0 μg), 按6.1方法操作,以峰高为纵坐标,以抗生素含量为横坐标,绘制工作曲线。
土霉素、金霉素和四环素的检测方法 1. 分析目标化合物土霉素,金霉素,四环素2. 仪器设备带荧光检测器的高效液相色谱仪。3.试剂除下列试剂外,使用附录2所列试剂。咪唑:特级咪唑缓冲溶液:将68.08g咪唑、0.37g EDTA 和10.72g乙酸镁溶解在800mL水中,用乙酸调节pH7.2后,加水至1,000mL。含有乙二胺四乙酸的柠檬酸缓冲溶液:第1液:将21.0g柠檬酸溶解在水中至1,000mL。第2液:将71.6g磷酸氢二钠溶解在水中至1,000mL。在1.86g乙二胺四乙酸中加入307mL第1液和193mL第2液混合的溶液溶解。苯乙烯二乙烯苯共聚物小柱 (265mg):在内径8~9mm 聚乙烯管中填充265mg柱色谱用苯乙烯二乙烯苯共聚物或具有同等分离特性的物质。4.标准品盐酸土霉素:本品1.000mg 含有土霉素0.850mg以上效价,分解点为190℃~194℃。盐酸金霉素:本品1.000mg含有盐酸金霉素0.900mg以上效价,分解点为210℃以上。盐酸四环素:本品1.000mg 含有盐酸四环素0.900mg以上效力,分解点为214℃以上。5.试验溶液的制备a 提取方法①肌肉、肝脏和肾脏
[color=#444444]在分离盐酸四环素的时候发现在色谱柱残留太大,冲不下来,有经验的大神能否给个建议。[/color][color=#444444] 溶剂A: water+5mM Ammonium Formate 溶剂B: MeOH[/color][color=#444444] 流动相梯度:[/color][color=#444444] Time(min) Flow Rate(mL/min) %A %B[/color][color=#444444] 0 0.3 95 5[/color][color=#444444] 10 0.3 0 100[/color][color=#444444] 15 0.3 0 100[/color][color=#444444] 18 0.3 95 5[/color][color=#444444]出峰时间在6.09min.[/color]
如题,因工作需求需要测定某成品中甲胺盐酸盐含量(含量很低,估计只有千分之几)的测定方法。经过文献调研,决定采用酸碱滴定的方法,首先拿较纯的甲胺盐酸盐(含量约90%),考虑到成品中该物质含量很低,所以将滴定液(氢氧化钠溶液)的浓度配的也很低,经标定后,氢氧化钠滴定标准溶液浓度为0.01mol/L。然后拿这个滴定液去滴定,分别采用了酚酞指示剂(变色范围8.0-10.0)和百里香酚酞指示剂(变色范围9.3-10.0)做指示剂,变色点都比化学计量点提前好多。本来计算着滴定液需要22ml的,实际滴3ml就会变色。。。。推测原因,是甲胺盐酸盐与氢氧化钠反应生成的甲胺,溶于水后呈碱性,使终点提前。然后,不知道怎么办了?换指示剂?还是怎么地?同样别人的参考文献是测甲氧胺盐酸盐的,同样的方法,用的是0.1mol/L的氢氧化钠滴定液和酚酞指示剂,文章表明该方法很好。。。为什么甲胺盐酸盐和甲氧胺盐酸盐差距会这么大呢?求高人指教!有没有其他的测量方法?多谢,拜托~
用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]MS分析四环素类物质,已经在流动相中加了5mmol的草酸,土霉素物质峰型还比较好,四环素有点前沿峰,金霉素和强力霉素拖尾特别严重,前面有好几个小峰中间一直分不开的。有没有分析过相关物质的大神帮忙分析一下,这种情况要怎么解决呢?

今天发现了2个 差向四环素 虽然都这么叫 但是结构式 分子式 完全不一样,有没有做过这个物质的老师,高手指点下,2者的区别,怎么选?http://ng1.17img.cn/bbsfiles/images/2014/04/201404031821_495334_2378824_3.jpg
[B]动物源性食品中四环素、沙星类[/B] 残留量的快速测定方法1 范围本方法规定了动物源性食品中四环素类、沙星类高效液相色谱的快速测定方法。本方法适用于动物源性食品中四环素类、沙星类高效液相色谱的快速测定。2.1 原理试样中的残留物经四环素类、沙星类快速检测前处理试剂盒处理,样液经四环素类专用层析柱净化、浓缩用高效液相色谱检测,外标法定量。2.2 试剂和材料除另有规定外,所有试剂均为分析纯,水为重蒸馏水。2.2.1 乙腈:色谱纯。2.2.2 甲醇:色谱纯。2.2.3 三乙胺(分析纯)2.2.4 磷酸(85%)(分析纯)2.2.5 磷酸氢二钠:优级纯。2.2.6 乙二胺四乙酸二钠。2.2.7 柠檬酸:分析纯。2.2.8 磷酸氢二钠溶液:0.2mol/L。称取28.41g磷酸氢二钠,用水溶解,定容至1000mL。2.2.9 柠檬酸溶液:0.1mol/L。称取21.01g柠檬酸,用水溶解,定溶至1000mL。2.2.10 Mcllvaine缓冲溶液:将1000mL0.1mol/L柠檬酸溶液与625mL0.2mol/L磷酸氢二钠溶液混合。2.2.11 Na2EDTA-Mcllvaine缓冲溶液:0.1mol/L。称取60.5g乙二胺四乙酸二钠放入1625mLMclllvaine缓冲溶液中,使其溶解,摇匀。2.2.12 标准品: 土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星纯度大于98 %。2.2.13 标准贮备溶液:分别称取土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星各10mg,用甲醇溶解并定溶于100 mL棕色容量瓶中,配制成100 µ g/mL的标准贮备液,置于-20℃保存,有效期三个月。2.2.14 混合标准工作溶液:用流动相稀释标准贮备溶液,配制成土霉素、四环素、金霉素、强力霉素、氧氟沙星、诺氟沙星、环丙沙星、单诺沙星、恩诺沙星均为10 µ g/mL的混合标准溶液。0~4℃避光保存。2.2.15 四环素、沙星类快速检测前处理试剂盒*。2.3 仪器和设备2.3.1 高效液相色谱仪:配紫外-可见光波长检测器。2.3.2 匀浆机。2.3.3 固相萃取机2.3.4 离心机:4000 r/min。2.3.5 调速多用振荡器。2.3.6 聚四氟乙烯离心管: 2.5 mL,50 mL,具塞。2.4 样品制备准确称取已捣碎的样品5.00 g于50 mL离心管中,先加入四环素、沙星类快速检测前处理试剂盒中的提取剂 (液体20mL),用调速多用振荡器150 rpm振荡3 min,,4000 r/min离心5 min,收集上清液10mL加入四环素专用层析柱中(使用前依次用5mL甲醇、5mL提取剂、5mL水激活)挤干,用2mL提取剂洗涤,用0.80mL甲醇洗脱,收集洗脱液,用0.2mL流动相定容至1.0mL。供仪器测定。2.5 测定2.5.1 液相色谱条件a) 色谱柱: C18柱,250 mm×4 mm(i.d.),粒度5µ m b) 流动相: 0.05 mol/L磷酸/三乙胺缓冲液(pH2.4)+乙腈(80+20,V/V) c) 流速: 1.0mL/min d) 柱温: 室温 e) 检测波长: 四环素类紫外检测器350 nm。沙星类 紫外检测器310nm;荧光检测器激发波长280nm,发射波长450nm。f) 进样量:50 uL。3.5.2 标准工作曲线绘制移取各移取四环素类、沙星类混和标准液,用流动相稀释成20 ng/mL、50 ng/mL、250 ng/mL、500 ng/mL标准工作溶液。按液相色谱条件(3.5.1)进行测定,以色谱峰的峰面积为纵坐标,与其对应的浓度为横坐标作图,绘制标准工作曲线,标准工作曲线范围:20.0~500 ng/mL。 3.5.3 试样测定 用微量进样器准确吸取试样溶液(3.4),按液相色谱条件(3.5.1)进行测定,记录色谱峰的保留时间和峰面积。3.6 结果计算按式(1)分别计算供试样品中的四环素类、沙星类残留量。 2×ci×Vω= …… (1)mω-水产品中四环素、沙星类残留量,μg/kg;ci -标准曲线上查出试样溶液中四环素、沙星类标准工作溶液的浓度,(μg/L);V-最终定容体积数,mL;2-换算常数;m-供试试料样品重量,g。本方法分别计算四环素类、沙星类结果。3.7 检测限本方法土霉素、四环素检测限为20µ g/kg;金霉素、强力霉素的检测限为50µ g/kg,沙星类为:5µ g/kg3.8 回收率 本方法土霉素、四环素、金霉素、强力霉素回收率为:75%~85%;沙星类回收率为:75%~85%相关谱图附件可见联 系 人:王 伦 手 机:13810239506 EMAIL:wwj613@sina.com







 我要推广仪器
我要推广仪器
 下载APP
下载APP