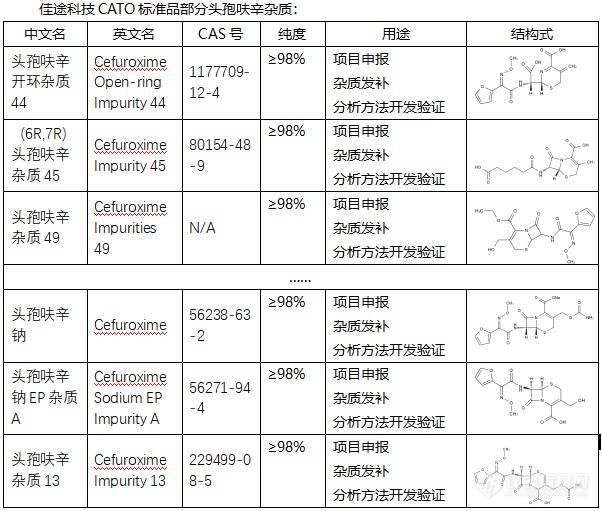
头孢呋辛是一种广泛使用的抗生素,主要用于治疗由敏感细菌引起的各种感染。在生产、储存和使用头孢呋辛的过程中,可能会产生一些杂质。这些杂质的存在可能会影响头孢呋辛的纯度和疗效,因此了解和控制这些杂质对于确保药物的安全性和有效性至关重要。头孢呋辛的杂质有多种,其中一些具有特定的CAS号、化学式和分子量。例如,头孢呋辛杂质33(Cefuroxime Impurity 33)的CAS号为929531-94-2,分子式为C16H16N4O9S,分子量为440.38。此外,还有其他一些头孢呋辛杂质,如头孢呋辛杂质A、B、C、D、E、F、G、H等。 CATO标准品提供的头孢呋辛全套的杂质,这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分。[img=,602,511]https://ng1.17img.cn/bbsfiles/images/2024/02/202402192104451830_7644_6381607_3.png!w602x511.jpg[/img] 广州佳途科技股份有限公司深知药物研发与质量控制的重要性,CATO标准品厂家,提供头孢呋辛全套的杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展,以满足客户在药物研发和质量控制方面的需求。[list][*]原创检测区[/list]◇头孢呋辛杂质头孢呋辛是一种广泛使用的抗生素,主要用于治疗由敏感细菌引起的各种感染。在生产、储存和使用头孢呋辛的过程中,可能会产生一些杂质。这些杂质的存在可能会影响头孢呋辛的纯度和疗效,因此了解和控制这些杂质对于确保药物的安全性和有效性至关重要。头孢呋辛的杂质有多种,其中一些具有特定的CAS号、化学式和分子量。例如,头孢呋辛杂质33(Cefuroxime Impurity 33)的CAS号为929531-94-2,分子式为C16H16N4O9S,分子量为440.38。此外,还有其他一些头孢呋辛杂质,如头孢呋辛杂质A、B、C、D、E、F、G、H等。CATO标准品提供的头孢呋辛全套的杂质,这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分。广州佳途科技股份有限公司深知药物研发与质量控制的重要性,CATO标准品厂家,提供头孢呋辛全套的杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展,以满足客户在药物研发和质量控制方面的需求。
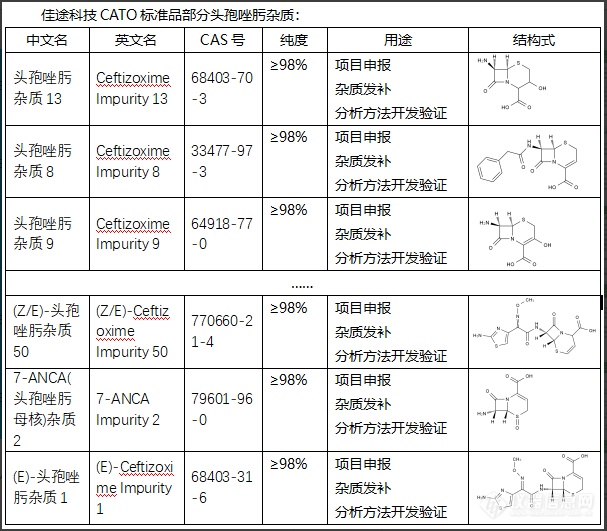
有关头孢唑肟杂质的作用,以下是要注意的一些可能性:1.负面作用:过多的杂质可能导致药物效力下降,并可能引发不良反应或副作用。例如,有些杂质可能导致过敏反应。2.毒性:某些杂质可能具有毒性。例如,某些杂质可能具有致癌性。3.影响药效:杂质可能会影响药物的生物利用度,即药物进入体内后能达到预期药效的能力。CATO标准品药品生产中的质量控制步骤非常重要,目的就是要尽可能减少杂质的存在。任何药品都必须经过严格的质量检测,确保其安全有效。[img=,607,531]https://ng1.17img.cn/bbsfiles/images/2024/02/202402041449269442_4660_6381668_3.png!w607x531.jpg[/img]
实验中用到冰醋酸配制的20%的醋酸溶液用于做萃取溶剂,但是测量时发现在300-400nm处有很强的荧光峰,请问如何除去冰醋酸中的杂质,或者有什么纯化的方法吗?非常感谢
请问冰醋酸中可能存在什么杂质,特别是一些荧光的杂质,怎么的纯化谢谢
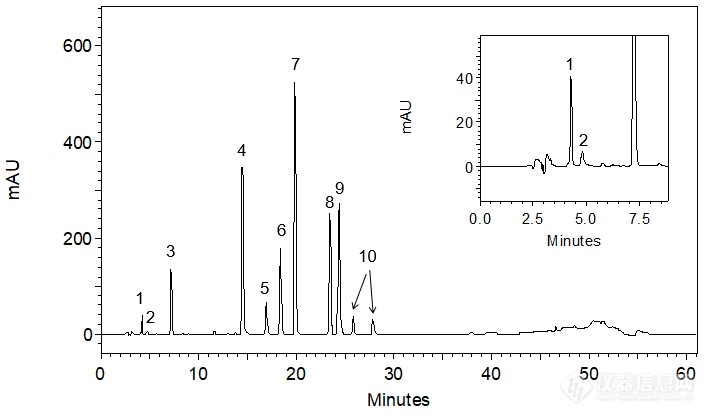
[align=center][b]头孢克洛有关物质——与9种杂质的共同分析[/b][/align]头孢克洛(cefaclor)为白色至微黄色粉末或结晶性粉末的化学品,微臭,本品在水中微溶,在甲醇、乙醇、三氯甲烷或二氯甲烷中几乎不溶,分子式:C15H14ClN3O4S。头孢克洛是β-内酰胺类抗生素,头孢菌素类药,是第二代头孢菌素,主要适用于敏感菌所致的急性咽炎、急性扁桃体炎、中耳炎、支气管炎、肺炎等呼吸道感染、皮肤软组织感染和尿路感染等。[align=center][img=,144,171]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140859582934_5220_2222981_3.gif!w144x171.jpg[/img][/align][align=center]头孢克洛[/align][align=center]M.W.: 367.81[/align]本实验对客户提供的头孢克洛原料药以及9种杂质(杂质A、B、C、D、E,7-ACCA,头孢克洛δ-3异构体,α-苯甘氨酸,苯甘氨酸甲酯盐酸盐)进行分析,希望得到杂质混合对照溶液及供试品溶液中各杂质的良好分离。客户反馈,将流动相磷酸盐体系的pH值由4.0提高到4.5可得到杂质混合对照溶液中7-ACCA和α-苯甘氨酸之间的良好分离,但头孢克洛与其相邻杂质E峰之间分离较难。客户前期使用了CAPCELL PAK C[sub]18 [/sub]MGII S3 4.6 mm i.d. × 250 mm色谱柱进行分析,在此基础上,我们尝试了其他填料的几款色谱柱进行分离尝试,分别为CAPCELL PAK C[sub]18[/sub] AQ(S3& S5)、CAPCELL PAK ADME(金刚烷基)、SUPERIOREX ODS、CAPCELL PAK PFP(五氟苯基)、CAPCELL PAK CN(氰基)。首先,参考客户提供的液相条件,使用高极性色谱柱[b]CAPCELL PAK C[sub]18 [/sub]AQ[/b]对杂质混合对照溶液进行分析尝试;为了得到杂质间的更好分离,粒径选择3 μm,如图1,[color=#2F5496]各杂质间均能得到良好的分离结果,头孢克洛与杂质[/color][color=#2F5496]E[/color][color=#2F5496]的分离度为[/color][color=#2F5496]2.70[/color][color=#2F5496],达到基线分离。[/color][color=#2F5496][/color][align=center][img=,690,405]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140902184290_9307_2222981_3.png!w690x405.jpg[/img][/align][align=center]图1 AQ S3 分析杂质混合对照溶液结果[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 头孢克洛 9. 杂质E [/color]10.杂质D[/align][color=#2F5496][img=,555,311]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140902187828_2715_2222981_3.png!w555x311.jpg[/img][/color]进一步分析供试品溶液,如图2,由于样品浓度较高,导致头孢克洛主峰向后展宽,进而将杂质E包于其中。[color=#2F5496][/color][align=center][color=#2F5496][img=,659,441]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140915544228_5404_2222981_3.png!w659x441.jpg[/img][/color][/align][align=center]图2 AQ S3 分析供试品溶液结果[/align][align=center][/align][align=left]为使头孢克洛和杂质E之间得到更好的分离,我们尝试对色谱条件进行调整。[/align][align=left][/align][align=left][b]1.调整柱温[/b][/align][align=left][b][/b]首先对温度进行调整:实验过程中发现柱温对头孢克洛与杂质E的出峰行为有较大影响——当柱温设置为20 ℃时,头孢克洛和杂质E之间能够得到良好分离;将温度提高到30℃时,杂质E向前移动趋势较大。为使杂质E峰出在头孢克洛峰前,避免由于供试品中头孢克洛峰的展宽而使杂质E被包于其内,进一步将柱温提高到40℃,发现头孢克洛与杂质E峰重合;最终,将柱温提高到45℃,此时杂质E峰移至头孢克洛峰前,但未能得到理想的分离结果。[/align][align=left][/align][align=center][img=,659,430]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140916597550_373_2222981_3.png!w659x430.jpg[/img][/align][align=center]图3 不同柱温条件下AQ S3分析杂质混合对照溶液结果[/align][align=center][/align][align=left][b]2.调整流动相[/b][/align][align=left][b][/b][/align][align=left]考虑到提高柱温对色谱柱寿命的影响,仍选择初始使用的20℃,对流动相梯度条件进行调整。在增强整体保留时间的同时,发现[color=#538135]头孢克洛和杂质[/color][color=#538135]E[/color][color=#538135]的出峰顺序发生了颠倒[/color],且[color=#538135]分离良好[/color],进而有效避免了杂质E被包于头孢克洛主峰中的问题;而在主峰后出峰的杂质D与头孢克洛之间分离度亦较高,即使供试品溶液中的头孢克洛峰展宽,也不会出现将杂质D包于其中的问题。[/align][align=left]因此我们在此梯度条件下进一步对供试品溶液进行分析,如图4,头孢克洛与各杂质峰之间均能得到良好的分离结果。[/align][align=left][/align][align=center][img=,679,417]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140917450308_6331_2222981_3.png!w679x417.jpg[/img][/align][align=center]图4 AQ S3分析杂质混合对照溶液及供试品溶液结果(调整梯度)[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 杂质E 9. 头孢克洛[/color] 10.杂质D[/align][align=left][img=,587,335]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918136074_9375_2222981_3.png!w587x335.jpg[/img][/align][align=left][/align][align=left]为使客户有更多的色谱柱选择,本实验室也尝试使用键合金刚烷基的高极性色谱柱CAPCELL PAK ADME分析杂质混合对照溶液和供试品溶液,如图5,在分析杂质混合对照溶液时,能够得到各组分的良好分离,同时发现杂质E和头孢克洛出峰顺序发生颠倒,但同时也发现头孢克洛峰与其后相邻杂质D峰之间分离度较低(Rs=1.71);因此,如图6,在分析供试品溶液时,由于色谱峰向后展宽,使得杂质D被包于头孢克洛主峰中,未能得到理想分离结果。[/align][align=left][/align][align=center][img=,690,426]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918484278_6616_2222981_3.png!w690x426.jpg[/img][/align][align=center]图5 ADME 分析杂质混合对照溶液结果[/align][align=center] [/align][align=center]1.α-苯甘氨酸 2. 7-ACCA 3. 杂质A 4. 杂质B 5. 苯甘氨酸甲酯盐酸盐 6.杂质C[/align][align=center]7. 头孢克洛δ-3异构体 [color=#ff0000]8. 杂质E 9. 头孢克洛[/color] 10.杂质D[/align][align=left][/align][align=center][img=,689,417]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140918485898_9906_2222981_3.png!w689x417.jpg[/img][/align][align=center]图6 ADME 分析杂质混合对照溶液结果[/align][align=left][img=,585,336]http://ng1.17img.cn/bbsfiles/images/2018/06/201806140919331328_5070_2222981_3.png!w585x336.jpg[/img][/align][align=left][/align][align=left][/align][align=left]之后,我们也尝试使用了CN(氰基柱)和PFP(五氟苯基)以及高碳载量的SUPERIOREX ODS色谱柱,在客户提供的色谱条件下对杂质混合对照溶液进行分析,均未能得到更理想的分离结果。[/align]
我公司现在使用GPC分析醋酸纤维素的分子量和分子量分布,但是由于杂质影响大,无法准确反应所需要的信息。所以象各位请教,还有什么仪器或设备可以测量醋酸纤维素的分子量、分子量分布,不胜感激!
进口兽药质量标准硫酸头孢喹肟注射液Liusuan Toubaokuiwo ZhusheyeCefquinome sulfate Injection本品为硫酸头孢喹肟与油酸乙酯等配制而成的混悬注射液。含头孢喹肟(C23H24N6O5S2)应为标示量的90.0%~105.0%。【性状】 本品为类白色至浅褐色混悬液体;久置分层。【鉴别】(1)含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品峰的保留时间一致。(2)取摇匀后的供试品2 ml,加水5 ml,稀盐酸1 ml,混匀,置超声浴中超声10分钟,弃去有机层,溶液显硫酸盐的鉴别反应(附录15页)。【检查】有关物质 照含量测定项下的方法。取摇匀后的供试品1.0 ml,加入流动相25.0 ml,置超声浴中超声5分钟,弃去有机层,取水层滤过,取续滤液10µ l,注入液相色谱仪,记录色谱图,2,3-环己基吡啶与头孢喹肟相对保留时间为0.20。按峰面积归一化法计算,2,3-环己基吡啶应不得过3.0%,其他单一杂质应不得过0.50%,杂质总量应不得过4.0%。水分 取本品,照水分测定法(附录58页,第一法)检查,含水分不得过0.2%。细菌内毒素 取摇匀后的供试品2 ml与细菌内毒素检查用水3 ml混匀,分成2等份,振摇30秒,离心15分钟(2000g),吸取水层1 ml,加1 mol/L氢氧化钠溶液0.06 ml调节pH值至6.5~7.5。用细菌内毒素检查用水按1:10稀释后,照细菌内毒素检查法(附录73页)检查,每1 mg头孢喹肟中含细菌内毒素的量应小于0.1 EU。无菌 取供试品8瓶,混合均匀,加入含6%吐温-80的蛋白胨缓冲液(1g/L)400ml,混匀,加入800×106单位青霉素酶(每1ml供试品溶液,加2×106单位青霉素酶),充分振摇,将供试品倒置,在37℃放置4小时;取供试品溶液,依法检查(附录79页,直接接种法),应符合规定。分散性 取本品1瓶,振摇30秒,将供试品转移置玻璃容器中,不得观察到结块或沉淀物。沉降 取本品1瓶,振摇30秒,取供试品10 ml置刻度试管中(内径1.0~1.5 cm),10分钟内不得沉淀。粒度 取摇匀后的供试品,置显微镜下检查,颗粒直径在5µ m以下应不得少于80%,10µ m以下不得少于90%,20µ m以下不得少于95%,50µ m以下不得少于100%。装量 按最低装量检查法(附录67页)检查,应符合规定。【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;取一水合高氯酸钠3.45g溶于1000 ml水中,加磷酸12 ml和乙腈90 ml,用三乙胺调节pH至3.6为流动相;检测波长为270 nm。取头孢噻肟约25 mg,溶于100.0 ml流动相中,另取头孢喹肟约25 mg,置25 ml量瓶中,精密加入上述头孢噻肟溶液1 ml,用流动相稀释至刻度。精密量取10µ l注入液相色谱仪,记录色谱图;计算头孢喹肟与头孢噻肟的分离度,应大于1.0。
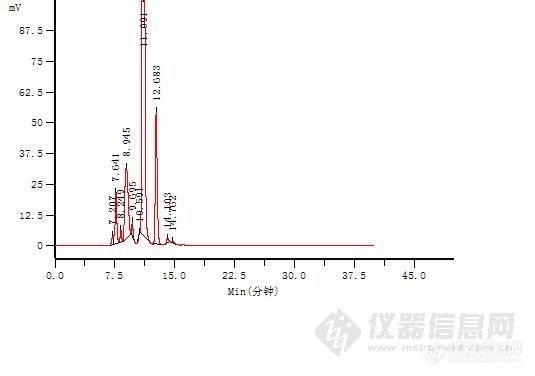
高效分子排阻色谱法测定注射用盐酸头孢替安高分子杂质头孢替安是杀菌性头孢菌素类广谱抗生素,头孢替安不但对革兰氏阳性菌有效,而且对革兰氏阴性菌。如流感嗜血杆菌,大肠杆菌、克雷白氏菌、奇异变形杆菌等的作用更强。对肠杆菌,枸橼酸杆菌、吲哚阳性变形杆菌等,也有抗菌作用头孢替安在肺中药物浓度较高,其它脏器和肌肉也有一定的浓度。临床应用于敏感菌所导致的感染,如肺炎、支气管炎、胆道感染、腹膜炎、尿路感染以及手术后或外伤引起的感染和败血症等。其基本结构同已上市的的头孢菌素类抗生素一样,头孢替安也会形成高分子聚合物,也会在临床使用中引发速发型过敏反应。对患者危害极大。已有的注射用盐酸头孢替安国家药品标准未将盐酸头孢替安高分子聚合物列为检定项目,国内的药学研究也未见头孢替安高分子聚合物的研究和报道。从临床用药安全性考虑,根据中国药典2010年版二部附录凝胶色谱原理。采用常用的葡聚糖凝胶G-10检测聚合物时由于头孢替安分子结构自身的原因,头孢替安不能完全缔合,因些我们采用高效分子排阻色谱法,以球状蛋白色谱用亲水硅胶为填充剂 TOSOH TSKgelG2000SW(7.5*300mm),测定注射用盐酸头孢替安高分子杂质1.仪器与试剂(1)仪器:岛津LC-10ATvp泵 岛津SPD-10AVP紫外可见光多波长检测器 浙大2010色谱数据工作站 色谱柱:TOSOH TSKgelG2000SW(7.5*300mm) (2)试剂: 乙腈 (色谱纯,天津市四友生物医学技术有限公司) 磷酸氢二钠(分析纯,北京化学试剂公司) 磷酸二氢钠(分析纯,北京化学试剂公司)双蒸水 (自制)2 色谱条件色谱柱:TOSOH TSKgelG2000SW(7.5*300mm)流动相:磷酸盐缓冲液(p H:6.8[/color
我需要购买棉酚和醋酸棉酚的标准品,用液相色谱测定棉酚含量。但是西域和sigma都没有卖。不知道哪里有卖的?知道的帮帮。谢谢。

回忆贴:远去了的GC-MS和不用再伺候的科技精英,这个是几年前的事了,当时没机会发表,现在没机会用GC-MS了,把以前的经历作写出来,虽然数据和图谱基本找不到了,但记忆还是非常深刻的,乘着今天加班无聊,完成这月的原创任务。以前单位的重点项目羰基合成醋酐,其中的杂质在研发阶段是别人做的(后来跳槽了,我也不知道她以前怎么做的,反正科研精英们也不会告诉我),生产了也就不关心里面的杂质了,质检的主任工程师闲得无聊,拿来一点产品让我分析,总离子流图出来了,但靠检索得到的数据都有点问题。http://ng1.17img.cn/bbsfiles/images/2012/12/201212301032_417223_1640192_3.jpg以上是GC-MS的总离子流图,醋酐保留时间为4.28min,杂质醋酸为3.60min、EDA为4.99min,而在醋酐和EDA中有多个小杂质,其中4.76min的杂质(以下称为A)质谱图三个明显的碎片为29,43,57,检索结果是2,3-戊二酮(原图没有了,只能用标准图代替了)http://ng1.17img.cn/bbsfiles/images/2012/12/201212301042_417224_1640192_3.jpg记得匹配率还是很好的,但我总觉得有点问题,因为醋酐沸点在139℃,EDA沸点为168℃,而2,3-戊二酮在我查到的资料中只有115℃,不该这么后面,更重要的是我写不出得到这个物质的机理(毕竟搞分析的不是科研精英),然后就跟送样的人商量,给我找各种中间馏分,终于拿到了一个A含量很高的物料,当时我想醋酐在水中会水解成醋酸并且和水混溶,而里面含量较高的杂质EDA微溶于水,通过萃取来看看A是水溶还是酯溶性的,可以推测大致结构,同时分开醋酐和EDA,相当于浓缩提纯。做法是:将物料加入一定量的水,搅拌至油相不再减少,加入乙酸乙酯进行振荡静置分离,用GC-MS分析两相的成分。另人意外的是两相中均不含有A,而出现了一定量的丙酸。虽然分离并不成功,但试验过程中的现象给了我重要的线索,A遇水很容易完全水解,显然要比一般的丙酸酯快的多,而且从质谱碎片来看它的结构应该是比较简单的,而从谱库中无法检索到匹配的物质则说明这个物质可能不在谱库中。因此我怀疑是丙醋酸酐,虽然没有该物质的物性资料,但它沸点应该在醋酸酐和丙酸酐之间,而丙酸酐的沸点是168℃,因此丙醋酸酐出峰位置应该在醋酐和EDA之间,而且乙酰基分子量为43,丙酰基分子量为57,乙基分子量29,和质谱图相符合。既然谱库没有,标样肯定是买不到了,只好自己合成了。虽然合成酸酐的方法很多,但用于验证的合成方法有其特殊性。虽然无需考虑成本,对产率也无要求。但采用的方法应当尽可能使用少量试剂,以免混合物中物质过多造成判断失误。而用于GC-MS分析,还要考虑不能有对仪器不利的物质进入仪器。显然用常规的浓酸催化或者酰氯钠盐反应生成的酸酐如果不进行分离是不能用于GC-MS分析的。由于丙醋酸酐是丙酸和醋酸脱水而成,而醋酐本身也有脱水功能,因此我就把丙酸和醋酸酐反应进GC-MS分析,没有使用其它催化剂。试验方法:将丙酸和醋酐等比例混合,在120度加热4小时,冷却,进GC-MS分析。反应液一共出现了非常明显的[/fo
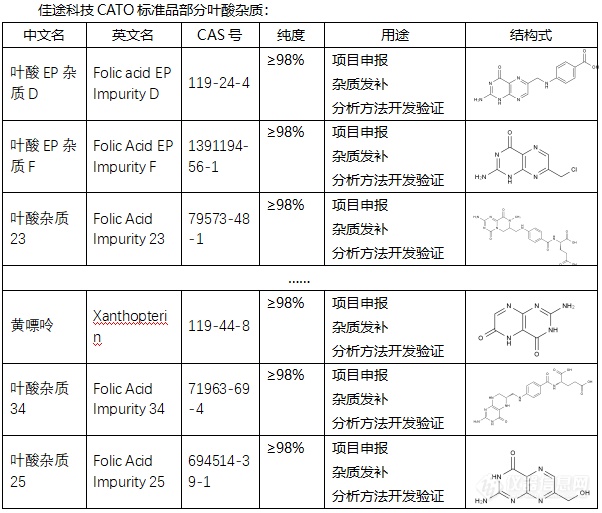
[font=宋体]◇叶酸杂质[/font][font='Segoe UI'][color=#05073b][font=Segoe UI] 叶酸杂质通常是指在叶酸的生产或保存过程中产生的非目标化合物。这些杂质可能会影响叶酸的纯度和效果,因此在叶酸的生产和质量控制过程中需要严格控制其含量。叶酸杂质有多种类型,每一种都具有不同的化学特性,如[/font]CAS号、分子式、分子量等。例如,有一种叶酸杂质CAS号为82778-08-3,分子式为C7H7ClN6HCl,分子量为247.08。另一种叶酸杂质G的CAS号为6810-75-9,英文名称为Folinic Acid Impurity G。此外,叶酸杂质5的CAS号为873397-19-4,纯度为98% HPLC。[/color][/font][font=宋体][font=Calibri] CATO[/font][font=宋体]标准品提供的叶酸全套的杂质[/font][/font][font=宋体],[/font][font=宋体]这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分[/font][font=宋体]。[img=,602,513]https://ng1.17img.cn/bbsfiles/images/2024/02/202402182015587706_5356_6381607_3.png!w602x513.jpg[/img][/font][font=宋体][color=#05073b][back=#fdfdfe] 广州[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]佳途科技[/back][/color][/font][font=宋体][color=#05073b][back=#fdfdfe]股份有限公司[/back][/color][/font][font='Segoe UI'][color=#05073b][back=#fdfdfe]深知药物研发与质量控制的重要性[/back][/color][/font][font=宋体][font=宋体],[/font][font=Calibri]CATO[/font][font=宋体]标准品厂家,提供叶酸全套[/font][/font][font=宋体]的[/font][font=宋体]杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展[/font][font=宋体],[/font][font=宋体]以满足客户在药物研发和质量控制方面的需求。[/font]
[em06] 各位大哥大姐:小妹急切寻找以下标准:工业甲醇,活性炭,醋酸乙酯,醋酸丁酯,叔丁胺的国家标准或行业标准.急急急.请大家帮帮忙啦!!!
请教一个技术问题,我现在做酸碱滴定,用0.1N NaOH滴定醋酸乙酯中的酸度,酚酞指示终点。样品体系为:70%醋酸2%硫酸10%水15%醋酸乙酯其他,杂志据说,使用自动电位滴定仪可以一次滴出醋酸和硫酸的含量,是真的吗?PS,由于体系中有不少的酯,样品制备时不能用水稀释,否则会引起酯的水解。
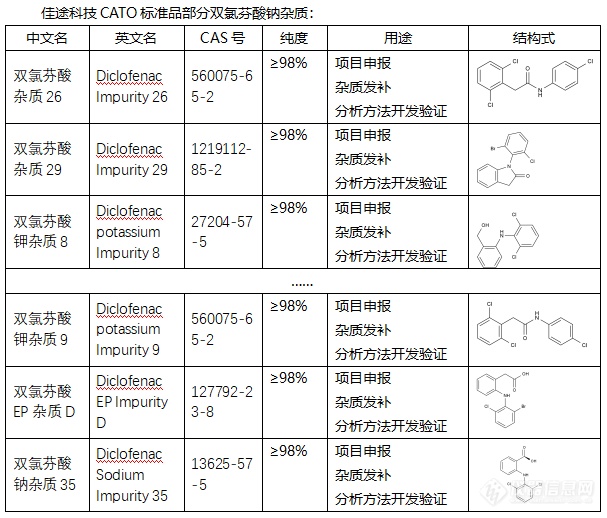
◇双氯芬酸钠杂质在双氯芬酸钠的生产和储存过程中,可能会产生一些杂质,双氯芬酸钠的杂质有多种,包括但不限于以下几种:双氯芬酸钠杂质A:这是一种具有特定CAS号(15362-40-0)和分子式(C14H9Cl2NO2)的杂质。其分子量为278.13,密度为1.4±0.1 g/cm3,沸点为488.6±45.0°C at 760 mmhg,熔点为115-119°C;双氯芬酸钠杂质(1-(2,6-DICHLOROPHENYL)INDOLIN-2,3-DIONE):这是一种具有CAS号的杂质,其化学式为C14H7Cl2NO2。双氯芬酸钠的其他杂质:除了上述两种杂质外,双氯芬酸钠还可能存在其他杂质,如乙酰氯芬酸杂质、醋氯芬酸杂质等。CATO标准品提供的双氯芬酸钠全套的杂质,这些杂质对于药物的纯度和稳定性研究至关重要,也是药物研发过程中不可或缺的一部分。[img=,607,518]https://ng1.17img.cn/bbsfiles/images/2024/02/202402192056045756_8062_6381607_3.png!w607x518.jpg[/img]广州佳途科技股份有限公司深知药物研发与质量控制的重要性,CATO标准品厂家,提供双氯芬酸钠全套的杂质,为客户提供更加精准、可靠的分析标准品,助力药物研发事业的快速发展,以满足客户在药物研发和质量控制方面的需求。
最近准备做维生素A,GB5009.82-2016中在标准溶液配制和分析结果表述中都是用维生素A表述的,分别用“准确称取25.0mg维生素A标准品”“X——试样中维生素A的含量,维生素A单位为微克每百克(μg/100g)”表述;我们买的标品是维生素A醋酸酯;我们的检验报告单又以“维生素A(以视黄醇计)”体现。我查到1IU维生素A=0.3μgRE 1IU维生素A=0.344μg维生素A醋酸酯等换算关系,但在实验过程中究竟该如何处理这些关系了,比如要准确称取25.0mg的维生素A标准品,那我该称多少的维生素A醋酸酯;维生素A醋酸酯需不需要皂化;维生素A又称视黄醇,那检验报告单中维生素A(以视黄醇计)作何理解,视黄醇和视黄醇当量有何异同;标准品和对照品有何异同等。拜托吧里大神赐教啊,万分感谢
头孢西丁钠有关杂质 头孢西丁钠有关杂质
哪位有醋酸汞的标准,能否分享下~~~是试剂级的标准~~谢谢~
在验收维生素E醋酸酯标准品的时候,不够仔细,我没有认真查看,现在要用了才发现是液体状的,可是小瓶子中却只有100mg,我素手无策了,又怕浪费,不知道该怎么称取那少的可怜的标准品了?我是初次接触高效液相色谱,仪器也是新买的,还烦请请各位赐教帮帮忙啊。在此先谢过了。。。
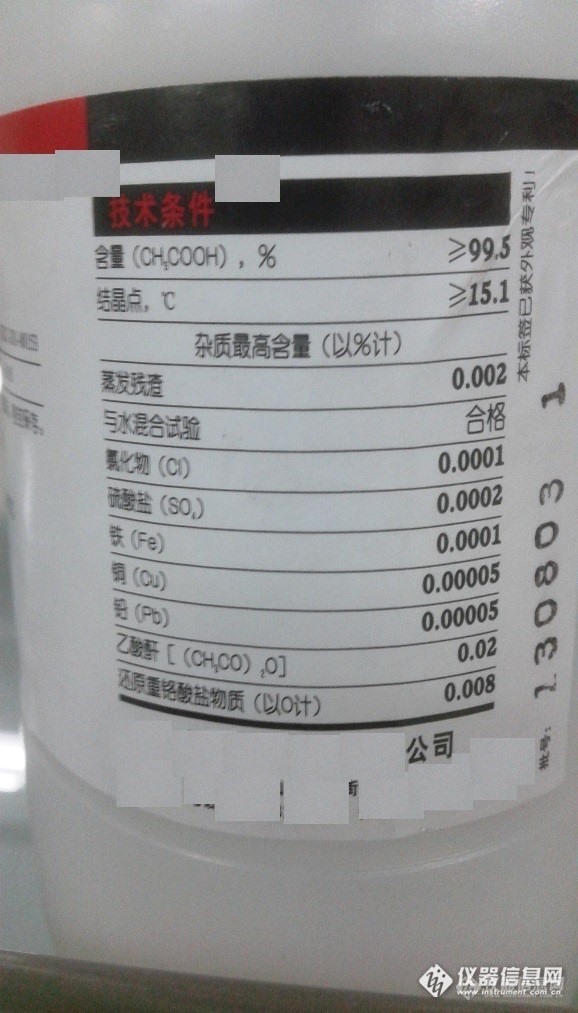
Merck EMSURE用户体验报告系列之一作者:山东某制药公司分析中心 在我公司某制剂有关物质和含量的检测过程中,使用到了醋酸铵(CH3COONH4)和冰醋酸(CH3COOH)两种化学试剂。其中,醋酸铵是有关物质和含量的流动相缓冲盐,冰醋酸是流动相的pH调节剂。这两种化学试剂的质量好坏,对检测结果具有较大的影响。 在试验中,作者发现,采用国内某品牌的醋酸铵和冰醋酸配制缓冲盐进行检测时,色谱图基线噪音较大,使检测方法的灵敏度明显降低。而采用Merck公司的EMSURE系列的优级纯醋酸铵和优级纯冰醋酸配制缓冲盐进行试验,基线噪音明显较小。见图1。http://ng1.17img.cn/bbsfiles/images/2015/02/201502111407_535432_2491887_3.jpg图1 试验色谱图(A.国内某品牌试剂测定图谱;B. Merck EMSURE优级纯测定图谱;C. 国内某品牌试剂测定局部放大图谱;D. Merck EMSURE优级纯测定局部放大图谱;) 试验之余,作者仔细对比了一下Merck EMSURE和国内品牌的醋酸铵和冰醋酸。我惊奇的发现,在这冰醋酸的质量控制指标中,Merck EMSURE优级纯的质控指标竟然多达29项。其中,Merck EMSURE的优级纯冰醋酸共有9种阴离子检测指标,15种金属离子检测指标;而国内某品牌的冰醋酸仅有11项质控指标,其中,4种阴离子检测指标,3种金属离子检测指标。无论是控制的重金属离子种类还是无机阴离子的数量,Merck EMSURE优级纯的质量明显高人一等。从质量控制指标的角度可以看出,Merck EMSURE优级纯的质量具有明显的优势。 空口无凭证,有图有真相。作者将冰醋酸试剂标签上的质控指标拍照展示,大家可以自己进行对比,请看图2。http://ng1.17img.cn/bbsfiles/images/2015/02/201502111408_535433_2491887_3.jpghttp://ng1.17img.cn/bbsfiles/images/2015/02/201502111408_535434_2491887_3.jpg图2 Merck EMSURE系列优级纯冰醋酸(左)和国内某品牌分析纯试剂冰醋酸(右) 众所周知,在色谱方法的检测过程中,流动相缓冲盐和pH调节剂等化学试剂的纯度和杂质情况对于检测结果有一定的影响。如果化学试剂中存在一些微量的无机阴离子,则试验所得色谱图的基线噪音会较大,影响检测的灵敏度。缓冲盐中如果存在极微量金属离子如铅、铜、镍、锌、钙、铁、铝等,则会对色谱柱的使用寿命和保留行为产生较大影响。同时,各种杂质的存在往往会对试验结果产生一定的干扰,引起结果的误判。因此,在试验过程中,选择质量更好的优级纯进行化学和色谱分析将会使结果更加准确。
醋酸甲地孕酮醋酸甲地孕酮 拼音名:Cusuan Jiadiyuntong 英文名:Megestrol Acetate 书页号:2000年版二部-1014 C24H32O4 384.52本品为6-甲基-17α-羟基孕甾-4,6-二烯-3,20-二酮17- 醋酸酯。按干燥品计算,含C24H32O4应为97.0%~103.0%。 【性状】 本品为白色或类白色的结晶性粉末;无臭,无味。 本品在氯仿中易溶,在丙酮或醋酸乙酯中溶解,在乙醇中略溶,在乙醚中微溶,在水中不溶。 熔点 本品的熔点(附录Ⅵ C)为213 ~220℃。 比旋度 取本品, 精密称定, 加氯仿溶解并定量稀释制成每 1ml中含 50mg 的溶液,依法测定(附录Ⅵ E),比旋度为+9°至 +12°。 【鉴别】 (1) 取本品约50mg,加乙醇制氢氧化钾试液2ml ,置水浴中,加热5 分钟,冷却,加硫酸2ml ,煮沸1 分钟,即发生醋酸乙酯的香气。 (2) 本品的红外光吸收图谱应与对照的图谱(光谱集545图)一致。 【检查】 杂质吸收度 取本品,精密称定,加无水乙醇溶解并定量稀释制成每1ml中约含10μg 的溶液,照分光光度法(附录Ⅳ A),在287nm的波长处有最大吸收,在240nm与287nm波长处的吸收度比值不得大于0.17。 其他甾体 取本品适量,精密称定,以无水乙醇为溶剂,配制成每1ml 含2mg 的溶液(1) 与每1ml 含0.04mg的溶液(2) 。用含量测定项下的方法和溶液,取10μl 注入液相色谱仪,调整仪器灵敏度,使主成分峰高度达记录仪的满量程。再分别取溶液(1) 和(2) 各10μl,进样。记录色谱图至主成分峰保留时间的2 倍。溶液(1) 显示的杂质峰数不得超过4 个,各杂质峰面积及其总和分别不得大于溶液(2) 主峰面积的1/2 和3/4 。干燥失重 取本品,在105 ℃干燥至恒重,减失重量不得过0.5%(附录Ⅷ L)。【含量测定】 照高效液相色谱法(附录Ⅴ D)测定。 色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;甲醇-水(70:30) 为流动相;检测波长为288nm 。理论板数按醋酸甲地孕酮峰计算应不低于1000,醋酸甲地孕酮峰和内标物质峰的分离度应符合要求。 内标溶液的制备 取炔雌醇约50mg,精密称定,置10ml量瓶中,以甲醇溶解并稀释至刻度,摇匀,即得。 测定法 取醋酸甲地孕酮对照品约20mg,精密称定,置25ml量瓶中,以甲醇溶解并稀释至刻度,摇匀;精密量取该溶液与内标溶液各2ml ,置10ml量瓶中,以甲醇稀释至刻度,摇匀,取10μl 注入液相色谱仪,记录色谱图;另取本品适量,同法测定。按内标法以峰面积计算,即得。 【类别】 孕激素类药。 【贮藏】 遮光,密封保存。 【制剂】 醋酸甲地孕酮片
[B][center]药物中杂质的来源及杂质限量检查[/center] [/B]药物只有合格品与不合格品;一般化学试剂分为4个等级(基准试剂、优级纯、分析纯、化学纯) [B]药物中一般杂质检查 [/B][B]氯化物为一指示性杂质。[/B] 通过对氯化物的控制,可同时控制与氯化物结合的一些阳离子以及某些同时生成的副产物。可从氯化物检查结果显示药物的纯度,间接考核生产、贮藏过程是否正常。 1. 原理 药物中微量的氯化物在硝酸酸性条件下与硝酸银反应,生成氯化银的胶体微粒而显白色浑浊,与一定量的标准氯化钠溶液在相同条件下产生的氯化银浑浊程度比较,判定供试品中氯化物是否符合限量规定。 Ag+ + Cl- → AgCl ↓ [B]硫酸盐检查法 [/B] 1. 原理 药物中微量的硫酸盐在稀盐酸酸性条件下与氯化钡反应,生成硫酸钡的微粒而显白色浑浊,与一定量的标准硫酸钾溶液在相同条件下产生的硫酸钡浑浊程度比较,判定供试品中硫酸盐是否符合限量规定。 [B]铁盐检查法 [/B]硫氰酸盐法 巯基醋酸法 砷盐检查法 1. 古蔡氏法 1. 原理 金属锌与酸作用产生新生态的氢,与药物中微量砷盐反应生成具挥发性的砷化氢,遇溴化汞试纸产生黄色至棕色的砷斑,与同条件下一定量标准砷溶液所生成的砷比较斑,判断砷盐的含量。 [B]硒、氟及硫化物检查法 [/B]1. 氧瓶燃烧法 适用于以共价键结合的卤素、硫、硒的有机药物。 本法系将有机药物防入充满氧气的密闭燃烧瓶中进行燃烧,将燃烧所产生的欲测组分吸收于适当的吸收液中,然后根据欲测组分的性质,选用合适的分析方法进行鉴别、检查或含量测定。 [B]注意事项及讨论 [/B]1. 根据被燃烧分解的样品量选用适宜大小的燃烧瓶。 2. 测定氟化物时应改用石英燃烧瓶。 1. 硒检查法 (1). 操作方法 样品与对照品液,调节Ph2.0±0.2,加盐酸羟胺,二氨基萘,比色。 [B]硫化物检查法 [/B] 方法同砷盐检查第一法,不装醋酸铅棉花,以醋酸铅试纸代替溴化汞试纸。 标准液取1ml 5/ml [B]澄清度检查法 [/B]将一定浓度的供试品溶液与浊度标准液分别置于配对的比浊用玻璃管,同置黑色背景上,在漫射光下观察。浊度标准液 硫酸肼与乌洛托品溶液混合分五个等级,未超过0.5等级即为澄清。BP98规定未超过1等级即为澄清。 [B]溶液颜色检查法 [/B]CHP2000 [B]1. 比色法[/B] 色调标准贮备液 黄色液 重铬酸钾液(BP98用氯化铁) 红色液 氯化钴液 蓝色液 硫酸铜液 配成各种色调色号标准比色液共50种。 [B]2. 分光光度法 [/B] [B]易碳化物检查法 [/B]检查药物中含有的遇硫酸易碳化或易氧化而呈色的有机杂质。 对照品液 样品液 加硫酸5后,加供试品。 [B]炽灼残渣检查法[/B] 取供试品1.0~2.0g或个药品项下规定的重量,置已炽灼至恒重的坩埚中,精密称定,缓缓炽灼至完全碳化,放冷至室温;除另有规定外,加硫酸使湿润,低温加热至硫酸蒸气除尽后,在700~800炽灼使完全灰化,移至干燥器内,放冷至室温,精密称定,再在700~800炽灼至恒重,即得。残渣限量一般为0.1~0.2% 一般应使炽灼残渣量为1~2mg 若需将炽灼残渣留作重金属检查时,炽灼温度必须控制在500~600。 [B]干燥失重测定 [/B]1. 常压恒温干燥法 2. 干燥剂干燥法 3. 减压干燥法 [B]水分测定法 [/B][B]费休氏法 [/B] 本法是根据碘和二氧化硫在吡啶和甲醇溶液中能与水起定量反应的原理以测定水分。 [B]甲苯法[/B] 在加热状态下,甲苯夹带着水分蒸出,收集蒸出的水分测定。 [B]药物中特殊杂质检查 [/B] [B]一、物理法 [/B] [B]二、化学反应法 [/B](一)容量分析法 (二)重量分析法 (三)比色法和比浊法 [B]三、色谱法 [/B]1.纸色谱法 薄层色谱法 TLC是药典中最常用的特殊杂质限量检查方法。 1.在一定供试品及检查条件下,不允许有杂质斑点存在 2.以待测杂质对照品检测 3.将供试品稀释到适当浓度作为杂质对照品溶液 4.选用质量符合规定的与供试品相同的药物作为杂质对照品 [B]高效液相色谱法 [/B] [B][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法 [/B] 1.面积归一化法 2.主成分自身对照法 3.内标法测定 4.内标法加校正因子法 5.外标法 有机溶剂残留量测定法 [B]分光光度法 紫外分光光度法 比色法 [url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]分光光度法[/B]
新手第一次做液相,测定醋酸含量,流动相甲醇:纯水20:80,检测波长210,为什么基线往上飘,还有那么多杂峰呢,这是标准品,按理没那么多峰呐!是我用的流动相不对吗,求各位大神指点!
首先制备杂质标准储备液ABCDE,然后取药品标准品约12.0mg,精密称定,转移至100ml容量瓶。加入流动相约50ml,超声溶解。将A、B、C、D、E溶液各转移3.0ml(用大容量吸管转移)至同一100ml容量瓶,加流动相稀释至刻度。该溶液的英文名称是impurity composite standard solution with drug大家觉得是翻译成含药品的杂质标准液好,还是翻译成含杂质的药品标准液好?谢谢!
替诺福韦杂质是一种化学物质,它是替诺福韦的同分异构体或相关化合物。替诺福韦是一种核苷酸逆转录酶抑制剂,用于治疗HIV和乙型肝炎。COTO标准品是一种高纯度的标准物质,用于测定替诺福韦及其杂质的纯度、含量和化学性质。通过与COTO标准品进行对比和分析,可以确定替诺福韦及其杂质的结构、组成和含量,从而保证替诺福韦的质量和安全性。在药物研发和生产过程中,COTO标准品的使用非常重要。它可以提供可靠的参照物,用于质量控制、药物分析和化学计量学研究。通过使用COTO标准品,可以确保替诺福韦及其杂质的准确性和可靠性,为药物的安全性和有效性提供保障。总的来说,COTO标准品在替诺福韦杂质的研究和控制中具有重要作用。通过使用COTO标准品,可以更好地了解替诺福韦及其杂质的性质和含量,从而确保药物的安全和有效性。同时,也需要加强生产过程中的管理和监督,加强质量标准和监管措施的执行力度,确保药物质量和安全。
如题,俺第一次测盐酸左氧氟沙星,做有关物质时杂质A与左氧保留时间完全重叠,排除了乙酸铵、高氯酸钠等试剂滴原因,实在没辙咧,请教大虾帮忙。盐酸左氧氟沙星有关物质测定方法(来源:中国药典2010年版第一增补本): 有关物质 取本品,精密称定,加0.lmol/L盐酸溶液溶解并定量稀释制成每1ml中约含1.2mg的溶液,作为供试品溶液,精密量取适量,用0.1mol/L盐酸溶液定量稀释制成每1ml中含2.4ug的溶液,作为对照溶液。另精密称取杂质A对照品约18mg,置100ml量瓶中,加6mol/L氨溶液1ml与水适量使溶解,用水稀释至刻度,摇匀,精密量取2ml,置100ml量瓶中,用水稀释至刻度,摇匀,作为杂质A对照品溶液。照高效液相色谱法(附录V D)测定,用十八烷基硅烷键合硅胶为填充剂;以醋酸铵高氯酸钠溶液(取醋酸铵4.0g和高氯酸钠7.0g,加水1300ml使溶解,用磷酸调节pH值至2.2)-乙腈(85 :15)为流动相A,乙腈为流动相B;按下表进行线性梯度洗脱。柱温为40°C;流速为每分钟1ml。称取左氧氟沙星对照品、环丙沙星对照品和杂质E对照品各适量,加0.1mol/L盐酸溶液溶解并稀释制成每1ml中约含左氧氟沙星1.2mg、环丙沙星和杂质E各6ug的混合溶液,取10ul注人液相色谱仪,以294nm为检测波长,记录色谱图,左氧氟沙星峰的保留时间约为15分钟。左氧氟沙星峰与杂质E峰和左氧氟沙星峰与环丙沙星峰的分离度应分别大于2.0与2.5。量取对照溶液10ul注人液相色谱仪,以294mn为检测波长,调节检测灵敏度,使主成分色谱峰的峰高约为满量程的20%。精密量取供试品溶液、对照溶液和杂质A对照品溶液各10ul,分别注人液相色谱仪,以294nm和238nm为检测波长,记录色谱图。供试品溶液色谱图中如有杂质峰,杂质A(238nm检测)按外标法以峰面积计算,不得过0.3%。其他单个杂质(294nm检测)峰面积不得大于对照溶液主峰面积(0.2%),其他各杂质(294nm检测)峰面积的和不得大于对照溶液主峰面积的2.5倍(0.5%)。供试品溶液色谱图中任何小于对照溶液主峰面积0.1倍的峰可忽略不计。时间(分钟) 流动相A(%) 流动相B(%) 0 100 0 18 100 0 25 70 30 39 70 30 40 100 0 50 100 0
本人急需醋酸钡的标准!谢谢!
求助冰醋酸中硫酸盐检测水浴蒸干时加入1ML碳酸钠的作用是什么?谢谢
标准品是异黄酮的4种组分,用的流动相是甲醇:水:醋酸=45:54:1,第一和第二种组分的峰型很好,但是第三和第四种组分的峰总是与杂质峰分不开。我也调整了流动相的比例,进样量也调整过了,但是问题依然存在。(我用的液相色谱仪比较老,只能进行等梯度洗脱)。我是新手,对液相色谱了解的也不是很多,现在很着急啊,连标准曲线都做不好。更别说将来要测量样品中的含量了。
哪位同行有《醋酸钴》的国家标准能否提供一下,急用?非常感谢,我在网上没找到。

头孢西丁钠有关杂质







 我要推广仪器
我要推广仪器
 下载APP
下载APP