在此请教各位老师,原子荧光测水汞时,样品和标准系列要不要国标方法中“溴化钾溴酸钾氧化汞为二价汞再滴加盐酸羟胺还原过量氧化剂”那一步??我看瑞利原子荧光仪器方法书中测水汞并没有那一步。是不是因为标液本身就是二价汞所制,不需要这一步;而样品就是甲基汞量会很少(超痕量),没有必要再氧化成二价无机汞。???大家有没有做这一步呢?这一步有必要吗?谢谢!!
本来想用锆标准溶液来检验EDTA 的浓度,但是却遇到了问题。是这样的:EDTA 的浓度已经用氧化锌标定过的,想用锆标准溶进行检验(EDTA是用来滴定原料中氧化锆的含量)。我之前也检验过,用的锆标准溶液是国家钢铁材料测试中心的,盐酸做介质,效果很好。但是现在买的锆标准溶液介质换了,变成了硝酸与氢氟酸的混合酸做介质,往里面加二甲酚橙指示剂时颜色稍变成黄色的了(正常的话应该是紫红色的 ),颜色不正常了,无法滴定。出现这种问题是不是二甲酚橙被混合酸氧化了,失去指示剂的作用?抑或是别的原因?
三氧化二氯铝的测定1.方法提要试样经强碱熔解过滤除去二氧化锰沉淀,铝留在溶液中。铝能和EDTA形成中强度络合物。在pH5.5—6.0下使铁铝铜锌铅鈦镍等例子和EDTA完全络合后用氯化锌溶液回滴过量的EDTA。然后加氟化钠置换EDTA-Al络合物,再用氯化锌标准溶液滴定释放出的EDTA,以二甲酚橙作指示剂,由黄变红为终点。2.试剂1.EDTA溶液 C=0.01mol/L 称取3.723g乙二胺乙酸二钠溶解于水中移入1000毫升容量瓶中用水定容。2.氯化锌标准溶液 称取1.2821克金属锌(99.9%)于150毫升烧杯中,加15毫升(1+1)盐酸加热蒸发至2毫升移入1000毫升容量瓶中用氨水(1+1)中和至甲基橙变黄,再用(1+1)盐酸滴至指示剂变红过量5滴,用水定容,此溶液每毫升等于1毫克三氧化二铝。3.乙酸—乙酸钠缓冲溶液 pH5.5-6.0 溶解200克结晶乙酸钠于500毫升水中加10毫升冰乙酸用水稀释至1000毫升。3.分析步骤称取0.1-0.5克试样于银坩埚中加1克过氧化钠4克氢氧化钠混均,于500-600℃熔融至红色透明,取下冷却洗净坩埚底置于250毫升烧杯中加50毫升水1克碳酸钠濅取,用盐酸调pH大于3,洗净坩埚,冷却定容100毫升。干过滤,吸取20毫升鱼300毫升三角瓶中,用(1+1)氨水中和至pH6左右,加过量的0.01molEDTA,再加20毫升乙酸—乙酸钠缓冲溶液在电炉上煮沸3分钟。冷却后加3-5滴二甲酚橙,用氯化锌标准溶液滴至换色恰好变为红色,不记读数,加1克氟化钠在电炉上煮沸3分钟,冷却后补加1滴二甲酚橙用氯化锌标准溶液滴至黄色恰好变为红色即为终点。计算: Al2O3=TV/m×100
化学需氧量测定中用到邻苯二甲酸氢钾溶液做标准溶液,进行校核实验。这种溶液是不是很容易被氧化,保存不了几天
我配制了一桶0.3mol/L氢氧化钠标准溶液,用基准物邻苯二甲酸氢钾标定出来的数据波动很大,根本不平行。可是用硫酸标准溶液标定出来却很平行(己排除了滴定管、基准物、烘箱的准确度因素),而且有个奇怪的现象,就是邻苯二甲酸氢钾标称样量一样数据就能平行,如果称样量不一样数据就不能平行,称样量越大标定出来的数据越高,称样量越小标定出来的数据就越低(比如称1.9g邻苯二甲酸氢钾标定得0.3050mol/L,称1.5g邻苯二甲酸氢钾标定得0.3000mol/L,如果8组都称1.9g邻苯二甲酸氢钾8组都标定得0.3050mol/L,如果8组都称1.5g邻苯二甲酸氢钾8组都标定得0.3000mol/L)这是为什么啊?
1、五氧化二钒铁含量的测定火焰原子吸收光谱法YB/T 4199-20092、五氧化二钒硫、磷、砷和铁含量的测定电感耦合等离子体原子发射光谱法YB/T 4200-20093、五氧化二钒五氧化二钒含量的测定过硫酸铵氧化--硫酸亚铁铵滴定法YB/T 4218-20104、五氧化二钒磷含量的测定铋磷钼蓝分光光度法YB/T 4219-20105、五氧化二钒氧化钾、氧化钠含量的测定电感耦合等离子体原子发射光谱法YB/T 4220-20106、五氧化二钒硫含量的测定硫酸钡重量法YB/T 5332-2009
GCMS测皮革中富马酸二甲酯标准中用中性氧化铝净化,为什么我们做的时候,浓缩液过氧化铝柱子的时候,流出液还是很浑浊,好像并没有起到净化的效果,进一步离心、过滤膜后,谱图的本底干扰也比较严重。反而用C18净化效果要好一点,回收率也不错,重复性有点差,还要进一步确认,有没有童鞋有同样的情况?
我配制了一桶20L的0.25mol/L氢氧化钠标注溶液,用邻苯二甲酸氢钾标定数据一点都不稳定,最高的数据0.2495,最低的0.2444,可是用硫酸标准溶液标定却平行得很好,为什么呀?不明白呀?知道的快快指导下吧,我快郁闷死了!
三丁基氧化锡除了DIN 38407-13:2001、ISO 17353:2004以及GB/T20385-2006之外还有其他什么相关的标准吗?二甲苯麝香的标准又有哪些呢?
大家好,我现在正在做一种亚铁和氨基酸的配合物,但是不知道如何防止二价铁氧化为三价铁,在文献上看到说可以加入抗氧化剂,但是没有说加哪一种,请问一下,加什么最好???????
[color=#DC143C]摘要:简述了绿色化学试剂碳酸二甲酯的特性、应用和合成方法。 [/color] 关键词:碳酸二甲酯;绿色试剂 文章编号:1005-6629(2006)12-0032-02中图分类号:O623.624文献标识码:E 在目前的化学工业生产中,仍然使用一些剧毒的原料,如光气、硫酸二甲酯等,为了人类的可持续发展,在化工生产过程中,迫切需要采用无毒或低毒的化学原料来代替有毒的原料,使用绿色试剂,淘汰有毒原料,是化学工业发展的必然趋势。 碳酸二甲酯(dimethyl carbonate,简称DMC),就是一种新的绿色基础化学试剂。1992年在欧洲作为非毒性物质注册登记,被称为二十一世纪绿色有机化学原料。近几年来,随着碳酸二甲酯生产工艺的突破,应用领域日益广泛。作为一种清洁有机化学试剂,碳酸二甲酯一方面可替代光气、硫酸二甲酯、氯甲烷及氯甲酸甲酯等剧毒或致癌物进行羰基化、甲基化、甲酯化及酯交换等反应生成多种重要化工产品;另一方面,以碳酸二甲酯为原料可以开发、制备多种高附加值的精细专用化学品,在医药、农药、合成材料、染料、润滑油添加剂、食品增香剂、电子化学品等领域具有广泛应用;第三,由于氧含量高、相溶性好,可用作低毒溶剂和燃油添加剂。因此,碳酸二甲酯具有重要的应用价值和广阔的市场前景。 1碳酸二甲酯的特性 碳酸二甲酯结构式(CH3O)2CO,分子量为90.08, 相对密度1.070,折射率1.3697,熔点4℃,沸点90.1℃。在常温下是一种无色透明、略有刺激性气味的液体,具有无毒、无腐蚀性、氧含量高、相溶性好等特点,其分子结构独特,结构中含有羰基、甲基、甲氧基等多种官能团,因而具有多种反应活性,在许多化学反应场合可替代光气、硫酸二甲酯(DMS)等化学品,作为重要的羰基化和甲基化试剂。由于碳酸二甲酯的化学性质非常活泼,可与醇、酚、胺、肼、酯等发生化学反应,故可衍生出一系列重要化工产品。其化学反应的副产物主要为甲醇和CO2,与光气、硫酸二甲酯等的反应副产物盐酸、硫酸盐或氯化物相比,危害相对较小。 2碳酸二甲酯的制备方法 目前合成碳酸二甲酯主要有光气法、酯交换法和甲醇氧化羰基合成法等,其中具有工业意义的工艺路线为后两种: 一是酯交换法,又称为石化路线。二是甲醇液相氧化羰基合成法,又称为煤化路线。

空气中硫酸二甲酯的测定方法 甲、1,2-萘醌-4-磺酸钠比色法1 原理硫酸二甲酯与亚硝酸钠作用,生成硝基甲烷,在氢氧化钙存在下,与1,2-萘醌-4-磺酸钠生成紫蓝色化合物,比色定量。2 仪器2.1 多孔玻板吸收管。2.2 抽气机。2.3 流量计,0~1L/min。2.4 具塞比色管,10ml。2.5 分光光度计3 试剂3.1 吸收液:无水乙醇。3.2 1,2-萘醌-4-磺酸钠溶液,5g/L。临用前配制。3.3 亚硝酸钠溶液,100g/L。3.4 饱和氢氧化钙溶液(若混浊应在使用前过滤)。3.5 标准溶液:于25ml量瓶中加入10ml吸收液,准确称量,加入3滴硫酸二甲酯,再准确称量,两次称量之差即为硫酸二甲酯的质量。加吸收液至刻度,计算1ml溶液中硫酸二甲酯的含量,使用时用吸收液稀释成1ml=100微克的标准溶液。此标准溶液须临用前配制,3h内稳定。4 采样串联两个各装10ml吸收液的多孔玻板吸收管,以0.5Lmin的速度,抽取8L空气。5 分析步骤5.1 对照试验:同采样。将吸收管装好吸收液带至现场,但不抽取空气,照样品分析。5.2 样品处理:用吸收管中的吸收液洗涤进气管内壁3次,自每个吸收管中各取5.0ml样品溶液分别放入比色管中。5.3 标准曲线的绘制:按表63配制标准管。向标准管中各加入0.1ml 1,2-萘醌-4-磺酸钠溶液(3.2),表63 硫酸二甲酯标准管的配制[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201503_52388_1625938_3.jpg[/img]摇匀,加0.5ml亚硝酸钠溶液(3.3),摇匀,在60℃水浴中加热5min,取出冷却后加1ml饱和氢氧化钙溶液(3.4),振摇1min,静置2min。各加水到10ml,混匀,于波长564nm下比色。以硫酸二甲酯含量对吸光度作图,绘制标准曲线。5.4 测定:样品管操作同标准管,比色后由标准曲线上查出硫酸二甲酯的含量。6 计算X=2(C1+C2)/V0式中:X——空气中硫酸二甲酯的浓度,mg/m3;C1、C2——分别为第1、第2吸收管所取样品溶液中硫酸二甲脂的含量,微克;V0——标准状况下的样品体积,L。7 说明7.1 采得的样品必须及时分析。7.2 当硫酸二甲酯浓度为10、20、30、40、50微克/5ml时,其变异系数分别为9.2%、6.7%、5.7%、2.6%、2.1%。--------------------------------------------------------------------------------乙、高效液相色谱法1 原理空气中硫酸二甲酯经硅胶吸附,丙酮解吸后,在碱性和加热的条件下与对硝基苯酚反应生成对硝基茴香醚。经ODSC18柱分离,用紫外检测器检测。以保留时间定性,峰面积定量。2 仪器2.1 硅胶管:在长80mm,内径3.5~4.0mm,外径6.0mm的玻璃管中装入100mg60~80目层析用硅胶,两端用玻璃棉固定,套上乳胶帽或熔封后保存。在装管前于120~130℃活化2h。2.2 采样器,0~1L/min。2.3 恒温水浴箱。2.4 具塞试管,10ml。2.5 分液漏斗,250ml。2.6 微量注射器,10微升。2.7 高效液相色谱仪,紫外检测器。1ng的硫酸二甲酯给出的信噪比不低于3∶1。色谱柱:柱长25cm,内径4.6mm,不锈钢柱。柱填料:ODSC18(5微米)柱温:55℃流动相:5+5甲醇流量:1ml/min紫外检测器波长:305nm。3 试剂3.1 硫酸二甲酯。3.2 对硝基苯酚。3.3 重蒸馏水。3.4 丙酮、乙醚、甲醇,重蒸馏提纯。3.5 氢氧化钠溶液,C(NaOH)=0.3mol/L。3.6 标准溶液:于100ml量瓶中加入10ml丙酮,准确称量,加入10滴硫酸二甲酯,再准确称量,两次称量之差即为硫酸二甲酯的质量。加丙酮至刻度,计算1ml溶液中硫酸二甲酯的含量。使用时,用丙酮稀释成浓度分别为5.0、30.0、50.0、100.0、150.0微克/ml的标准溶液。此溶液须临用前配制,4h内稳定。4 采样在采样现场打开硅胶管(2.1),以0.2~0.3L/min的速度采集10L以上空气。采样后将管的两端套上胶帽。5 分析步骤5.1 对照试验:取2支未采过样的硅胶管(2.1),按照样品处理过程同样处理作为空白对照。5.2 样品处理:将样品管中的硅胶倾入具塞试管(2.4)中,加2ml丙酮(3.4),0.4g对硝基苯酚,8ml氢氧化钠溶液(3.5),以下按标准曲线操作。5.3 标准曲线的绘制:取6支试管(2.4),按表64配制标准管,充分混匀。在40℃水浴中保温1h,取出冷至室温。用10ml乙醚在分液漏斗中提取3min,静置分层。用微量注射器(2.6)取5微升乙醚提取液进样。每种浓度重复3次,取峰面积的平均值,以硫酸二甲酯含量对峰面积作图,绘制标准曲线,保留时间为定性指标。表64 硫酸二甲酯标准管[img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201504_52389_1625938_3.jpg[/img]5.4 测定 取5微升乙醚提取液进样,以保留时间定性,峰面积定量。硫酸二甲酯的色谱图见图54。6 计算X=C*2000/V0式中:X——空气中硫酸二甲酯的浓度,mg/m3;C——由标准曲线上查出的硫酸二甲酯的含量,微克;V0——标准状况下的样品体积,L。7 说明7.1 本法的检测限为1ng(进样5微升液体样品)。测定范围为0.25~30.0mg/m3。在此范围内变异系数低于5.2%。7.2 100mg硅胶对硫酸二甲酯的穿透容量为630微克。丙酮解吸效率不低于85.0%。7.3 样品在常温下可稳定两天。7.4 硅胶管要放在干燥器内保存。现场如果湿度过大,将影响采样结果。7.5 生产现场未见干扰物存在。7.6 使用不同厂家、不同型号、不同批号的硅胶时,应重新测定穿透容量和解吸效率。 [img]http://ng1.17img.cn/bbsfiles/images/2007/05/200705201505_52390_1625938_3.jpg[/img]
我想的到硫酸二甲酯的质量标准麻烦大家帮帮忙,小第先谢谢了哈
用食品整治办【2009】5号公告,附件里面的方法测富马酸二甲酯对高脂肪类样品(譬如沙琪玛),按标准,称取10g沙琪玛,加了中性氧化铝10g,除脂肪效果不好,最终氮吹浓缩剩余1mL以上的油脂。直接加氯仿定容也不合适,回收率也不好对低脂类样品进行实验,结果正常,回收率都有80%以上请教各位老师,有什么更好的替换方法除油脂,让富马酸二甲酯检测更好
食品中富马酸二甲酯残留量的测定 1 范围 本方法规定了食品中富马酸二甲酯残留量的GC测定方法。 本方法适用于粮食、糕点、水果等食品中富马酸二甲酯残留量的测定。 本方法的检测限(LOD)为:25mg/kg,最低检出浓度为25ug/ml。 2 规范性引用文件 下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。 GB/T 6682 分析实验室用水规格和试验方法 3 原理 样品中富马酸二甲酯(DMF)经提取净化后,用附氢火焰离子检测器的[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]进行分离测定,与标准系列比较定量 4 试剂和材料 4.1除非另有说明,所有试剂均为分析纯。水为符合GB/T 6682规定的一级水 4.2氯仿 4.3无水硫酸钠。 4.4中性氧化铝(层析用60-80目)。 4.5标准溶液贮备液:0.1g富马酸二甲酯(含量99.9%),用少量氯仿溶解,转移到100ml容量瓶中,用氯仿稀释至刻度,该标准溶液含富马酸二甲酯1mg/ml。 4.6标准溶液使用液:分别吸取标准溶液5、10、15、20、25、30ml于100ml容量瓶中,用氯仿稀释至刻度,富马酸二甲酯浓度分别为50、100、150、200、250、300ug/ml。 5 仪器与设备 5.1[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url],附氢火焰离子检测器 5.2匀浆机。 5.3粉碎机。 6 分析步骤 6.1样品制备 6.1.1 粮食、糕点、及含水分少低脂类的固体食品 称取5.0g或10.0g粉碎样品,置于250ml具塞三角烧瓶中,加30ml氯仿,振摇30min,用定性滤纸过滤,取10ml滤液,吹入氮气使浓缩至1ml,备用。 6.1.2 含脂肪较多的样品 称取粉碎样品10.0g,加中性氧化铝5-10g(视脂肪多少而定),以下按6.1.1“加30ml氯仿…”起,依法操作。 6.1.3 水果类 将水果去皮,切成碎片,加等量蒸馏水于匀浆机中匀浆后,称取20.0g匀浆液(相当于10g样品),加氯仿30ml,振摇30min,用定性滤纸过滤于125ml分液漏斗中,待分层后,用无水硫酸钠过滤,取滤液10ml,吹入氮气浓缩至1ml,待测。 6.2 测定 6.2.1色谱参考条件 6.2.1.1色谱柱: 玻璃柱(内径3mm,长2m),内装涂以2%OV-101和6%OV-210混合固定液的60-80目Chromosorb W.AW DMCS(HP) 6.2.1.2气流速度:氮气50ml/min 空气500ml/min 氢气35ml/min 。 6.2.1.3温度:气化室及检测器200℃,柱温155℃。. 6.2.1.4进样量:1μL。 6.2.2 测定 注入1uL标准系列中各浓度标准使用液于[url=https://insevent.instrument.com.cn/t/Mp][color=#3333ff]气相色谱仪[/color][/url]中,测得不同浓度富马酸二甲酯的峰高,以浓度为横坐标,相应的峰高值为纵坐标,绘制标准曲线。同时注射一定体积样品溶液,测得峰高与标准曲线比较定量。 6.2.3 阳性样品的确证 按照上述条件测定试样和标准工作溶液,如果试样中的质量色谱峰保留时间与标准工作溶液一致(变化范围在±2.5%之内) 条件许可可以通过GC—MS定性 6.2.4 空白实验 除不称取样品外,均按上述测定条件和步骤进行。 6.2.5 允许差 在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。 7. 结果计算 样品中富马酸二甲酯残留量按照下式计算:[align=center][img]http://img.vogel.com.cn/2013/0426/0858274456.jpg[/img][/align] 7. 1相关技术参数 方法最低检出限:25mg/kg。回收率在88.9%~94.2%范围内,其相对标准偏差在4.32%~9.07%的范围内。
气相测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
五氧化二磷钼酸铵分光光度法 GBZ/T 160.30-2004(改进)一、 原理空气中的五氧化二磷或三氯化磷用吸收液采集,生成的磷酸与钼酸铵和氯化亚锡反应生成磷钼蓝,在680nm 波长下测量吸光度,进行定量。二、 仪器1、 多孔玻板吸收管。2、 空气采样器,流量0~3L/min。3、 具塞试管,10ml。4、 恒温水浴。5、 分光光度计。三、 试剂 实验用水为去离子水。1、 硫酸,ρ20=1.84g/ml2、 硫酸溶液,5mol/L:28.8ml 硫酸慢慢注入水中,定容至100ml。3、 吸收液:水。4、 氯化亚锡溶液:溶解2.5g 氯化亚锡于100ml 丙三醇中,室温下可使用1个月。(注:配置氯化亚锡溶液时要加热溶解或者置于90℃中水浴中溶解。)5、 钼酸铵溶液,50g/L。(注:应该采用四水合钼酸铵。)6、 五氧化二磷标准溶液:准确称取0.2454g 干燥过的磷酸氢二钾(K2HPO4),溶于水中,定量转移入1000ml 容量瓶中,再稀释至刻度,此溶液为100*g/ml 标准贮备液。临用前,用水稀释成10.0*g/ml 五氧化二磷标准溶液。或用国家认可的标准溶液配制。四、分析步骤1、 对照试验:将装有10.0ml 吸收液的多孔玻板吸收管带至采样点,除不连接空气采样器采集空气样品外,其余操作同样品,作为样品的空白对照。2、 样品处理五氧化二磷样品的处理:用采过样的吸收液洗涤吸收管进气管内壁3 次,将吸收液倒入具塞比色管中,摇匀。于沸水浴中加热15min,取出冷却。吸取5.0ml 放入另一具塞比色管中,供测定。若样品液中五氧化二磷浓度超过测定范围,可用吸收液稀释后测定,计算时乘以稀释倍数。3、五氧化二磷标准曲线的绘制:按表1配置标准管。向各标准管中加入0.5ml 硫酸溶液,摇匀;加0.2ml 钼酸铵溶液,混匀;加1 滴氯化亚锡溶液,摇匀;(应该定容至10mL,否则显色重现性很差)放置15min。以0标准管为参比,于680nm 波长(1cm比色皿)下测量吸光度,以五氧化二磷含量(ug)对相应吸光度绘制标准曲线。 表1 P2O5标准管 管号 10 ug/mL P2O5(mL) 超纯水(mL) P2O5含量(ug)1 0.00 5.00 02 0.20 4.80 23 0.40 4.60 44 0.60 4.40 65 0.80 4.20 86 1.00 4.00 10 五、结果讨论1、标准上说磷钼络合物还原成磷钼蓝必须在一定的酸度下进行,酸度过低则空白管呈蓝色。以氯化亚锡为还原剂时,最适宜的硫酸溶液浓度为0.80~0.95mol/L,以前采用的是;加入的量应该一致。显色达到稳定后,应尽快测定。实验证明定容至10mL,硫酸浓度降低并不影响显色和吸光度。反而是使显色更快更完全。2、 改进后的标准曲线直接安装标准来做无法做出标准曲线,经过改进后按表1测定五氧化二磷标准曲线,线性关系r=0.9997回归方程Y=0.004700+0.027200X 式中X为五氧化二磷含量,Y为五氧化二磷的吸光度值。3、改进后的方法的精密度和最低检出限用2、4、8 ug的五氧化二磷标准分别平行测定6次吸光度A值,RSD在3.70%~7.6%之间。另做10次0管样计算出10次中最低0零管A值对应的五氧化二磷含量,得出五氧化二磷最低检出限为0.2ug/mL。结果显示改进后的方法的精密度及最低检出限都满足比色分析的需要。完全按标准做无法得到满意结果4、改进后的方法准确度取一样品含量的五氧化二磷本底值,将溶液均分为9份,每3份一组。分别加入低、中、高3种浓度的五氧化二磷标准溶液,计算回收率。结果见表2.,证实改进的方法准确性是可靠的。 表2改进法测定 P2O5回收率(n=3) 样品 P2O5 含量(ug) P2O5 (ug) 平均回收率 (%) 加入量 测得量 1 5.13 2.00 2.10 1052 5.13 4.00 3.87 96.83 5.13 10.00 9.89 98.9 六、小结GBZ/T 160.30-2004只是GBZ/T 160标准中的相对有代表性的一个,其他标准很多地方也同样存在表述不详细甚至模棱两可的地方,需要我们自己做实验去确证和改进。这个实验关键就是最后定容至10mL,一旦这么做了,这个实验重复性不好,线性不好,准确性不可靠就迎刃而解了。
我做的草酸二甲酯中测定酸值,但在测定时草酸二甲酯在水中易分解。用乙醇溶解后,用碱滴加测定时用的是氢氧化钾,但时在终点时,过几秒中又变色了,说明氢氧化钾对产品分解,导致酸值偏高,请问哪位高手知道怎么测才能准确呢?
附件2食品中富马酸二甲酯残留量的测定 ([url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法)1 范围 本方法规定了食品中富马酸二甲酯残留量的GC测定方法。本方法适用于粮食、糕点、水果等食品中富马酸二甲酯残留量的测定。本方法的检测限(LOD)为:25mg/kg,最低检出浓度为25ug/ml。2规范性引用文件下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。GB/T 6682 分析实验室用水规格和试验方法3 原理样品中富马酸二甲酯(DMF)经提取净化后,用附氢火焰离子检测器的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]进行分离测定,与标准系列比较定量4 试剂和材料4.1除非另有说明,所有试剂均为分析纯。水为符合GB/T 6682规定的一级水4.2氯仿4.3无水硫酸钠。4.4中性氧化铝(层析用60-80目)。4.5标准溶液贮备液:0.1g富马酸二甲酯(含量99.9%),用少量氯仿溶解,转移到100ml容量瓶中,用氯仿稀释至刻度,该标准溶液含富马酸二甲酯1mg/ml。4.6标准溶液使用液:分别吸取标准溶液5、10、15、20、25、30ml于100ml容量瓶中,用氯仿稀释至刻度,富马酸二甲酯浓度分别为50、100、150、200、250、300ug/ml。5 仪器与设备5.1[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url],附氢火焰离子检测器5.2匀浆机。5.3粉碎机。6 分析步骤6.1样品制备6.1.1 粮食、糕点、及含水分少低脂类的固体食品 称取5.0g或10.0g粉碎样品,置于250ml具塞三角烧瓶中,加30ml氯仿,振摇30min,用定性滤纸过滤,取10ml滤液,吹入氮气使浓缩至1ml,备用。6.1.2 含脂肪较多的样品 称取粉碎样品10.0g,加中性氧化铝5-10g(视脂肪多少而定),以下按6.1.1“加30ml氯仿…”起,依法操作。6.1.3 水果类 将水果去皮,切成碎片,加等量蒸馏水于匀浆机中匀浆后,称取20.0g匀浆液(相当于10g样品),加氯仿30ml,振摇30min,用定性滤纸过滤于125ml分液漏斗中,待分层后,用无水硫酸钠过滤,取滤液10ml,吹入氮气浓缩至1ml,待测。6.2 测定6.2.1色谱参考条件6.2.1.1色谱柱: 玻璃柱(内径3mm,长2m),内装涂以2%OV-101和6%OV-210混合固定液的60-80目Chromosorb W.AW DMCS(HP) 6.2.1.2气流速度:氮气50ml/min;空气500ml/min 氢气35ml/min;。6.2.1.3温度:气化室及检测器200℃,柱温155℃。.6.2.1.4进样量:1μL。6.2.2 测定注入1uL标准系列中各浓度标准使用液于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]中,测得不同浓度富马酸二甲酯的峰高,以浓度为横坐标,相应的峰高值为纵坐标,绘制标准曲线。同时注射一定体积样品溶液,测得峰高与标准曲线比较定量。6.2.3 阳性样品的确证按照上述条件测定试样和标准工作溶液,如果试样中的质量色谱峰保留时间与标准工作溶液一致(变化范围在±2.5%之内)条件许可可以通过GC—MS定性6.2.4 空白实验除不称取样品外,均按上述测定条件和步骤进行。6.2.5 允许差在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。7. 结果计算样品中富马酸二甲酯残留量按照下式计算: X:样品中富马酸二甲酯残留量,mg/kgA:测定样品液中富马酸二甲酯含量,ug/mlV1:浓缩用样品提取液体积,mlV2:样品氯仿提取液总体积,mlV3:样品浓缩后的体积,mlV4:标准溶液进样体积,ulV5:样品溶液进样体积,ulm:样品重量,g7. 相关技术参数方法最低检出限:25mg/kg。回收率在88.9%~94.2%范围内,其相对标准偏差在4.32%~9.07%的范围内。
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
求助环境空气五氧化二磷测定的钼蓝分光光度法,HJ546-2015的标准曲线,扩项的方法证实,没查到标准曲线的斜率要求,望做过的前辈提供一下对应浓度的吸光值对比一下。急急急,谢谢各位。
[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]测3.5-二甲氧基苯甲酸甲酯要用什么标准物做外标
油漆、胶粘剂行业的新型溶剂---- 碳酸二甲酯碳酸二甲酯(简称DMC)是近年来受到国内外广泛关注的环保型绿色化工产品。1992年DMC在欧洲通过了非毒性化学品(Non toxic substance)的注册登记,属于无毒或微毒化工产品。因此其非反应性用途如作为涂料(油漆、油墨)、胶粘剂等行业的溶剂、溶媒正在实现工业化。目前,碳酸二甲酯作为一种新型的低毒溶剂在油漆、胶粘剂等行业在国内市场已经成熟应用并实现工业化,碳酸二甲酯在硝基漆、家具漆、车用漆、热塑性丙烯酸漆、丙烯酸聚氨酯漆、醇酸聚氨酯漆、氯化橡胶马路标线漆等多种油漆中均能应用,可取代目前使用的甲苯、醋酸乙酯、醋酸丁酯、丙酮或丁酮等溶剂,配制的油漆完全符合现有油漆的技术标准。碳酸二甲酯在胶粘剂行业亦可替代醋酸乙酯、醋酸丁酯、丙酮或丁酮等溶剂,应用前景看好。碳酸二甲酯在油漆、涂料和胶粘剂行业的应用优势在于: 1、碳酸二甲酯是一种无毒溶剂。2、碳酸二甲酯与其他有机物相溶性好。3、碳酸二甲酯的脱酯能力比较高4、碳酸二甲酯的溶沸点范围窄,表面张力大,粘度低,介电常数小。5、碳酸二甲酯具有较高的蒸发温度和较快的蒸发速度。6、碳酸二甲酯还具有闪点高、蒸汽压低,空气中爆炸下限高等特点因此是集清洁性和安全性于一身的绿色溶剂。7、碳酸二甲酯的价格较低,替代醋酸乙酯、醋酸丁酯等溶剂可大幅降低生产成本。目前国内众多的油漆、胶粘剂生产厂家对于碳酸二甲酯在溶剂方面的替代已经实现工业化,国际上诸多著名的油漆、胶粘剂的生产工厂亦正在使用碳酸二甲酯来代替传统的各种溶剂。在全球原油价格持续上涨和环境保护日益重视的情况下,碳酸二甲酯作为一种环保型的绿色溶剂将会有越来越广阔的市场应用。
今年4月1日,我国向世贸组织技术性贸易壁垒委员会通报了国家质检总局和国家标准化管理委员会联合发布的化妆品用二氧化钛中国国家标准(编号ICS:71.060.20)规范文件。 该标准由中国石油和化学工业协会提出,中国化学标准化技术委员会无机化工分会(SAC/TC63/SC1)归口,上海、天津、江苏及河南四家化工企业共同起草。 标准规定了化妆品用二氧化钛的分类、要求、试验方法、检验规则、标志、标签、包装、运输和贮存,适用用于化妆品生产制造的二氧化钛粉体,该物品在化妆品中主要起遮盖、改善肤色、增白及屏蔽紫外线的作用。标准第5章涉及的重金属、砷、铅、汞四项指标以及第8章标志、标签要求为强制性,其他为推荐性要求。 化妆品用二氧化钛应分别符合表1 和表2 要求:表1 I类产品的要求项目指标锐钛型(A)金红石型(R)二氧化钛(TiO2)w/% ≥9898干燥减量 w/% ≤0.50.5灼烧矢量 w/% ≤0.50.5水溶物 w/% ≤0.50.3重金属(以Pb计)w/% ≤0.00200.0020砷(As)w/% ≤0.00050.0005铅(Pb)w/% ≤0.00100.0010汞(Hg)w/% ≤0.00010.0001pH 6.5~8.56~8白度(Wh) ≥9090细度(45 μm) ≤0.10.1

[b][/b][align=center][b]二氧化双环戊二烯反应液的高效液相色谱分析[/b][/align][align=center] 摘要:采用高效液相色谱建立了快速分析二氧化双环戊二烯反应液的新方法,分析该反应液中的溶剂异丙苯、氧化剂过氧化氢异丙苯和反应副产物2-苯基异丙醇。以Agilent Eclipse XDB C18色谱柱(4*250mm)为分离柱,乙腈/0.1%磷酸为流动相,梯度淋洗,流量1.0 mL/min。实验结果表明,目标组分分离效果良好,且各目标化合物在各自配制的浓度范围内呈现良好的线性关系,回归系数均大于0.999,各目标组分的最低检出限为0.15~0.25 mg/L。实际试样中的加标回收率为101.94%~111.62%,对标准溶液、加标样品溶液及实际试样都进行了重复测定,其相对标准偏差均小于等于2.37%,定量结果准确可靠,数据精密度良好。将高效液相色谱应用于二氧化双环戊二烯反应液的分析,为二氧化双环戊二烯生产企业提供了一种简便、快速、准确的分析方法。[/align][b][/b] 关键词:高效液相色谱;过氧化氢异丙苯;异丙苯;2-苯基异丙醇;二氧化双环戊二烯二氧化双环戊二烯(DCPDDO),是一种重要的脂环族特种环氧化物,其耐热性和电绝缘性良好,且具有较高的硬度,被广泛应用于耐高温浇铸料、玻璃钢、粘合剂及电子器件封装等方面,在国内具有良好的市场前景和应用价值,极具开发潜力[sup][/sup]。二氧化双环戊二烯是由双环戊二烯(DCPD)经环氧化反应制得。目前,工业上一般采用卤醇法、过氧酸法和氢化过氧化物催化环氧化法等方法制备二氧化双环戊二烯,但这些方法对设备腐蚀比较严重,同时也会造成严重的环境污染,且副产物多,产物收率低[sup][/sup]。近年来国外都在开发以清洁氧源过氧化氢作为氧化剂,以固体杂多酸为催化剂的环氧化工艺[sup][/sup]。过氧化氢异丙苯(Cumene Hydroperoxide,CHP)为无色或淡黄色液体,可作为链式自动氧化反应和聚合反应的引发剂,有机化合物的氧化剂,已经广泛用于精细化工、高分子材料和有机合成等领域。苏如孟[sup][/sup]将钛硅分子筛用于催化过氧化氢异丙苯氧化丙烯反应,在最佳的反应条件下,过氧化氢异丙苯的有效利用率可达到72.75%。故考虑以过氧化氢异丙苯作为氧化剂氧化双环戊二烯,异丙苯(Isopropyl Benzene,IPB)为溶剂,钛硅分子筛作为催化剂,制备二氧化双环戊二烯,反应温度控制在50℃—100℃。 氧化反应中主要副反应产物是2-苯基异丙醇(2-Dimethyl Phenyl Carbinol,2-DPC)。[img=,603,136]https://ng1.17img.cn/bbsfiles/images/2019/08/201908121646479566_2467_1617661_3.png!w603x136.jpg[/img]目前,测定异丙苯、过氧化氢异丙苯和2-苯基异丙醇的方法主要有高效液相色谱(HPLC)法、[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url](GC)法和碘量法等。刘俊彦等[sup][/sup]使用超高效液相色谱仪,采用BEH C18反相色谱柱,以乙腈/水为流动相,流量0.4 mL/min,采用梯度洗脱,建立了准确可靠的快速分析异丙苯中过氧化氢异丙苯与酚类杂质的方法。刘岳树等[sup][/sup]建立了[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]-氢火焰离子化检测器同时测定过氧化氢异丙苯中异丙苯和苯乙酮含量的方法。郭阳等[sup][/sup]采用毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法建立了同时测定埃索美拉唑镁原料药中异丙苯、2-苯基异丙醇、乙醇等8种有机溶剂残留量的方法。该方法使用HP-1色谱柱,载气为氦气,流速为4.0 mL/min,分流比为10:1,程序升温,检测器为氢火焰离子化检测器,结果表明该方法灵敏度好。王华等[sup][/sup]利用I[sub]2[/sub]的氧化性和I[sup]-[/sup]的还原性来对过氧化氢异丙苯进行滴定,从而测定其浓度,并将碘量法与液相色谱测得结果比较,相差不大。综上所述,目前虽已开发了分别测定异丙苯、过氧化氢异丙苯和2-苯基异丙醇的方法,却未开发过同时测定异丙苯中过氧化氢异丙苯和2-苯基异丙醇的方法。本文建立了高效液相色谱法同时测定二氧化双环戊二烯反应液中异丙苯、过氧化氢异丙苯和2-苯基异丙醇的分析方法。本法简便、快速,可用于二氧化双环戊二烯产品的质量控制。[b]1 实验部分1.1 仪器与试剂[/b]Agilent 1260 SL 型高效液相色谱仪,配DAD检测器,自动进样器、柱温箱及二元高压泵; Mettler Toledo XS 205型分析天平;Milli-Q Advantage A10型超纯水机。乙腈(ACN,色谱纯),西班牙萨劳化工有限公司;磷酸(H[sub]3[/sub]PO[sub]4[/sub],分析纯),上海永华化学试剂有限公司;2-苯基异丙醇(99%),阿拉丁;异丙苯(99%),Adamas-beta;过氧化氢异丙苯(80%),阿拉丁;双环戊二烯(99%),广州市宏巨化工有限公司;钛硅分子筛TS-1,南京先丰纳米材料科技有限公司;样品由过氧化氢异丙苯氧化双环戊二烯制得。[b]1.2 色谱条件[/b]分析柱:Agilent Eclipse XDB C18色谱柱(4*250mm),稀释剂:乙腈;进样量:20μl,柱温:30℃,流速:1.0ml/min,检测波长为210 nm。梯度洗脱程序:[table][tr][td][align=center]Time/min[/align][/td][td][align=center]ACN /%[/align][/td][td][align=center]0.1% H[sub]3[/sub]PO[sub]4[/sub]/%[/align][/td][/tr][tr][td][align=center]0.01[/align][/td][td][align=center]30[/align][/td][td][align=center]70[/align][/td][/tr][tr][td][align=center]8.00[/align][/td][td][align=center]70[/align][/td][td][align=center]30[/align][/td][/tr][tr][td][align=center]10.00[/align][/td][td][align=center]90[/align][/td][td][align=center]10[/align][/td][/tr][tr][td][align=center]15.00[/align][/td][td][align=center]90[/align][/td][td][align=center]10[/align][/td][/tr][tr][td][align=center]15.10[/align][/td][td][align=center]30[/align][/td][td][align=center]70[/align][/td][/tr][tr][td][align=center]20.00[/align][/td][td][align=center]30[/align][/td][td][align=center]70[/align][/td][/tr][/table][b]1.3 溶液的配制[/b]1.3.1 对照品储备液的配制分别精密称取异丙苯标准品46.00 mg,过氧化氢异丙苯标准品31.94 mg,2-苯基异丙醇标准品23.44 mg,分别置于50 ml容量瓶中,加乙腈溶解并稀释至刻度,摇匀,最后得异丙苯对照品储备液(920.0 mg/L)、过氧化氢异丙苯对照品储备液(511.0 mg/L)和2-苯基异丙醇对照品储备液(468.8 mg/L),三种储备液都是单独配置,未混合。1.3.2 标准溶液的配制将上述对照品储备液用乙腈精密稀释适当倍数,各自配成4.60、18.40、46.00、92.00、184.00 mg/L系列异丙苯标准溶液,0.51、5.11、12.77、25.55、51.10 mg/L系列过氧化氢异丙苯标准溶液,0.47、4.69、11.72、23.44、46.88 mg/L系列2-苯基异丙醇标准溶液。1.3.3 样品溶液的配制精密称取实际样品61.90 mg,置50 ml容量瓶中,加乙腈溶解并稀释至刻度,摇匀,配成1238 mg/L样品溶液;精密量取约为1238 mg/L样品溶液1.25 ml于10 ml容量瓶中,加入乙腈定容,摇匀作为样品溶液(155 mg/L)。[b]2 结果与讨论[/b]2.1 [b] 色谱条件的优化[/b] 当使用乙腈与水为流动相时,过氧化氢异丙苯与2-苯基异丙醇的保留时间非常接近,即使调低有机相比例也无法将这两种物质很好的分离,即在等度的条件下,过氧化氢异丙苯与2-苯基异丙醇无法分离。故考虑将超纯水换成0.1%的磷酸溶液,并采用梯度淋洗,具体条件见1.2,使用该色谱条件时,2-苯基异丙醇与过氧化氢异丙苯的保留时间分别为6.8min和7.8min,且异丙苯的保留时间为13.1min,三种目标化合物能得到较好的分离。由于2-苯基异丙醇标样中含有异丙苯,过氧化氢异丙苯中含有2-苯基异丙醇和异丙苯,故考虑将三种标样分开测定,不测定混合标样。异丙苯、过氧化氢异丙苯和2-苯基异丙醇在210nm紫外吸收波长下的色谱图如图1所示。[img=,434,337]https://ng1.17img.cn/bbsfiles/images/2019/08/201908121647263745_2090_1617661_3.png!w434x337.jpg[/img]2.1 [b]标准溶液的线性关系与检出限[/b]实际试样测得结果中IPB,CHP和2-DPC的浓度分别为100.06,13.97,14.75 mgL[sup]-1[/sup],将实际试样中所测得浓度大致作为线性范围的中间点,以保证实际试样中三种目标化合物的浓度都在线性范围内,所以确定IPB,CHP和2-DPC的线性范围为4.60 - 184.00,0.51 - 51.10,0.47 - 46.88 mgL[sup]-1[/sup]。每份标准溶液测定6次,计算峰面积并取平均值,目标化合物的线性关系、检出限和定量限如表1所示。[align=center][b]表1 目标化合物的线性关系、检出限和定量限[/b][/align][align=center][b]Table 1 Linear relationship, detection limit and limit of quantitation of target compounds[/b][/align][table][tr][td][align=center][b]Component[/b][/align][/td][td][align=center][b]Linear range/(mgL[sup]-1[/sup])[/b][/align][/td][td][align=center][b]Correlation coefficient[/b][/align][/td][td][align=center][b]Regression equation [/b][/align][/td][td][align=center][b]Detection limit /(mgL[sup]-1[/sup])[/b][/align][/td][td][align=center][b]Limit of quantitation/(mgL[sup]-1[/sup])[/b][/align][/td][/tr][tr][td][align=center]IPB[/align][/td][td][align=center]4.60 - 184.0[/align][/td][td][align=center]0.999[/align][/td][td][align=center]Y=17.41X+15.60[/align][/td][td][align=center]0.25[/align][/td][td][align=center]0.60[/align][/td][/tr][tr][td][align=center]CHP[/align][/td][td][align=center]0.51 - 51.10[/align][/td][td][align=center]0.999[/align][/td][td][align=center]Y=18.17X+1.967[/align][/td][td][align=center]0.15[/align][/td][td][align=center]0.50[/align][/td][/tr][tr][td][align=center]2-DPC[/align][/td][td][align=center]0.47 - 46.88[/align][/td][td][align=center]0.999[/align][/td][td][align=center]Y=22.11x+4.028[/align][/td][td][align=center]0.17[/align][/td][td][align=center]0.47[/align][/td][/tr][/table][b]2.3 方法加标回收率[/b]精密移取5.00 ml浓度为155 mg/L的样品溶液于10 ml的容量瓶中,再加入一定量的对照溶液,定容,配置成回收率溶液。按上述条件连续进样,所得结果如下表2。由表可知异丙苯,过氧化氢异丙苯和2-苯基异丙醇的回收率分别在104.2%—111.6%,101.9%—107.2%,102.1%—108.4% 之间,在100.0%~115.0% 之间;RSD分别为为均小于2.50%,说明本方法的准确度较好。[align=center][b][img=,375,290]https://ng1.17img.cn/bbsfiles/images/2019/08/201908121648504751_7688_1617661_3.png!w375x290.jpg[/img][/b][/align][align=center][b]表2 异丙苯,过氧化氢异丙苯和2-苯基异丙醇的加标回收率(n=3)[/b][/align][align=center][b]Table 2 Recoveries of IPB , CHP and 2-DPC(n=3)[/b][/align][table][tr][td=1,2][align=center][b]Component[/b][/align][/td][td=4,1][align=center][b]Concentration/(mgL[sup]-1[/sup])[/b][/align][/td][td=1,2][align=center][b]Average Recovery/%[/b][/align][/td][td=1,2][align=center][b]RSD/%[/b][/align][/td][/tr][tr][td][align=center][b]Original[/b][/align][/td][td=2,1][align=center][b]Added[/b][/align][/td][td][align=center][b]Measured[/b][/align][/td][/tr][tr][td=1,3][align=center][b]IPB[/b][/align][/td][td][align=center]50.03[/align][/td][td][align=center]22.77[/align][/td][td=2,1][align=center]81.26[/align][/td][td][align=center]111.6%[/align][/td][td][align=center]1.25[/align][/td][/tr][tr][td][align=center]50.03[/align][/td][td][align=center]45.54[/align][/td][td=2,1][align=center]102.1[/align][/td][td][align=center]106.8%[/align][/td][td][align=center]0.65[/align][/td][/tr][tr][td][align=center]50.03[/align][/td][td][align=center]91.08[/align][/td][td=2,1][align=center]147.0[/align][/td][td][align=center]104.2%[/align][/td][td][align=center]0.13[/align][/td][/tr][tr][td=1,3][align=center][b]CHP[/b][/align][/td][td][align=center]7.37[/align][/td][td][align=center]2.56[/align][/td][td=2,1][align=center]10.65[/align][/td][td][align=center]107.3%[/align][/td][td][align=center]0.70[/align][/td][/tr][tr][td][align=center]7.37[/align][/td][td][align=center]6.39[/align][/td][td=2,1][align=center]13.94[/align][/td][td][align=center]101.3%[/align][/td][td][align=center]2.37[/align][/td][/tr][tr][td][align=center]7.37[/align][/td][td][align=center]12.77[/align][/td][td=2,1][align=center]20.53[/align][/td][td][align=center]101.9%[/align][/td][td][align=center]1.98[/align][/td][/tr][tr][td=1,3][align=center][b]2-DPC[/b][/align][/td][td][align=center]6.98[/align][/td][td][align=center]2.34[/align][/td][td=2,1][align=center]10.10[/align][/td][td][align=center]108.4%[/align][/td][td][align=center]1.94[/align][/td][/tr][tr][td][align=center]6.98[/align][/td][td][align=center]5.86[/align][/td][td=2,1][align=center]13.53[/align][/td][td][align=center]105.4%[/align][/td][td][align=center]1.79[/align][/td][/tr][tr][td][align=center]6.98[/align][/td][td][align=center]11.72[/align][/td][td=2,1][align=center]19.10[/align][/td][td][align=center]102.1%[/align][/td][td][align=center]0.19[/align][/td][/tr][/table][b]2.4 进样重复性[/b]取异丙苯、过氧化氢异丙苯和2-苯基异丙醇测定线性关系中浓度分别为46.00,12.77,11.72 mgL[sup]-1[/sup]的标准溶液作为进样重复性溶液,连续测定6次,记录峰面积。结果显示异丙苯、过氧化氢异丙苯和2-苯基异丙醇的RSD分别为0.20%,0.35%,0.85%(n=6),说明该方法的重复性良好。[b]2.5 样品测定[/b]2.5.1 精密度实验取配制好的样品溶液(155 mg/L),按上述色谱条件,对实际反应液样品进行分析,连续进样8次,记录峰面积。实际反应液样品在210nm紫外吸收波长下的色谱图见图2。实际样品中异丙苯、过氧化氢异丙苯和2-苯基异丙醇测定结果见表3。从表3可看出,定量分析结果的重复性良好。[align=center][b]表3 实际试样的测定结果(n=8)[/b][/align][align=center][b]Table 3 The results of actual sample (n=8)[/b][/align][table][tr][td][align=center][b]Component[/b][/align][/td][td][align=center][b]IPB[/b][/align][/td][td][align=center][b]CHP[/b][/align][/td][td][align=center][b]2-DPC[/b][/align][/td][/tr][tr][td][align=center][b]Concentration/(mgL[sup]-1[/sup])[/b][/align][/td][td][align=center]100.1[/align][/td][td][align=center]14.75[/align][/td][td][align=center]13.97[/align][/td][/tr][tr][td][align=center][b]Content[/b][/align][/td][td][align=center]64.55%[/align][/td][td][align=center]9.53%[/align][/td][td][align=center]9.03%[/align][/td][/tr][tr][td][align=center][b]RSD[/b][/align][/td][td][align=center]0.15%[/align][/td][td][align=center]0.47%[/align][/td][td][align=center]0.28%[/align][/td][/tr][/table]2.5.2 连续测定不同时间段的反应液取反应中不同时间段(间隔1小时)的样品分别配制样品溶液(500 mg/L),按上述色谱条件,对实际反应产物试样进行分析,记录峰面积。不同样品中过氧化氢异丙苯,2-苯基异丙醇和异丙苯的测定结果见表4,含量变化趋势见图3。[align=center][b]表4 连续多个样品的测试结果[/b][/align][align=center][b]Table 4 The results of multiple consecutive samples[/b][/align][table][tr][td=1,2][align=center][b]Component[/b][/align][/td][td=2,1][align=center][b]IPB[/b][/align][/td][td=2,1][align=center][b]CHP[/b][/align][/td][td=2,1][align=center][b]2-DPC[/b][/align][/td][/tr][tr][td][align=center][b]Concentration/[/b][/align][align=center][b](mgL[sup]-1[/sup])[/b][/align][/td][td][align=center][b]Content[/b][/align][/td][td][align=center][b]Concentration/[/b][/align][align=center][b](mgL[sup]-1[/sup])[/b][/align][/td][td][align=center][b]Content[/b][/align][/td][td][align=center][b]Concentration/[/b][/align][align=center][b](mgL[sup]-1[/sup])[/b][/align][/td][td][align=center][b]Content[/b][/align][/td][/tr][tr][td][align=center][b]0h[/b][/align][/td][td][align=center]216.8[/align][/td][td][align=center]43.01%[/align][/td][td][align=center]94.97[/align][/td][td][align=center]18.48%[/align][/td][td][align=center]9.57[/align][/td][td][align=center]1.92%[/align][/td][/tr][tr][td][align=center][b]1h[/b][/align][/td][td][align=center]225.6[/align][/td][td][align=center]44.79%[/align][/td][td][align=center]61.46[/align][/td][td][align=center]11.96%[/align][/td][td][align=center]44.64[/align][/td][td][align=center]8.96%[/align][/td][/tr][tr][td][align=center][b]2h[/b][/align][/td][td][align=center]223.6[/align][/td][td][align=center]44.37%[/align][/td][td][align=center]59.10[/align][/td][td][align=center]11.50%[/align][/td][td][align=center]49.26[/align][/td][td][align=center]9.89%[/align][/td][/tr][tr][td][align=center][b]3h[/b][/align][/td][td][align=center]227.9[/align][/td][td][align=center]45.22%[/align][/td][td][align=center]57.09[/align][/td][td][align=center]11.11%[/align][/td][td][align=center]50.23[/align][/td][td][align=center]10.09%[/align][/td][/tr][tr][td][align=center][b]4h[/b][/align][/td][td][align=center]236.7[/align][/td][td][align=center]46.96%[/align][/td][td][align=center]58.94[/align][/td][td][align=center]11.47%[/align][/td][td][align=center]54.65[/align][/td][td][align=center]10.97%[/align][/td][/tr][tr][td][align=center][b]5h[/b][/align][/td][td][align=center]215.9[/align][/td][td][align=center]42.83%[/align][/td][td][align=center]51.83[/align][/td][td][align=center]10.08%[/align][/td][td][align=center]49.53[/align][/td][td][align=center]9.95%[/align][/td][/tr][/table][img=,582,236]https://ng1.17img.cn/bbsfiles/images/2019/08/201908121648194131_8651_1617661_3.png!w582x236.jpg[/img]根据不同时间段反应液中三种化合物的变化趋势,可知在该反应中,作为溶剂的异丙苯含量变化不大,基本维持在40.0%—47.0%,在反应1小时后,作为氧化剂的过氧化氢异丙苯的含量从18.48%降至11.96%,反应副产物2-苯基异丙醇的含量从1.92%升至8.96%,随后氧化剂和副产物的含量基本稳定,变化不大,说明该反应主要在前1小时内进行。2 [b]结论[/b]上述实验结果表明,通过高效液相色谱梯度淋洗法能准确地分析二氧化双环戊二烯反应液中异丙苯、过氧化氢异丙苯和2-苯基异丙醇的含量,此方法灵敏度高、稳定性好、重复性满足实验要求。此外,可使用该方法对不同时间段的二氧化双环戊二烯反应液中不同化合物含量进行实时监测,获得该反应过程中化合物的变化趋势,对进一步探究和完善二氧化双环戊二烯的合成方法有重大意义。[b]参考文献:[/b] 何红振,范阳阳,李韶峰,等. 特种环氧树脂二氧化双环戊二烯的合成与应用. 化学推进剂与高分子材料,2017,15(5):29-39. 李丽,阎丽静,彭军,等. 高性能环氧树脂二氧化双环戊二烯的制备. 精细石油化工,2007,24(3):24-27. 于浩,沃善康,李丽娟,等. 脂环族环氧化物的合成与应用(四):二氧化双环戊二烯. 热固性树脂,2000,15(1):36-40. 张术栋,徐成华. 烯烃环氧化及其催化剂的研究进展. 合成化学,2003,11(4):294-299. Mizuno N,Yamaguchi K,Kamata K. Epoxidation of olefins with hydrogen peroxide catalyzed by polyoxometalate. Coor Chem Rev, 2005,249(17,18):1944-1956. 薛经纬. 二氧化双环戊二烯制备新工艺研究.山东:山东理工大学,2011. 徐强,杜咏梅,李春迎,等. 二氧化双环戊二烯的合成. 工业催化,2010,18(12):52-54. 苏如孟. 钛硅分子筛催化过氧化氢异丙苯氧化丙烯反应. 大连:大连理工大学,2018. 刘俊彦,李继文,王川. 超高效液相色谱法快速分析异丙苯中的过氧化氢异丙苯与酚类化合物. 石油化工,2017,46(7):934-937. 刘岳树,马武生. [url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法同时测定过氧化氢异丙苯中异丙苯和苯乙酮. 分析科学学报,2010,26(6):738-740. 郭阳,冯敏,陈玉洁. 毛细管[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法同时测定埃索美拉唑镁原料药中8种有机溶剂的残留量. 中国药房,2017,28(36):5160-5163. 王华. 两种不同方法对过氧化氢异丙苯产品浓度的分析. 数码设计(上),2018(6):205.
各位老师,NYT1723标准测食品中富马酸二甲酯。缓冲溶液用的乙酸钠,溴化四丁基铵,乙酸调PH=6.0。实验室现没有溴化四丁基铵,用别的缓冲液可以吗?有代替方法吗?感谢!!
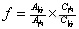
[align=center]医用胶中碳酸二甲酯含量的测定[/align][align=center]西安国联质量检测技术股份有限公司[/align][align=center]食品事业部:王军浩[/align][b]1.原理[/b] 试样经丙酮溶解定容,采用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]测定,保留时间定性,峰面积外标法定量。实验方法参考熊冬生等[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法测定碳酸二甲酯的含量。[b]材料与方法[/b]2.1仪器设备 [url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url]:配FID检测器,分析天平:感量为0.0001g 。2.2试剂丙酮:优级纯;正丁醇:分析纯碳酸二甲酯标准品[b]试样处理[/b]3.1 样品配制:取1.0mL样品,用丙酮定容至100mL,准确加入8μL正丁醇作为内标物于10mL容量瓶中,用碳酸二甲酯样品的丙酮溶液稀释至刻度,摇匀。3.2[b] [/b]色谱条件:3.2.1色谱柱:Rtx-Wax(30m×0.25mm×0.25μm),3.2.2流速:1.32mL/min ;3.2.3进样体积1.0μL;分流比:10:13.2.4柱温:程序升温,初始温度60℃保持1 min,以1℃/min的速率升温至63℃,保持1 min,再以60℃/min的速率升温至123℃,保持2min;进样口温度:250℃;检测器:250℃。3.3外标法计算公式:[align=center][img=,79,29]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091749325898_9906_2904018_3.png!w79x29.jpg[/img][/align][align=center][img=,107,29]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091749423558_5154_2904018_3.png!w107x29.jpg[/img][/align]式中: X-样品中碳酸二甲酯的含量,% -碳酸二甲酯的相对校正因子A[sub]内、[/sub]A[sub]标、[/sub]A[sub]样[/sub]--表示内标物、标准品、样品的出峰面积(峰高)C[sub]内、[/sub]C[sub]标、[/sub]C[sub]样[/sub]--表示内标物、标准品、样品的浓度(相对密度) 两次测试结果的相对误差小于10%即为测试平行[b]4实验结果[/b]4.1外标法标准曲线线性的确定用微量注射器分别精密量取碳酸二甲酯标准品2、4、8、16、32、64μL于10mL容量瓶中,准确加入8μL正丁醇作为内标物,用丙酮稀释至刻度,摇匀,使碳酸二甲酯的体积分数分别为:0.02%、0.04%、0.08%、0.16%、0.32%、0.64%。测碳酸二甲酯体积分数与峰面积的相关性,确定相关系数及线性范围,标准曲线见图1、图1-1。可见,碳酸二甲酯体积分数在0.02-0.64%范围内,Y=2.27751e+006X-32112.5,R[sup]2[/sup]=0.9932927;体积分数与色谱峰面积呈显著的线性关系,可满足定量分析的需要。[align=center][img=,624,399]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091749568659_4069_2904018_3.png!w624x399.jpg[/img] [/align][align=center]图1 碳酸二甲酯标准曲线[/align][align=center][img=,449,438]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091750081589_8599_2904018_3.png!w449x438.jpg[/img] [/align][align=center]图1-1 6种体积浓度碳酸二甲酯色谱图比较[/align]4.2检出限取0.02%标准溶液梯度稀释进样,至S/N=3±1,确定出碳酸二甲酯的最低检出限0.1nL即0.0001%。4.3加标回收及重复性 对样品进行加标回收实验,加标浓度设0.08%,回收率结果见图2,可见对样品进行的加标回收率在95%左右。对样品进行重复性实验结果见图3,结果可见,RSD为0.190%,由图2和图3结果表明本实验方法能够满足分析要求。[table][tr][td][align=center] [/align][/td][td][align=center]加入浓度%[/align][/td][td][align=center]测定值%[/align][/td][td][align=center]回收率/%[/align][/td][/tr][tr][td][align=center]样品[/align][/td][td][align=center]——[/align][/td][td][align=center]0.015[/align][/td][td][align=center]——[/align][/td][/tr][tr][td][align=center]样品加标[/align][/td][td][align=center]0.08[/align][/td][td][align=center]0.091[/align][/td][td][align=center]95[/align][/td][/tr][/table][align=center]图2 碳酸二甲酯样品加标回收率结果[/align][align=center] [/align][align=center][img=,448,460]http://ng1.17img.cn/bbsfiles/images/2018/07/201807091750234959_4333_2904018_3.png!w448x460.jpg[/img] [/align][align=center]图3 碳酸二甲酯加标样品重复性实验[/align][align=center] [/align][b]5.结论[/b]综上所述:医用胶中碳酸二甲酯的测定方法学从线性、重复性、回收率、准确度、最低检出限均符合分析要求。本方法的碳酸二丁酯的检出限为0.1nL即0.0001%,本方法可以用于医用胶中碳酸二甲酯的测定。
GB/T6432-94标准中4.6 盐酸标准溶液:邻苯二甲酸氢钾法标定,按GB601制备。平常都是用碳酸钠标定盐酸,用邻苯二甲酸氢钾标定氢氧化钠,这里却.......该如何理解呢?
1 范围 本标准规定了氧化铝中三氧化二铁含量的测定方法。 本标准适用于氧化铝中三氧化二铁含量的测定。测定范围:0.005%-0.100%, 2 方法原理 三价铁用盐酸经胺还原为二价铁,在乙酸一乙酸钠缓冲溶液中加人邻二氮杂菲使形成络合物,于分光光度计波长 510nm处测量其吸光度,借以测定三氧化二铁含量。 3 试剂 3.1 硼酸:优级纯。 3.2 无水碳酸钠:优级纯。 3.3 硝酸(3.00mol/L), 3.4 盐酸羟胺溶液(10g/L) 3.5邻二氮杂菲溶液(1g/L):称取1g邻二氮杂菲溶于1.5mL--2.5mI冰乙酸中(ρl.05g/mL),移入1000mL容量瓶中,用水稀释至1000mL,混匀。 3.6 缓冲溶液(pH=4.9):称取272g乙酸钠(CH3COONa3H20)溶于500mL水中,加人240mL 冰乙酸(pl.05g/mL),用水稀释至1000mL,混匀。 3.7 三氧化二铁标准贮存溶液:称取0.50g三氧化二铁(含量)99.99%,预先于600℃灼烧2h,并于干燥器(4.4)中冷却至室温)置于150mL烧杯中,沿杯壁加人20mL盐酸(ρ.19g/mL),盖上表皿微热使全部溶解,冷却至室温,将溶液移人1000mL容量瓶中,用水洗净烧杯,洗液并人容量瓶中,用水稀释至刻度,混匀。此溶液1mL含0.5mg三氧化二铁。 3.8 三氧化二铁标准溶液:移取25.00mL三氧化二铁标准贮存溶液(3.7)于500mL容量瓶中,加人30.0ml硝酸((3.3),用水稀释至刻度,混匀。此溶液1mL含0.025mg三氧化二铁,用时配制。 4 仪器、装置及器具 4.1铂坩埚:30mL,带盖。 4.2 分光光度计。 4.3 电热板:用调压器控制加热温度不高于250℃ 4.4 干燥器:用新活性氧化铝作干燥剂。 5 试样 5.1 试样应通过0.125mm孔径筛网。 5.2试样预先在300℃士100℃烘干2h,置于干燥器((4.4)中冷却至室温。 6 分析步骤 6.1 试料 称取约0.50g试样(5),精确至0.0001g 6.2 测定次数 独立地进行两次测定,取其平均值。 6.3 空白试验 随同试料 (6.1)做空白试验 6.4 测定 6.4.1将试料(6.1)置于铂坩埚(4.1)中,加人0.500g硼酸(3.1)和1.300g碳酸钠(3.2),用铂勺搅匀。盖卜坩埚盖,置于约700℃的高温炉中,升温至1000℃熔融20min,取出稍冷 空白试验直接在1000℃熔融2min-3min,取出稍冷。 6.4.2 向坩埚中加人沸水,加热至近沸,使熔块全部溶解,将溶液移人预先盛有22.3mL(随同试料空白则为12.6mL)硝酸(3.3)的150m工聚四氟乙烯烧杯中,坩埚用热水冲洗二次,用聚四氟乙烯棒搅拌使沉淀尽量溶解,坩埚用3ml一硝酸(3.3)和热水充分洗净,洗涤液并人烧杯中,盖上表皿,置电热板(4.3)上加热至沉淀全部溶解,取下,置冷水槽中冷却至室温。将溶液移人100ml容量瓶中,用水洗净烧杯,洗液并人容量瓶中,并用水稀释至刻度,混匀(此溶液也可用以测定二氧化硅量)。 6.4.3 分取50.00 mL试液于100mL容量瓶中[当三氧化二铁含量大于0.05%时,取25.00m工试液,补加1.5mI硝酸(3.3)加水至约50mL。并随同做对应的空白试验)。 6.4.4 向容量瓶中加人5.0mL盐酸经胺溶液(3.4),混匀。加人5.0mL邻二氮杂菲溶液(3.5)和25.0mL缓冲溶液((3.6),用水稀释至刻度,棍匀,放置10min。 6.4.5 将部分溶液移人3cm吸收池中,以水为参比,于分光光度计波长510nm处,测量其吸光度,将测得的吸光度减去随同试料空白试验溶液的吸光度后,从工作曲线上查出相应的三氧化二铁量。 6.5 工作曲线的绘制 于一组100mL容量瓶中,分别加人 0, 1.00, 2.00 ,3.00, 4.00, 5.00ml三氧化二铁标准溶液(3.9),加人3.0mI.硝酸((3.3),用水稀释至体积约50mL,混匀,以下按6.4.4进行。将部分系列标准溶液移人3cm吸收池中,以水为参比,于分光光度计波长510nm处,测量系列标准溶液的吸光度。以所测吸光度减去试剂空白溶液的吸光度,以三氧化二铁量为横坐标,吸光度为纵坐标,绘制工作曲线。 7 分析结果的计算 按下式计算三氧化二铁含量w(Fe2O3)(%): 式中:m1-----自工作曲线上查得的三氧化二铁量,单位为毫克(mg) m0-----试样的质量,单位为克(g) V1------分取试液的体积,单位为毫升(mL) V0------试液的总体积,单位为毫升(mL), 8.1 重复性 在重复性条件下获得的两次独立测试结果的测定值,在以下给出的平均值范围内,这两个测试结果的绝对差值不超过重复性限((r),超过重复性限(r)的情况不超过5%,重复性限((r)按以下数据采用线性内插法求得: w(Fe2O3) (%) 0.0087 0.0222 0.046 8 重复性限r (%) 0.0005 0.0003 0.001 3 8.2 允许差 实验室之间分析结果的差值应不大于表1所列允许差。 w(Fe2O3) 允 许 差 0.005--0.015 0.004 0.015--0.030 0.005 0.030--0.050 0.006 0.060--0.100 0.010 9 质量保证与控制 应用标准样品或控制样品,每月至少对本标准的有效性校核一次。当失效时应找出原因。纠正后重新进行校核。
3-甲基戊烯二酸经BSTFA衍生可生成3-甲基戊烯二酸(1), 3-甲基戊烯二酸(2), 3-甲基戊烯二酸(3), 3-甲基戊烯二酸(4)4种产物,其中前3种分子量为288,另一种为360。只知一种分子量为288的结构式的可能,那另两种分子量288可能是什么结构,还是空间结构的不同?分子量360又是何结构?求高手帮忙!请见附件。







 我要推广仪器
我要推广仪器
 下载APP
下载APP