进口兽药质量标准硫酸头孢喹肟注射液Liusuan Toubaokuiwo ZhusheyeCefquinome sulfate Injection本品为硫酸头孢喹肟与油酸乙酯等配制而成的混悬注射液。含头孢喹肟(C23H24N6O5S2)应为标示量的90.0%~105.0%。【性状】 本品为类白色至浅褐色混悬液体;久置分层。【鉴别】(1)含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品峰的保留时间一致。(2)取摇匀后的供试品2 ml,加水5 ml,稀盐酸1 ml,混匀,置超声浴中超声10分钟,弃去有机层,溶液显硫酸盐的鉴别反应(附录15页)。【检查】有关物质 照含量测定项下的方法。取摇匀后的供试品1.0 ml,加入流动相25.0 ml,置超声浴中超声5分钟,弃去有机层,取水层滤过,取续滤液10µ l,注入液相色谱仪,记录色谱图,2,3-环己基吡啶与头孢喹肟相对保留时间为0.20。按峰面积归一化法计算,2,3-环己基吡啶应不得过3.0%,其他单一杂质应不得过0.50%,杂质总量应不得过4.0%。水分 取本品,照水分测定法(附录58页,第一法)检查,含水分不得过0.2%。细菌内毒素 取摇匀后的供试品2 ml与细菌内毒素检查用水3 ml混匀,分成2等份,振摇30秒,离心15分钟(2000g),吸取水层1 ml,加1 mol/L氢氧化钠溶液0.06 ml调节pH值至6.5~7.5。用细菌内毒素检查用水按1:10稀释后,照细菌内毒素检查法(附录73页)检查,每1 mg头孢喹肟中含细菌内毒素的量应小于0.1 EU。无菌 取供试品8瓶,混合均匀,加入含6%吐温-80的蛋白胨缓冲液(1g/L)400ml,混匀,加入800×106单位青霉素酶(每1ml供试品溶液,加2×106单位青霉素酶),充分振摇,将供试品倒置,在37℃放置4小时;取供试品溶液,依法检查(附录79页,直接接种法),应符合规定。分散性 取本品1瓶,振摇30秒,将供试品转移置玻璃容器中,不得观察到结块或沉淀物。沉降 取本品1瓶,振摇30秒,取供试品10 ml置刻度试管中(内径1.0~1.5 cm),10分钟内不得沉淀。粒度 取摇匀后的供试品,置显微镜下检查,颗粒直径在5µ m以下应不得少于80%,10µ m以下不得少于90%,20µ m以下不得少于95%,50µ m以下不得少于100%。装量 按最低装量检查法(附录67页)检查,应符合规定。【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;取一水合高氯酸钠3.45g溶于1000 ml水中,加磷酸12 ml和乙腈90 ml,用三乙胺调节pH至3.6为流动相;检测波长为270 nm。取头孢噻肟约25 mg,溶于100.0 ml流动相中,另取头孢喹肟约25 mg,置25 ml量瓶中,精密加入上述头孢噻肟溶液1 ml,用流动相稀释至刻度。精密量取10µ l注入液相色谱仪,记录色谱图;计算头孢喹肟与头孢噻肟的分离度,应大于1.0。
有谁做过头孢噻肟钠 俄罗斯标准的,溶解度和化学鉴别中的试药是什么?
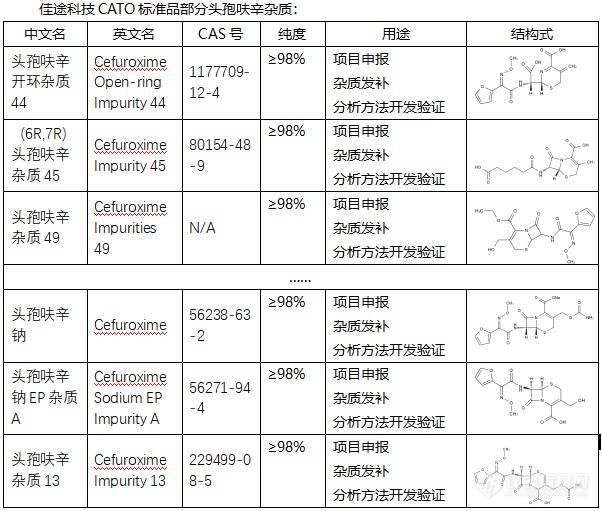
头孢呋辛是一种广泛使用的抗生素,主要用于治疗由敏感细菌引起的各种感染。在生产、储存和使用头孢呋辛的过程中,可能会产生一些杂质。这些杂质的存在可能会影响头孢呋辛的纯度和疗效,因此了解和控制这些杂质对于确保药物的安全性和有效性至关重要。头孢呋辛的杂质有多种,其中一些具有特定的CAS号、化学式和分子量。例如,头孢呋辛杂质33(Cefuroxime Impurity 33)的CAS号为929531-94-2,分子式为C16H16N4O9S,分子量为440.38。此外,还有其他一些头孢呋辛杂质,如头孢呋辛杂质A、B、C、D、E、F、G、H等。 CATO标准品提供的头孢呋辛全套的杂质,这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分。[img=,602,511]https://ng1.17img.cn/bbsfiles/images/2024/02/202402192104451830_7644_6381607_3.png!w602x511.jpg[/img] 广州佳途科技股份有限公司深知药物研发与质量控制的重要性,CATO标准品厂家,提供头孢呋辛全套的杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展,以满足客户在药物研发和质量控制方面的需求。[list][*]原创检测区[/list]◇头孢呋辛杂质头孢呋辛是一种广泛使用的抗生素,主要用于治疗由敏感细菌引起的各种感染。在生产、储存和使用头孢呋辛的过程中,可能会产生一些杂质。这些杂质的存在可能会影响头孢呋辛的纯度和疗效,因此了解和控制这些杂质对于确保药物的安全性和有效性至关重要。头孢呋辛的杂质有多种,其中一些具有特定的CAS号、化学式和分子量。例如,头孢呋辛杂质33(Cefuroxime Impurity 33)的CAS号为929531-94-2,分子式为C16H16N4O9S,分子量为440.38。此外,还有其他一些头孢呋辛杂质,如头孢呋辛杂质A、B、C、D、E、F、G、H等。CATO标准品提供的头孢呋辛全套的杂质,这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分。广州佳途科技股份有限公司深知药物研发与质量控制的重要性,CATO标准品厂家,提供头孢呋辛全套的杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展,以满足客户在药物研发和质量控制方面的需求。
哪里有头孢噻呋标准品?
求购头孢地尼E-异构体对照品头孢地尼做分析方法验证,其中有关物质头孢地尼E-异构体标准品买不到。很多销售公司都有但是资质不全我不能买他们的。请教各位大侠们,帮个忙吧。急用!!谢谢!!!!
美国药典标准测定头孢地尼含量色谱条件溶液A:14.2mg/ml无水磷酸氢二钠溶液B:13.6mg/ml磷酸二氢钾缓冲溶液:将溶液A与溶液B按2:1混合(pH7.0)溶液C:0.1%四甲基氢氧化铵水溶液,用1/10稀磷酸调节pH至5.5溶液D:37.2mg/mlEDTA二钠流动相:乙腈、甲醇、溶液C、溶液D (350:200:4500:2)系统适应性溶液:用缓冲溶液配制每0.2mg/ml头孢地尼RS和0.5mg/ml头孢地尼有关物质A RS标准溶液:用缓冲溶液配置0.2mg/ml头孢地尼RS标准品样品溶液:用缓冲溶液配制0.2mg/ml头孢地尼样品色谱柱:C18柱(5um,4.6*150mm)推荐:YMC-Pack ODS-AM(P/N:AM12S05-1546WT)检测波长:254nm柱温:40摄氏度流速:1.0ml/min进样量:5ul系统适应性样品:系统适应性溶液和标准溶液(注:头孢地尼有关物质A RS有4个色谱峰)拖尾因子:以头孢地尼峰计不得超过1.5(系统适应性溶液)分离度:头孢地尼有关物质A RS的第二个峰与头孢地尼峰的分离度不得小于1.2
新人有难,还请各位大仙帮助。我需头孢妥仑匹酯片的进口标准,有货源的给新人说声,感谢!我的邮箱ahwuyan@live.cn。
对照品:用于鉴别、检查、含量测定和校正检定仪器性能的标准物质;对照品由国家药品检定机构审查认可,其标准应不低于制品的质量标准。 标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。 对照品与标准品概念不清?对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质。标准品:用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示。文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已,造成错误的原因,可能是有的药品既有对照品,又有标准品。 例如:当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品。即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的。
药品质量标准中鉴别项目设置的几点考虑 审评三部 张哲峰 摘要:本文简要介绍了药品质量标准中常用的几种鉴别方法,并对常用鉴别方法的优势和局限进行了分析,针对鉴别项目设置中需注意之处提出了一些看法。 关键词:质量标准 鉴别项目 药品质量标准中鉴别是用以判定某已知药品的真伪而不是对未知药物进行结构确证,所以鉴别方法应以专属性好、简便易行为宜,尤其能将结构相似的同类药品加以区别为主要考虑因素。如新鱼腥草素钠及制剂标准中仅用化学法和UV法作鉴别,难以与结构类似物鱼腥草素钠及制剂相区分,质量标准不具备应有的专属性,可能给此后的市场监督造成混乱。 常用的鉴别方法包括色谱法、光谱法、化学法和生物学方法等,可根据药品具体特点加以选用。 色谱法(TLC法或HPLC法)利用不同物质在不同色谱条件下,各自色谱行为(比移值或保留时间)的不同,与对照品在相同色谱条件下进行色谱分离,比较其色谱行为的一致性,来鉴别药品的真伪。这类方法的运用使得结构相似化合物、同系物等的区分变得简单易行。HPLC法虽然主要用于定量,但如果运用得当,尤其在含量测定或有关物质项下已采用本法的情况下,利用对照品与供试品保留时间相同的特性作为鉴别依据,不必专门增加实验以提高鉴别的专属性,是非常可取的。值得注意的是色谱系统的稳定性要好,同一物质不同进样时保留时间的重现性必须有保证。这就要求流动相与固定相相匹配,C18链在水相环境中不易保持伸展状态,故在C18柱的反相色谱系统中,流动相有机溶剂比例通常不应低于5%,否则C18链的随机卷曲将造成色谱系统不稳定导致组份保留值波动,不利于此种鉴别。即便如此,在实际操作中有时依然能遇到同一物质在完全相同的色谱系统中保留时间不一致的情况,尤其梯度洗脱时此种现象更为常见。药典中对保留时间的一致性未予具体规定,此时,操作中可增加供试品溶液与对照品溶液等量混合,进样后出现单一色谱峰作为鉴别依据,可以弥补该法之不足,此操作可列入质量标准。在含量和有关物质未采用HPLC法的情况下,一般不单独采用本法作鉴别。 TLC法除色谱行为外,还可将斑点颜色作为鉴别依据,可由两个因素把握供试品与对照品的同一性,而且简便易行,堪称一个很好的鉴别方法。但由于薄层板质量、边缘效应等因素的影响,实际操作中有时也会遇到同一物质在同一块薄层板上的Rf值不一的情况,可比照HPLC的情况,操作中增加供试品溶液与对照品溶液等量混合,点样后出现单一斑点作为鉴别依据,此点在2005年版药典中已有体现。也有人提议明确Rf值偏差不超过5%,作为鉴别要求,但其可行性有待考察。单独使用TLC鉴别时,要有色谱系统适应性试验内容,如要求几种结构相似化合物的混合溶液色谱展开后应显示相应的几个斑点或最难分离物质对能够分开的情况下,供试品溶液与对照品溶液主斑点的颜色与位置应一致。 在中国药典2005年版中,对TLC鉴别法在斑点的颜色与位置明确规定的基础上对斑点大小也做出明确要求:供试品与同浓度对照品溶液颜色与位置应一致,斑点大小应大致相同;或供试品与对照品等体积混合,应显示单一,斑点紧密;或供试品溶液的主斑点与上述混合溶液的主斑点的颜色与位置一致,大小相似;或选用与供试品化学结构相似药物对照品,两者的比移植应不同(例如芬布芬与酮洛芬,地塞米松磷酸钠与泼尼松龙磷酸钠,醋酸氢化可的松与醋酸可的松,泼尼松龙与氢化可的松,甲睾酮与睾酮,左旋多巴与酪氨酸);或上述两种溶液等体积混合,应显示两个清晰分离的斑点。 光谱法中IR法因可反映较多的结构信息,在组份单一、结构明确的原料药鉴别中作为首选, 药物存在多晶型现象又无可重复转晶方法时一般不采用此法,但如果药物存在多晶型现象,且需鉴别其有效晶型,IR图谱可以反映其有效晶型特点时,本法又是一种有效而简便易行的鉴别方法。制剂中则因辅料影响、提取过程可能导致晶型变化而一般不采用IR法,而采用所受影响因素较少的UV法。 常用的UV鉴别方法有:测定最大吸收波长,或同时测定最小吸收波长;规定一定浓度的供试液在特定吸收波长(最大吸收或最小吸收)处的吸收度;经化学处理后,测定其反应产物的吸收光谱特征;规定几个特定吸收波长及其吸收度比值(峰-峰、峰-谷、谷-谷);规定几个特定吸收波长及其吸收系数。因末端吸收所受影响因素较多,UV法鉴别时,一般不宜用220nm以下波长的吸收特性作鉴别;因反映的结构信息少,一般也尽量不用单一吸收峰作鉴别依据;为提高专属性,可将上述几个方法结合起来使用。 化学鉴别法一般是特定官能团或特定结构化合物的特性反应,与其它鉴别方法结合使用,可以使得鉴别的专属性更加突出。化学鉴别法具有专属性较强、反应迅速、现象明显的特点才有使用价值。包括在适当条件下产生颜色、荧光,发生沉淀反应或产生气体等现象。 1.呈色反应:即向供试品溶液中加入适当试剂,在一定条件下发生化学反应,生成易于观测的有色产物。常见的反应类型有:[/c
头孢泊肟酯的有关物质的分析,请问各路高人做过没有,色谱条件是什么,提前感谢了
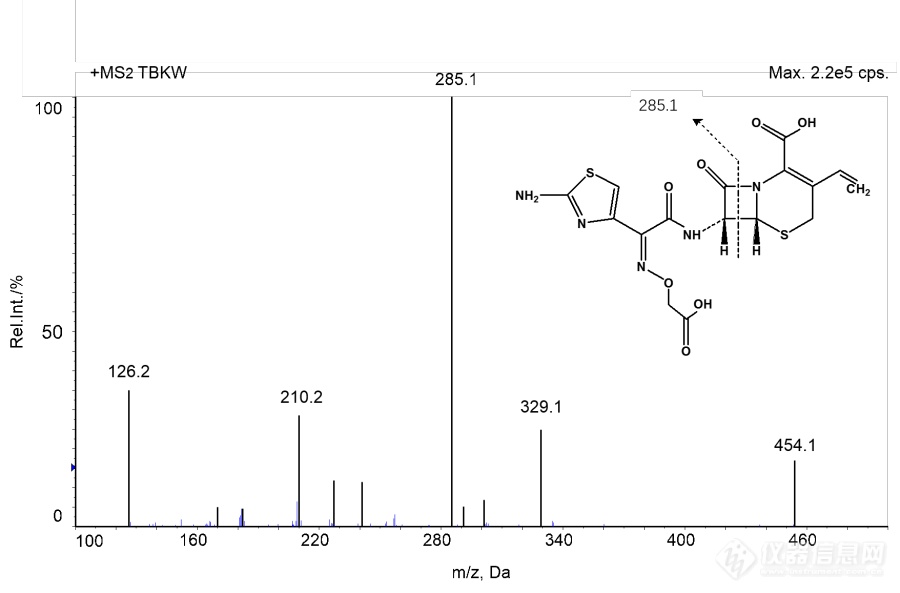
[align=left][b][size=32px][color=#ff0000]本篇文章暂不授权任何公众号发布[/color][/size][/b][/align][align=center][b]高脂肪饮食对头孢克肟片的药动学影响 [/b][/align][align=left][b]摘要:[/b]目的:研究比较空腹和高脂餐后单剂量口服用头孢克肟的药代动力学和生物利用度。方法:采用双交叉给药实验设计,12名健康男性受试者空腹及餐后单剂量口服1mg头孢克肟片,以头孢他美为内标,HP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS法测定血药浓度,DAS 2.0软件处理药动学参数。结果:头孢克肟空腹和餐后单剂量给药的主要药动学参数为:Cmax分别为([color=black]3.805 ±0.710[/color])μg/ml和([color=black]1.604 ±0.483[/color]) μg/ml,Tmax 分别为([color=black]4.792±0.582[/color]) h和([color=black]4.000±1.225[/color]) h,AUC[sub]0-t [/sub]分别为([color=black]32.923±7.804[/color]) μghmL[sup]-1[/sup]和([color=black]12.785±4.688[/color]) μghmL[sup]-1[/sup],AUC[sub]0-[/sub][sub][color=#231f20]∞[/color][/sub]([color=black]33.955±8.484[/color]) μghmL[sup]-1[/sup]和([color=black]13.082±4.932[/color]) μghmL[sup]-1[/sup]。两种给药方案的Cmax和AUC取自然对数后经方差分析,Tmax经非参数检验,发现Cmax,AUC和Tmax的差异有统计学意义(P<0. 05)。结论;与空腹组给药相比,餐后组吸收速率减慢,消除半衰期延长,生物利用度降低。[/align][align=left][b]关键词:[/b]头孢克肟;药动学;空腹餐后;生物利用度;[/align][align=left]头孢克肟(Cefixime)是一种重要的头孢菌素类抗生素,属于可口服的第三代头孢菌素类抗生素,临床上应用于敏感菌引起的肺炎、支气管炎、泌尿道感染、淋病、胆囊炎、胆管炎、猩红热、中耳炎、副鼻窦炎等。本研究参考有关人血浆中头孢克肟的定量方法,志愿者在空腹和餐后两种情况下服用进口头孢克肟片后取血,采用灵敏的[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析技术评价饮食对头孢克肟在人体内的药动学影响,为临床用药提供指导。[/align][align=left][b]材料与方法[/b][/align][align=left][b]1 仪器与试药[/b][/align][align=left]API4000型三重四级杆串联离子肼质谱仪,美国Applied Biosystem Sciex公司;Aglient 1260型液相色谱系统,包括G1312B二元泵,G1322A在线脱气机,G1316A柱温箱,Aglient Technology公司; NASCA F5100型自动进样器,日本SHISEIDO公司;资生堂CAPCELL PAC ADME柱(2.1*100mm,3mm,日本SHISEIDO公司);保护柱:Phenomenex C18(4*3.0mm,5mm,Torrance,CA,USA);梅特勒-托利多AG135电子天平,梅特勒-托利多仪器上海有限公司;Heraeus Multifuge XIR型离心机,美国Thermo Fisher公司;IKA VIBRAX VXR型振荡器;KQ5200DE型数控超声清洗机,昆山市超声仪器有限公司;Eppendorf可调式及自动[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]电动[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url],德国Eppendorf公司。[/align][align=left]头孢克肟标准品(批号:130503-201716,纯度:89.2%,中国食品药品检定研究院);头孢他美(批号:130564-201601,纯度>99.8%,中国食品药品检定研究院);甲醇:美国Fisher公司,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url] 级,批号:LOT180821;甲酸:美国MREDA公司,色谱纯,批号:LOT095224;乙腈:美国Fisher公司,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url] 级,批号:LOT180821;超纯水:屈臣氏重蒸水,批号:20180815.[/align][align=left][b]2 试验方法[/b][/align][align=left][b]2.1 试验方案[/b][/align][align=left][b]2.1.1 受试对象[/b][/align][align=left]12名健康男性受试者,年龄在18-45岁,体重指数在19-24范围内,健康,无心血管、肝脏、肾脏、消化道、精神神经等疾病史,无药物过敏史。试验前详细询问既往病史,作全面的体格检查及实验室检查,心电图、血压、肝肾功能及血尿常规检查均正常,试验前两周未用任何药物。所有受试者均签署知情同意书,试验方案经本院伦理委员会审批同意。[/align][align=left][b]2.1.2 给药方案与样品采集[/b][/align][align=left] 试验采用双交叉设计。12名受试者随机分成两组,每组6人。1组为空腹组,另1组为餐后组。受试者在试验前1 d的17:00进人I期临床试验病房,晚上统一清淡饮食,并禁食10h,但不禁水。次日晨1组空腹口服药物,用250mL水送服。另1组则统一进食高脂标准餐,进餐时程为30 min,后即用250 ml温开水送服试验药物。0, 0.5,1,1. 5,2,2.5,3,3.5,4,4.5,5,6.5,7,8,10,12,15和24小时,抽取静脉血4 ml,置肝素化抗凝试管中,分离血浆于-80℃贮存,待测。[/align][align=left][b]2.2 色谱及质谱条件[/b][/align][align=left]色谱条件:流动相:乙腈(0.5%甲酸):水(0.5%甲酸)=40:60等度洗脱;柱温:40℃;流速:0.2 mL/min;进样量:5 mL;运行时间:4min。洗针程序:50%甲醇洗针5s,超声洗针5s(纯水),再次用50%甲醇洗针5s。[/align][align=left]质谱条件:离子源:电喷雾(ESI);扫描方式:多反应监测(MRM);离子化方式:正离子;检测离子对:TBKW m/z454.2/285.1;内标:389.2/241.1.离子源电压:4800V;离子源温度:400℃;气帘气:15psi;碰撞气:4psi 雾化气:60psi;辅助气:55psi;解簇电压:70V;碰撞诱导解离电压:TBKW:28V,TBTM :21V。[/align][align=left][b]2.3 溶液配制及样品处理[/b][/align][align=left][b]2.3.1溶液配制[/b][/align][align=left]对照品溶液:精密量取头孢克肟对照品11.21 mg,置于15 mL EP管中,用自动[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url][/color][/url]精密加入10 mL甲醇溶解,得到1 mg/mL的对照品储备液,置于-20℃备用。用50%甲醇-水配制成系列头孢克肟标准工作液,浓度分别0.5,1,5,10,20,50,100,160 mg/mL。4℃冰箱避光保存。[/align][align=left]质控工作液:精密量取头孢克肟对照品11.15 mg,置于15 mL EP管中,用自动[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url][/color][/url]精密加入10 mL甲醇溶解,得到1 mg/mL的对照品储备液,置于-20℃备用。用50%甲醇-水配制成系列头孢克肟标准工作液,浓度分别1,5, 20,130mg/mL。4℃冰箱避光保存。[/align][align=left]内标工作液:精密量取头孢他美对照品10.02 mg,置于15 mL EP管中,用自动[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液枪[/color][/url][/color][/url]精密加入10 mL甲醇溶解,得到1 mg/mL的内标储备液,置于-20℃备用。用纯甲醇稀释至500 ng/mL,4℃冰箱避光保存。[/align][align=left][b]2.3.2生物样本处理方法[/b][/align][align=left]a.血浆标准品及质控样品处理方法:取药前血浆190 uL,加10 uL适当浓度的标准溶液,混匀,加400 uL甲醇(含内标500 ng/mL),涡旋2min,离心(4℃,14000r/min)15min, 取100 uL上清液,加流动相A相200uL混匀,取5μL进样。[/align][align=left]血浆标准曲线范围: 0.025,0.05,0.25,0.5,1,2.5,5,8 u g/ml。[/align][align=left]质控样品浓度为: 0.075,1,6.5μg/ml 最低定量限: 0.075 ng/mL。[/align][align=left]b.血浆样品处理方法:取给药血浆200 uL,加400 uL甲醇(含内标500ng/mL),涡旋2min,离心(4℃, 14000 r/min) 15min, 取100 μL上清液,加流动相A相200μL混匀,取5μL进样。[/align][align=left][b]2.3.3数据统计处理方法[/b][/align][align=left]使用AB Secix公司Analyst 1.5.2软件采集处理数据。[/align][align=left][b]3方法学确证[/b][/align][align=left]对建立的方法进行方法验证, 参照中国药典2015版《生物样品定量分析方法指导原则》(草案),从方法的选择性、标准曲线和定量下限、精密度和准确度、稳定性、回收率、基质效应、残留效应和同位素效应等各方面进行方法验证。[/align][align=left][b]3.1选择性 [/b]分别取6份不同来源的人空白血浆样品以及相应人空白血浆配制的LLOQ 样品进行[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS分析测定,考察空白血浆中的内源性物质是否干扰待测物及其内标的测定。接受标准为:空白血浆中待测物保留时间处色谱峰面积不高于LLOQ色谱峰面积的20%;空白血浆中内标保留时间处色谱峰面积不高于内标色谱峰面积的5%。[/align][align=left][b]3.2 标准曲线[/b] 标准曲线设计8个浓度点, 头孢克肟血浆浓度分别为0.025, 0.05,0.25,0.5,1,2.5,5,8 u gmL-1, 按“血浆标准品及质控样品处理方法”项下操作, 以每个待测物浓度为横坐标, 待测物与内标物的峰面积比值为纵坐标, 用加权 ([i]W [/i]= 1/[i]x[/i]2) 最小二乘法进行回归运算, 求得的直线回归方程即为标准曲线。接受标准为:校正标样回算的浓度一般应该在标示值的±15%以内,定量下限处应该在±20%内;至少75%校正标样,含最少6个有效浓度,应满足上述标准标准;曲线的相关系数的值r≥0.990。[/align][align=left][b]3.3 残留考察 [/b]在标准曲线最高浓度点后连续进样2个空白样品, 考察样品与内标的残留效应。接受标准为:空白血浆中待测物保留时间处色谱峰面积不高于LLOQ色谱峰面积的20%;空白血浆中内标保留时间处色谱峰面积不高于内标色谱峰面积的5%。[/align][align=left][b]3.4 定量下限 [/b]根据血浆标准品处理方法,配制头孢克肟定量下限浓度的血浆样品6份(2.50ng/mL) ,考察其准确度和精密度。[/align][align=left][b]3.5 精密度与准确度[/b] 按“血浆标准品及质控样品处理方法”项下操作,配制最低定量限、低、中、高4个浓度的血浆样品,每浓度进行6个样本分析,分别在3日内测试,根据当日标准曲线计算质控样品的测得浓度,根据结果计算本法的日内、日间精密度与准确度。接受标准:批内精密度:低、中、高浓度批内变异系数≤15%,定量下限的变异系数≤20%。批间精密度:低、中、高浓度批间变异系数≤15%,定量下限的变异系数≤20%。准确度:所有质控样品准确度均值一般应在标示值的±15%之内,定量下限准确度应在标示值的±20%之内。[/align][align=left][b]3.6 回收率和基质效应 [/b]取6份不同来源的空白人血浆,加入人脂肪,制备不同来源的含20%的高脂肪血浆,另取同样6份不同来源的空白人血浆,加入人全血,制备不同来源的含4%的溶血血浆,按比例加入内标溶液和沉淀蛋白溶剂,混匀,离心,取上清,将基质提取出来,分别加入低,中,高浓度质控溶液,经流动相稀释进样分析,测定血浆,高脂血浆,溶血血浆三种处理后的基质下的低、中、高浓度质控样品TBKW及其内标的峰面积。 [/align][align=left]取6份不同来源的空白人血浆及6份水,按“血浆标准品及质控样品处理方法”项下操作,测定空白血浆,标准溶液状态下的低、中、高浓度质控样品TBKW及其内标的峰面积。 [/align][align=left]基质因子(效应)=处理后的空白血浆归一化后的峰面积/标准溶液归一化的峰面积*100% [/align][align=left]提取回收率=未处理空白血浆归一化后的峰面积/处理后的空白血浆归一化后的峰面积*100%。[/align][align=left]接受标准:提取回收率不一定需要满足接近100%,但是必需是稳定而可重现的。基质因子不一定需要接近100%,但是在不同个体的基质中应是稳定而可重现的。不同来源的内标归一化基质因子精密度应≤15%。[/align][align=left][b]3.7 溶液稳定性 [/b]按“血浆标准品及质控样品处理方法”项下操作,新鲜配制头孢克肟储备液并用50%甲醇稀释到低,高质控溶度,每个浓度各3个;取-20℃存放7天,30天的储备液稀释到高质控浓度3个以及4℃对应存放7天,30天储存的低,高浓度质控工作液各3个,进样分析,分别评价储备液和工作液溶液7天,30天的稳定性。接受标准:各储备条件下和对照(新鲜配制)的溶液的归一化峰面积的比值偏差在±15%之间。[/align][align=left][b]3.8 血浆样品稳定性 [/b]室温放置:取多份空白血浆适量,分别加入相应浓度的TBKW标准溶液,配制低、中、高三个浓度血浆样品室温放置5h、处理后的样本室温放置6h、冰箱4°C放置20h,按“血浆标准品及质控样品处理方法”项下操作,进样分析,考察血浆质控样品室温放置稳定性。另取精密度试验第二批三个浓度血浆样品,测定后于自动进样器(10°C)中放置16h后再进样分析,考察血浆质控样品于自动进样器(10℃)中放置16h后稳定性。[/align][align=left]冻融:取多份空白血浆适量,分别加入定量的TBKW标准溶液,配制低、中、高三个浓度血浆样品放置-30°C至少12h,分别经1次冻融、2次冻融、3次冻融冷冻解冻循环,每次溶解时流动自来水溶解不超过15min,放置室温15 min后冷冻,按“血浆标准品及质控样品处理方法”项下操作,处理后进样分析,观察各次循环后稳定性。[/align][align=left] 长期冷冻:取多份空白血浆适量,分别加入定量的TBKW标准溶液,配制低、中、高三个浓度血浆样品,于-30°C放置3天、8天和27天,按“血浆标准品及质控样品处理方法”项下操作,处理后进样分析,观察-30°C条件下冷冻保存稳定性。接受标准:各储备条件下每一浓度水平测定值的精密度≤15%,准确度应在±15%之内。[/align][align=left][b]3.9 批分析 [/b]将精密度实验组样本中最低定量限、低、中和高四个浓度的样品进行了8轮进样,在实验样品开始前证实生物分析方法的效能。[/align][align=left][b]4实验结果[/b][/align][align=left][b]4.1 质谱结果 [/b]由于 TBKW 的结构中既含有碱性氮原子, 也含有羧基, 所以本实验比较了正、负两种离子检测。结果发现, TBKW 的甲醇-水 (50∶50) 溶液 (在正离子模式下响应约为负离子的10倍, 故优先选择ESI 源下正离子检测方式。在正离子检测模式下,TBKW 及内标 TBTM 分别主要生成 m/z454.2 和m/z389.2 的 [sup]+[/sup]峰, 选择性对 [sup]+[/sup]峰进行产物离子扫描分析, TBKW 生成的主要碎片离子分别为[i]m/z [/i]285.1 ,[i]m/z[/i]126.1和[i]m/z[/i]210.1,TBTM生成的主要碎片离子有[i]m/z [/i]241.1。但实验过程中发现,[i] m/z[/i]126.1和[i]m/z[/i]210.1 的碎片离子响应弱, 且噪音高, 故最终选择[i]m/z [/i]285.1 和[i]m/z [/i]241.1 分别作为TBKW 及内标 TBTM 定量分析时的产物离子。待测物及内标的[sup]+[/sup]产物离子全扫描质谱图及相应出峰时间见图1-4。[/align][align=center] [img=,562,319]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241116590213_28_3255306_3.png!w562x319.jpg[/img] [/align][align=center]图1 TBKW二级特征碎片质谱图[/align][align=center][img=,555,315]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241117114646_9141_3255306_3.png!w555x315.jpg[/img] [/align][align=center]图2 TBTM二级特征碎片质谱图[/align][align=center][img=,559,296]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241117239993_3223_3255306_3.png!w559x296.jpg[/img] [/align][align=center]图3 TBKW色谱质谱图[/align][align=center][img=,567,326]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241117339148_8729_3255306_3.png!w567x326.jpg[/img] [/align][align=center]图4 TBTM色谱质谱图[/align][align=left][b]4.2 方法学验证结果[/b][/align][align=left][b]4.2.1 选择性 [/b]结果表明, 空白人血浆中的内源性物质不干扰头孢克肟和内标头孢他美的测定, 且同位素内标头孢他美不干扰头孢克肟的测定。[/align][align=left][b]4.2.2 标准曲线[/b] 用加权 ([i]W [/i]= 1/[i]x[/i][sup]2[/sup])最小二乘法进行回归运算, 求得的标准曲线的相关系数 ([i]r[/i][sup]2[/sup]) 均大于0.99。根据标准曲线,头孢克肟的线性范围为0.025~8 ug/ml。典型标准曲线如下所示: [i]y [/i]= 0.828 [i]x [/i]+4.35e[sup]-3[/sup]([i]r[/i][sup]2[/sup]= 0.9994)。[/align][align=left][b]4.2.3 残留考察 [/b]结果显示,样品与内标保留时间处均未出现干扰杂质峰, 在本实验选择的色谱和质谱条件下, 待测物及内标无残留。[/align][align=left][b]4.2.4 定量下限[/b] 其准确度和精密度见“精密度与准确度”项下。[/align][align=left][b]4.2.5 精密度与准确度[/b] 头孢克肟每一浓度水平的QC样品的日内、日间平均准确度均在15%之内,相对标准偏差均<15%,日内、日间精密度与准确度结果符合生物样本测定要求。相关数据见表1。[/align][align=center]Table 1 Precision and accuracy of TBKW (n=18).[/align][align=center] [img=,610,133]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241121176922_9541_3255306_3.png!w610x133.jpg[/img][/align][align=left][b]4.2.6 回收率和基质效应 [/b]待测物TBKW基质效应的平均值为27.7-42.9% 内标TBTM基质效应的平均值为2.1-52.1%,经内标归一化计算后得出TBKW内标归一化基质效应平均为59.1-62.8%,RSD符合要求。待测物TBKW高脂血浆基质效应的平均值为25.3-28.4% 内标TBTM的高脂血浆基质效应平均为41.9-47.6%,经内标归一化计算后得出TBKW内标归-化高血脂基质效应平均值为59.0-60.4%,RSD均符合要求。待测物TBKW溶血血浆基质效应的平均值为22.0-25.5%,内标TBTM的高血脂基质效应平均为38.6-43.9%,经内标归一化计算后得出TBKW内标归一化高血脂基质效应平均值为55.9-62.6%,RSD均符合要求。表明在试验选择的样品处理、色谱与质谱条件下,待测物和内标均表现出较强的基质效应,均表现出较强的离子抑制作用,特别是TBKW,但该机制效应对待测物和内标作用方向致、稳定,经内标校正后,虽然还存在基质效应,但对分析结果不会产生明显影响。TBKW及其内标TBTM的提取回收率结果,分别见表7-3。待测物TBKW在低、中、高三个QC浓度水平上提取回收率的平均值在138.2%-148.2%之间,内标的提取回收率的平均值在124.0%-130.4%之间,内标校正后提取回收率的平均值在110.4-114.1%之间,RSD符合要求。待测物和内标物在不同浓度水平的提取回收率结果是精密和可重现的,相关数据见表2。[/align][align=center]Table 2 Extraction recoveryand matrix effect of TBKW (n = 6).[img=,631,234]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241121399474_1836_3255306_3.png!w631x234.jpg[/img][/align][align=left][b]4.2.7 溶液稳定性[/b] 用甲醇配制的TBKW储备液1mg/mL-20℃冻存7、22天后的含量均值为新鲜配制的TBKW的106.5%和106.0%,未见降解;TBKW工作液储备液75、6500ng/mL 4℃冷藏7、30天后的含量均值分别为新鲜配制的TBKW的94.5%,113.4% 和89.8%,109.2%,不存在明显降解。[/align][align=left][b]4.2.8 血浆样品稳定性 [/b]TBKW血浆样品在室温放置5h、处理好的血浆样本室温放置6h、自动进样器放置16h,冻融3次、长期冷冻(27天,高于待测样本保存时间)试验条件下,各浓度质控样本的准确度均值在85-115%之间,RSD%均小于15%,表明TBKW血浆样品在上述所考察的情况下稳定。血浆样品稳定性测试结果满足人体血浆样本检测需求。[/align][align=left][b]4.2.9 批分析 [/b]对8轮试验数据进行了比较。结果表明在同一进样批中,每浓度含 48个样本,共计192个样本,其中LLOQ中有6个点准确度20%,少于总样品数的20% (12.5%),且LLOQ准确度和精密度平均值均15%,QC-L、 QC-M 和QC-H的准确度和精密度平均值均15%,总进样时间约18小时。即除血浆标准曲线外和质控点,同批次测定 180个样本能够满足生物样本分析要求.[/align][align=left][b]4.3 药时曲线[/b] 12名志愿者单剂量空腹及餐后口服300 mg头孢克肟胶囊后,平均药-时曲线见图5[/align][align=center][img=,507,279]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241122298854_2799_3255306_3.png!w507x279.jpg[/img] [/align][align=center]图5 12名健康受试者在空腹和餐后口服单剂量头孢克肟后平均药-时曲线 [/align][align=left][b]4.4药代动力学参数及统计结果 [/b]见表3。[/align][align=center]Table 3 Mainpharmacokinetic parameters of cefixime capsules taken on an empty stomach andafter meals[/align][align=center] [img=,546,175]https://ng1.17img.cn/bbsfiles/images/2019/10/201910241122478527_4468_3255306_3.png!w546x175.jpg[/img][/align][align=left][b]5 讨论[/b][/align][align=left]比较空腹和高脂餐后口服头孢克肟的血药浓度的达峰时间[color=black]Tmax,[/color]达峰浓度[color=black]Cmax[/color][color=black]及曲线下面积[/color]AUC的数值,发现餐后口服头孢克肟的达峰时间推迟约0.792 h,达峰浓度约为空腹时的42.2%,餐后相对于空腹给药的相对生物利用度为39%,证明食物对头孢克肟的吸收速率、消除半衰期及吸收总量等药动学参数有显著的影响,故推荐头孢克肟片剂饭前空腹服用。[/align][align=left]饮食因素对口服药物的影响常为病人所忽视,为了避免药物对胃肠道的刺激常喜饭后服药,但餐后服药易受食物等因素的影响。本文结果提示:头孢克肟片不宜餐后服药,较合理的给药方案应是在空腹或饭前2h服用。[/align][align=left]食物降低头孢克肟的吸收速率及吸收程度的原因可能有以下几点: ①食物的存在降低了胃排空速率,使药物在胃中停留时间延长,使胃中的药物浓度降低,药物的吸收减慢,从而导致达峰时间延长 ②头孢克肟为弱酸性药物,主要在小肠吸收,食物会使肠液的 pH 减小,降低了头孢克肟的溶解性和溶出度,从而导致头孢克肟的吸收量降低。这些研究结果为头孢克肟片在临床安全合理用药提供重要的参考依据。[/align][align=left][b]参考文献[/b][/align][align=left]张羽长,李明铭,隋欣蕙,赵春杰.头孢克肟胶囊人体药动学研究及生物等效性评价.药学服务与研究,2008,8(06):429-431.[/align][align=left] Meng, Fang, et al."Sensitive liquid chromatography-tandem mass spectrometry method for thedetermination of cefixime in human plasma: Application to a pharmacokineticstudy." Journal of Chromatography B 819.2 (2005): 277-282.[/align][align=left]KREMER JM,WESTHOVENSR ,LEON M ,et al .Treatment of rheumatoid arthritis by selective inhibition ofT-cell activation with fusion protein CTLA4lg. N Engl J Med ,2003,349(20):1907 - 1915.生物样品定量分析方法指导原则(草案),中国药典,2015。[/align]
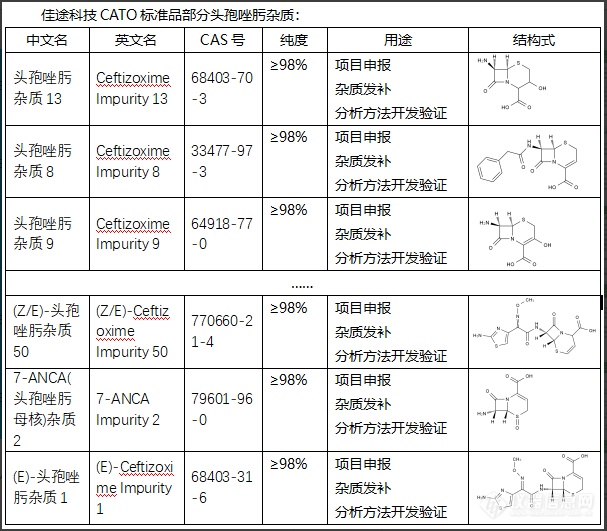
有关头孢唑肟杂质的作用,以下是要注意的一些可能性:1.负面作用:过多的杂质可能导致药物效力下降,并可能引发不良反应或副作用。例如,有些杂质可能导致过敏反应。2.毒性:某些杂质可能具有毒性。例如,某些杂质可能具有致癌性。3.影响药效:杂质可能会影响药物的生物利用度,即药物进入体内后能达到预期药效的能力。CATO标准品药品生产中的质量控制步骤非常重要,目的就是要尽可能减少杂质的存在。任何药品都必须经过严格的质量检测,确保其安全有效。[img=,607,531]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041449269442_4660_6381668_3.png!w607x531.jpg[/img]
[color=#333333]对照品与标准品概念[/color][color=#333333]对照品与标准品是2个不同的概念,中国药典凡例中已有明确的定义:对照品系指用于鉴别、检查、含量测定和校正检定仪器性能的标准物质,而标准品系指用于生物检定、抗生素或生物药品中含量或效价测定的标准物质,以效价单位(U)表示.文献中常将2种概念混淆,认为对照品就是标准品,是1种物质2种提法而已[1,2],造成错误的原因,可能是有的药品既有对照品,又有标准品.例如,当用微生物法测定头孢克罗效价时,用头孢克罗标准品,用HPLC或UV法测定时,则用对照品;非那西丁当用作熔点校准物质时,用熔点标准品,测定含量时,用对照品.即使是同一种物质的标准品和对照品,它们的规格、标定方法以及用途都可能是不同的.[/color]
求头孢西丁钠、头孢泊肟钠红外图谱
求头孢西丁钠、头孢泊肟钠红外图谱

Venusil XBP C18(L)分析头孢噻肟钠的分析报告摘要:本实验按照头孢噻肟钠2010版中国药典方法进行测定,以含量测定和有关物质检测方法共同进行色谱柱的筛选。在选定的色谱柱上,进一步考察了该色谱柱用于头孢噻肟钠系统适用性、含量、有关物质等项测试的结果,并初步考察了色谱柱的使用寿命,结果表明VenusilXBP C18(L)适用于头孢噻肟钠的分析:(1)系统适用性溶液中共检出7个杂质,分离度均符合标准要求;(2)对照品平行进样6针RSD为0.13%,保留时间17.039分钟,柱效5068,拖尾因子1.116,均符合标准要求;(3)含量测定中,头孢噻肟钠保留时间16.936分钟,柱效4779,拖尾因子1.118,符合标准要求;(4)有关物质测定中共检出6个杂质,分离度均符合标准要求;(5)用于头孢噻肟钠含量测试时,连续进样300针,保留时间、柱效和拖尾因子均无显著变化,表明VenusilXBP C18(L)对该样品和流动相有较好耐受性。关键词:头孢噻肟钠;VenusilXBP C18(L);2010版药典;液相色谱法前言头孢噻肟钠为中国药典2010版二部收录品种,本实验按照该标准进行测试,通过测试得出VenusilXBP C18(L)适用于头孢噻肟钠分析,其测试结果令人满意。实验部分试剂材料超纯水、甲醇、无水磷酸氢二钠、磷酸高效液相色谱柱:Venusil XBP C18(L);5 μm,150 Å,4.6 × 150mm样品制备系统适用性溶液制备:取头孢噻肟对照品适量,加流动相溶解并稀释制成每1 ml约含1 mg的溶液,作为系统适用性试验溶液。实验结果系统适用性测定结果表1. Venusil XBP C18(L)用于头孢噻肟钠系统适用性溶液测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410220596_01_2864683_3.png表2.Venusil XBP C18(L)用于头孢噻肟钠对照品溶液测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410251562_01_2864683_3.png含量测定结果表3. Venusil XBP C18(L)用于头孢噻肟钠含量测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410281917_01_2864683_3.png有关物质测定结果表4.Venusil XBP C18(L)用于头孢噻肟钠有关物质测定结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410305808_01_2864683_3.png色谱柱批次验证结果表5. Venusil XBP C18(L)批次验证结果http://ng1.17img.cn/bbsfiles/images/2017/10/2015082410355084_01_2864683_3.png色谱柱寿命测试结果表6.Venusil XBP C18(L)用于头孢噻肟钠寿命测试结果http://ng1.17img.cn/bbsfiles/images/2015/08/201508241038_562414_2864683_3.png

我用液质联用检测抗生素,用中检所的标准品,单标居然出现两个峰,大家帮分析下是啥问题。目标物是阿莫西林,溶剂是甲醇/水(50/50),液相流动相是乙腈/水,梯度洗脱,20%-80%,时间18min。用头孢氨苄和头孢拉定的单标进样也都出现两个峰,纯度写的是95%。两张图分别为阿莫西林单标的质谱离子流图和两个峰处的碎皮离子峰图,两个峰的碎皮离子峰图是一样的。http://ng1.17img.cn/bbsfiles/images/2012/04/201204110831_360531_2424544_3.jpghttp://ng1.17img.cn/bbsfiles/images/2012/04/201204110832_360532_2424544_3.jpg
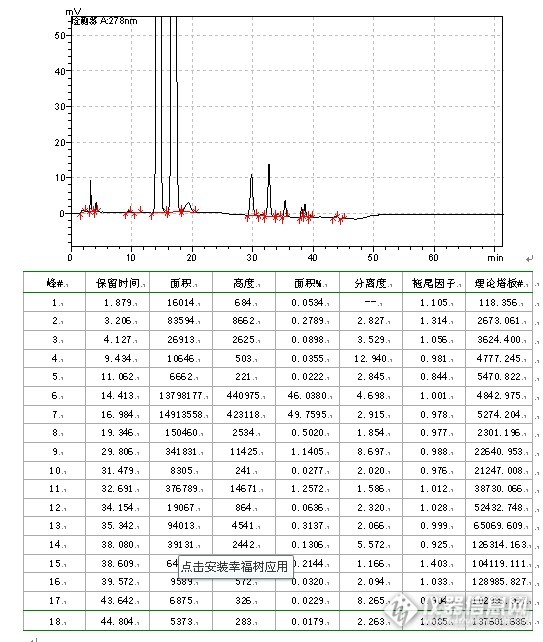
“色”路蹒跚,不拘一格,头孢呋辛酯干混悬剂有关物质流动相摸索部分。我主要工作是仿6,在遇到国家标准,我一般持研究态度,因为仿制药提倡的是仿制其质量而不是标准,一般会进行些比较。例如该品种在中国药典2010年班二部有收载,用的是等度洗脱方式,在进行研究的时候,发现梯度洗脱更具优势,所以就改变了系统方式。其具体研究方式如下:6.4 有关物质6.4.1方法的选择参照中国药典2005年版二部收载的头孢呋辛酯原料药质量标准、美国药典USP32-NF27收载的头孢呋辛酯混悬剂质量标准和新药转正标准第60册收载的头孢呋辛酯干混悬剂质量标准WS1-(X-391)-2004Z有关物质检查项,本品选择高效液相色谱法作为有关物质检测方法。6.4.2方法学验证6.4.2.1 试验材料仪器:LC-10AT、20AT VP泵(SHIMADZU CORPORATION)SPD-10A、20A、M20A VP紫外检测器(SHIMADZUCORPORATION)工作站:LC solution(SHIMADZU CORPORATION) 色谱柱:C18色谱柱,粒度5um,规格250mm×4.6mm,wel518425,LN:W1801.19,SN:1021096.主要试剂:甲醇(色谱纯)、磷酸二氢铵(分析纯)、6.4.2.2 波长选择根据专属性试验结果并参照中国药典2010年版(二部)收载的头孢呋辛酯原料药质量标准、美国药典USP32-NF27收载的头孢呋辛酯混悬剂质量标准和新药转正标准第60册收载的头孢呋辛酯干混悬剂质量标准WS1-(X-391)-2004Z有关物质检查项,本品选择高效液相色谱法作为有关物质检测方法。波长选择为278nm。6.4.2.3 流动相选择参照中国药典2010年版(二部)收载的头孢呋辛酯原料药质量标准、美国药典USP32-NF27收载的头孢呋辛酯混悬剂质量标准和新药转正标准第60册收载的头孢呋辛酯干混悬剂质量标准WS1-(X-391)-2004Z有关物质检查项色谱条件:用十八烷基硅烷键合硅胶为填充剂;流速为每分钟1.0ml,检测波长278nm,进样量20μl。6.4.2.3.1 等度洗脱流动相配制:①0.2mol/L磷酸二氢铵溶液-甲醇(60:40);②0.2mol/L磷酸二氢铵溶液-甲醇(50:50);③0.2mol/L磷酸二氢铵溶液-甲醇(55:45)。供试样品配制:称取头孢呋辛酯对照品适量(约相当于头孢呋辛11mg),置20ml量瓶中,加2ml甲醇超声溶解,分别用相应的流动相稀释至刻度,摇匀,滤过,精密量取20μl注入液相色谱仪并记录色谱图。试验结果见表10-6,色谱图见图37~39。表10-6等度洗脱试验结果 流动相异构体保留时间 (min.)拖尾因子与相邻峰 分离度理论板数出峰个数①异构体B15.7570.9922.189487111异构体A18.6880.9673.030[f
本人正在做中药的生物碱提取,我之前看了资料,对样品进行了初步提取(最后一步是氯仿萃取),然后选择改良碘化铋钾、碘-碘化钾和磷钼酸进行定性,但是当我把这三种试剂分别滴到样品之后,问题来了:1、 理论说这三种试剂如果有阳性反应的话,是呈橘红色、棕黄色的,但我滴下去后,出现的是分层了,而没有沉淀出现。为什么大家都说加进去后会有沉淀呢?样品的溶剂是氯仿,而沉淀试剂的溶剂是稀冰醋酸,这两者是不相溶的,那何来的反应出现沉淀呢?2、我要提取的生物碱,目前还没有标准品可以购买,对于这种没有标准品的生物碱,要拿什么做对照品呢?请大家多多指教。我查了文献,也有其他人在做同样这种情况的实验(没有标准品的生物碱提取),但他们是用其他的生物碱作为对照品来鉴别的,可以这样的吗?这样做的依据是什么呢?好迷茫啊,希望大家多多回复啊!!!!谢谢了!
头孢泊肟酯 残留溶剂 照残留溶剂测定法(中国药典2010年版二部附录Ⅷ P)测定。甲醇、乙腈、丙酮、二氯甲烷、异丙醇、丁酮、乙酸乙酯、四氢呋喃、乙酸丁酯、1,2-二氯乙烷、乙酸异丙酯、苯、四氯化碳、环己烷、二氧六环、甲基异丁基酮、吡啶、甲苯色谱条件与系统适用性试验 采用DB-624毛细管柱(或与之类似的柱),柱温40℃,维持22min,以100℃/min的速度升温至120℃,维持10min。氢火焰离子化检测器(FID),检测器温度为250℃,进样口温度为200℃,顶空温度为70℃,顶空时间为30min,载气为氮气,取系统适用性溶液顶空进样,按正丙醇(内标)、丁酮、乙酸乙酯的顺序出峰,各峰间的分离度均应符合规定……要顶空进样甲烷气体,记录甲烷的保留时间作为色谱系统的死时间(t0)再顶空进样供试品溶液,记录色谱图,色谱图中如有色谱峰出现,计算供试品溶液色谱图中诸色谱峰的保留时间(tR)相对于正丙醇保留时间(tR(正丙醇))的相对调整保留时间(RART);现在甲烷气体已经让人去买了,估计是钢瓶。怎么装进顶空瓶呢,然后进甲烷气体平衡温度、传输管温度设多少合适呢?
本人正在做中药的生物碱提取,我之前看了资料,对样品进行了初步提取(最后一步是氯仿萃取),然后选择改良碘化铋钾、碘-碘化钾和磷钼酸进行定性,但是当我把这三种试剂分别滴到样品之后,问题来了:1、 理论说这三种试剂如果有阳性反应的话,是呈橘红色、棕黄色的,但我滴下去后,出现的是分层了,而没有沉淀出现。为什么大家都说加进去后会有沉淀呢?样品的溶剂是氯仿,而沉淀试剂的溶剂是稀冰醋酸,这两者是不相溶的,那何来的反应出现沉淀呢?2、我要提取的生物碱,目前还没有标准品可以购买,对于这种没有标准品的生物碱,要拿什么做对照品呢?请大家多多指教。我查了文献,也有其他人在做同样这种情况的实验(没有标准品的生物碱提取),但他们是用其他的生物碱作为对照品来鉴别的,可以这样的吗?这样做的依据是什么呢?好迷茫啊,希望大家多多回复啊!!!!谢谢了!
请问哪位能提供一下头孢布烯和其制剂的质量标准内容?将不胜感激!
[size=5][b][size=3]硫酸头孢喹肟注射[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]量标准[/size][size=3]进口兽药质量标准[/size][size=3]硫酸头孢喹肟注射液[/size][size=3]Liusuan Toubaokuiwo Zhusheye[/size][size=3]Cefquinome sulfate Injection[/size][size=3] [/size][size=3]本品为硫酸头孢喹肟与油酸乙酯等配制而成的混悬注射液。含头孢喹肟(C23H24N6O5S2)应为标示量的90.0%~105.0%。[/size][size=3]【性状】 本品为类白色至浅褐色混悬液体;久置分层。[/size][size=3]【鉴别】(1)含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品峰的保留时间一致。[/size][size=3](2)取摇匀后的供试品2 ml,加水5 ml,稀盐酸1 ml,混匀,置超声浴中超声10分钟,弃去有机层,溶液显硫酸盐的鉴别反应(附录15页)。[/size][size=3]【检查】有关物质 照含量测定项下的方法。[/size][size=3]取摇匀后的供试品1.0 ml,加入流动相25.0 ml,置超声浴中超声5分钟,弃去有机层,取水层滤过,取续滤液10µ l,注入液相色谱仪,记录色谱图,2,3-环己基吡啶与头孢喹肟相对保留时间为0.20。按峰面积归一化法计算,2,3-环己基吡啶应不得过3.0%,其他单一杂质应不得过0.50%,杂质总量应不得过4.0%。[/size][size=3]水分 取本品,照水分测定法(附录58页,第一法)检查,含水分不得过0.2%。[/size][size=3]细菌内毒素 取摇匀后的供试品2 ml与细菌内毒素检查用水3 ml混匀,分成2等份,振摇30秒,离心15分钟(2000g),吸取水层1 ml,加1 mol/L氢氧化钠溶液0.06 ml调节pH值至6.5~7.5。用细菌内毒素检查用水按1:10稀释后,照细菌内毒素检查法(附录73页)检查,每1 mg头孢喹肟中含细菌内毒素的量应小于0.1 EU。[/size][size=3]无菌 取供试品8瓶,混合均匀,加入含6%吐温-80的蛋白胨缓冲液(1g/L)400ml,混匀,加入800×106单位青霉素酶(每1ml供试品溶液,加2×106单位青霉素酶),充分振摇,将供试品倒置,在37℃放置4小时;取供试品溶液,依法检查(附录79页,直接接种法),应符合规定。[/size][size=3]分散性 取本品1瓶,振摇30秒,将供试品转移置玻璃容器中,不得观察到结块或沉淀物。[/size][size=3]沉降 取本品1瓶,振摇30秒,取供试品10 ml置刻度试管中(内径1.0~1.5 cm),10分钟内不得沉淀。[/size][size=3]粒度 取摇匀后的供试品,置显微镜下检查,颗粒直径在5µ m以下应不得少于80%,10µ m以下不得少于90%,20µ m以下不得少于95%,50µ m以下不得少于100%。[/size][size=3]装量 按最低装量检查法(附录67页)检查,应符合规定。[/size][size=3]【含量测定】 照高效液相色谱法(附录24页)测定。[/size][size=3]色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;取一水合高氯酸钠3.45g溶于1000 ml水中,加磷酸12 ml和乙腈90 ml,用三乙胺调节pH至3.6为流动相;检测波长为270 nm。取头孢噻肟约25 mg,溶于100.0 ml流动相中,另取头孢喹肟约25 mg,置25 ml量瓶中,精密加入上述头孢噻肟溶液1 ml,用流动相稀释至刻度。精密量取10µ l注入液相色谱仪,记录色谱图;计算头孢喹肟与头孢噻肟的分离度,应大于1.0。[/size][size=3]对照品溶液的制备 精密称取硫酸头孢喹肟对照品适量(约相当于头孢喹肟25 mg),置250 ml量瓶中,用流动相溶解并稀释至刻度,摇匀,即得。[/size][size=3]供试品溶液的制备 精密量取摇匀后的供试品适量(约相当于头孢喹肟50 mg),精密加入流动相100 ml,置超声浴中超声5分钟,弃去油层,滤过;精密量取续滤液5 ml,置25 ml量瓶,加流动相稀释至刻度,摇匀,即得。[/size][size=3]测定法 精密量取对照品溶液和供试品溶液各10µ l,分别注入液相色谱仪,记录色谱图。按外标法以峰面积计算,即得。[/size][size=3]【作用与用途】抗生素类药。主要用于治疗大肠杆菌引起的奶牛乳房炎,多杀性巴氏杆菌或胸膜肺炎放线杆菌引起的猪呼吸道疾病[/size][size=3]【用法与用量】肌内注射 一次量 每1 kg体重 牛1 mg 一日1次 连用2日 猪2~3 mg 一日1次 连用3日[/size][size=3]【休药期】牛 5日;猪 2日;弃奶期 1日。[/size][size=3]【规格】 50ml:1.25g[/size][size=3]【贮藏】遮光,25℃以下保存。[/size][size=3]【有效期】 2年。[/size][size=3]【生产企业】英特威国际有限公司(Intervet International GmbH)[/size][size=3] [/size][size=3] [/size][size=3]注:[/size][size=3]蛋白胨缓冲液(1g/L)[/size][size=3]蛋白胨(酪蛋白胨或肉蛋白胨) 1g[/size][size=3]KH2PO4 3.6g[/size][size=3]Na2HPO4• 2H2O 7.2g[/size][size=3]NaCl 4.3g[/size][size=3]最终pH为7.0±0.2[/size][size=3]溶液应无色澄清[/size][/b][/size]

各位好!有没有做过头孢替坦酸的?按照日本药典方法做头孢替坦酸有关物质实验的时候主峰前面出现了一个小峰,谱图如下:http://ng1.17img.cn/bbsfiles/images/2015/08/201508051144_559190_1866875_3.jpg实验条件:检测器:VWD;检测波长:254nm;色谱柱:C18色谱柱(L=250mm,5μm,φ=4.6mm);0.1mol/L磷酸溶液:取11.53g磷酸加水1000ml,摇匀,即得;流动相:0.1mol/L磷酸-色谱甲醇-色谱乙腈-色谱冰乙酸=1700:105:105:100。(此流动相临用新制)流速:2.0ml/min;柱温:40℃。样品前处理:取甲醇10ml,注入10μl于液相色谱仪。如果样品溶解用甲醇和水的话前面的峰就没有了,不知道什么情况。求各位大侠支招!
[color=#444444]请教大神,走头孢喹肟血浆样品紫外高效液相色谱,标准品样能走出来,用血浆经前处理后的样就走不出来了,血浆前处理过程主要是用乙腈重复提两次,取上清液,然后氮吹干,进样,请问走不出峰是什么原因呢?急求,多谢![/color]
最近在做头孢检测,方法回收率一直不好,以下几个标准有做过的吗?怎么样?讨论下吧GB-T 21314-2007 动物源性食品中头孢匹林、头孢噻呋残留量检测方法 液相色谱-质谱/质谱法GBT 22942-2008 蜂蜜中头孢唑啉 头孢匹林 头孢氨苄 头孢洛宁 头孢喹肟残留量的测定 液相色谱-串联质谱法SNT 1988-2007 进出口动物源食品中头孢氨苄、头孢匹林和头孢唑啉残留量检测方法
随着国内燕窝市场蓬勃发展,燕窝及其制品缺失国家标准,进而带来监管失力、燕窝市场鱼龙混杂、食品安全难保障的矛盾愈发凸显。昨天获悉,国家相关部门目前正在为制定燕窝国家标准甚至国际标准做准备,今年由广东检验检疫局和中国检科院共同承担的“燕窝及其制品的真假鉴别方法研究”科技项目已顺利通过鉴定。专家指出,这意味着对于燕窝真伪有了权威的鉴定方法,为我国建立燕窝的国家、国际产品标准及卫生标准提供了技术支撑和科研保证。 从广东检验检疫部门获悉,国家质检总局早已布署攻关“燕窝及其制品的真假鉴别方法研究”科技项目,并由该局和中国检科院共同承担。据介绍,为攻关燕窝及其制品的真假鉴别方法,课题小组人员历时多年,深入印尼等燕窝产地,了解原料采集、生产加工及其市场流通等情况,并采集了大量原料燕窝以及市售的燕窝样品,进行了多次测试。最终研究出鉴定真假燕窝的方法。据悉,该鉴别方法可有效分辨人为加入的掺假物质和天然存在的营养物质,而且还可用于大量样品的快速测定。
GB/T 22942-2008 蜂蜜中头孢唑啉、头孢匹林、头孢氨苄、头孢洛宁、头孢喹肟残留量的测定 液相色谱-串联质谱法
大家有没有用过中检所的抗生素标准品的?如头孢氨苄、阿莫西林等,我正在用,结果用液质联用检测出两个色谱峰,质谱结果显示,这两个峰都是同一种物质,猜测标准品不纯,是同分异构体的混合物,大家有没有类似的遭遇?
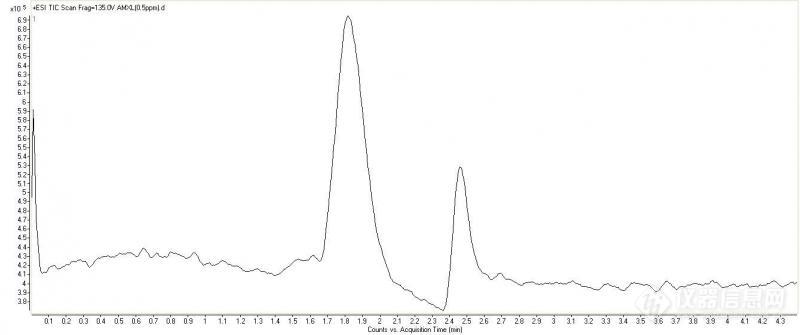
[color=#444444]我用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质联用[/color][/url]检测抗生素,用中检所的标准品,单标居然出现两个峰,大家帮分析下是啥问题。目标物是阿莫西林,溶剂是甲醇/水(50/50),液相流动相是乙腈/水,梯度洗脱,20%-80%,时间18min。用头孢氨苄和头孢拉定的单标进样也都出现两个峰,纯度写的是95%。[/color][color=#444444]两张图分别为阿莫西林单标的质谱离子流图和两个峰处的碎皮离子峰图,两个峰的碎皮离子峰图是一样的。[/color][color=#444444][img=,690,288]https://ng1.17img.cn/bbsfiles/images/2019/07/201907091139146787_4268_1701336_3.jpg!w690x288.jpg[/img][img=,690,216]https://ng1.17img.cn/bbsfiles/images/2019/07/201907091139153614_7712_1701336_3.jpg!w690x216.jpg[/img][/color]







 我要推广仪器
我要推广仪器
 下载APP
下载APP