测定乙酸乙酯残留,溶剂为二甲基亚砜,测定精密度,发现有一些溶剂峰没有出现,而有的溶剂峰却出现了。请教大家是怎么回事,应该主溶剂的峰都是应该出现的啊,而且很大。溶剂峰和乙酸乙酯峰分离度很好。乙酸乙酯出峰在10分钟,二甲基亚砜出峰在17分钟。
异氰基乙酸乙酯和N,N-二甲基苯胺是否可以用[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]检测?有没有方法条件呢?
苯乙酸是医药、农药、香料等有机合成的中间体。在医药工业中用于青霉素、地巴唑等药物的生产。苯乙酸经氯化、酯化得到α-氯代苯乙酸乙酯,用于稻丰散和乙基稻丰散的生产,这两种农药是广谱性有机磷杀虫剂。苯乙酸本身也是农药植物生长激素。苯乙酸广泛存在于葡萄、草莓、可可、绿茶、蜂蜜等中。苯乙酸在低浓度时具有甜蜂蜜味,在1ppm以下仍具有甜味,是一种重要的香料成分。苯乙酸还具有很强的杀菌作用。
如题,在2.9min时乙酸乙酯出峰,在17min左右N,N-二甲基乙酰胺出峰,出峰时间为10min,峰形特别不好,四分之一圆形,我用的是岛津-2010,进样口和检测器温度均为200度,柱温为70度,分流比为10,急求!!!谢谢各位前辈!!
如题,在合成中有一步反应是用邻羟基苯乙酸制备其二钠盐,其中产物中可能含有的成分有邻羟基苯乙酸、邻羟基苯乙酸的一钠盐、二钠盐,请问如何建立检测方法将其分离呢?谢谢! 试过液相的方法,但是分不开,也试过双相滴定,但是里面还有过量的NaOH,影响结果,也试过用酚羟基的显色反应,但这个又太灵敏了,无法定量。请大家指导一下吧。
[color=#444444]甲酰基苯乙酸甲酯的色谱含量测试中。由于甲酰基苯乙酸甲酯会有醇酮互变性质,在液相色谱(乙腈:水=50:50)下出现三个峰。但是在LC——MS下从第一个峰开始到第三个峰这之间所有的时间都出现了M+1峰(包括峰之间的)。谁能告诉我这是怎么回事。[/color][color=#444444]有哪个大侠做过甲酰基苯乙酸甲酯的含量测试的,求指导。[/color]
求助各位同行:苯乙酸的国标或行标(HGB3444-62)全文.多谢了.我的邮箱:zjp9933@163.com
[color=#d40a00][size=2]维权声明:本文为[font=Times New Roman]11093661[/font]原创作品,本作者与仪器信息网是该作品合法使用者,该作品暂不对外授权转载。其他任何网站、组织、单位或个人等将该作品在本站以外的任何媒体任何形式出现均属侵权违法行为,我们将追究法律责任。[/size][/color] 青霉素发酵过程中利用液相分析对发酵液中的苯乙酸,效价检测存在着很是矛盾的主体,那就是发酵过程中的效价检测是批量检测,在检测过程中青霉素效价有高有低,这样就不利于药典中规定的标准品与待测品的含量大致相同的规定,这就不可能每做一个样品就做一个标准,这样不实际也不利于节约成本的。那么就需要我们做一个线性范围来使在实际检测的过程中的效价范围在线性范围类,这样就有利于测样的准确性。本人通过长期反复工作实践发现在青霉素发酵过程中发酵液的稀释倍数在100倍以上时,出现了这样一个情况:在检测效价的过程中对苯乙酸检测的结果很不稳定,特别是长期工作的液相更是如此,本人在工作中试验发现原因主要是基线的漂移造成了这个现象。而检测苯乙酸对生产发酵中的地位相当重要,苯乙酸是青霉素发酵过程中的主要原材料之一,而苯乙酸的多少又决定了发酵水平高低。所以说苯乙酸的检测也同样重要。 而效价,苯乙酸是同时检测出来的,如果稀释倍数大于100后解决了效价检测的准确性得到了提高,但是苯乙酸的检测准确性也就低了。所以这个矛盾主体也就出现了。本人在长期的检测中实践发现如果分开来检测,也就是两次检测,而对于两次检测过程中的效价与苯乙酸两种物质稀释倍数采取不同的稀释倍数这样有利于检测结果的准确性。或者对苯乙酸检测采取[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]检测同时稀释倍数根据苯乙酸残量的多少采取不同的稀释倍数这样有利于检测结果的准确性,重现性。而根据本人了解某特大型青霉素发酵公司也就是只是对效价采取的稀释倍数的不同,这样提高了效价的准确性而导致了苯乙酸残量的检测的准确性也就降低了,导致发酵水平无法与成都某特大型青霉素发酵公司的水平相比较。这就说明了苯乙酸检测尤为重于效价检测。而分两次检测或气,液相同时检测这样不利于成本降低,本人通过长期的摸索,比较发现利用苯乙酸的多少来决定稀释倍数后,再根据效价的高低采取不同的标准品的含量,这样就利于检测的准确性与成本的降低——相当于分段检测。 本人事先声明这个纯属个人自己摸索试验得出的结论,在实际生产,检测中虽然得到了应用,但由于自己的试验结果,水平有限所以具体过程没有完全介绍,十分抱歉。
4-氯乙酰乙酸乙酯的检测标准及检测方法N,N-二乙基乙二胺、N,N-二甲基乙二胺、N-乙基乙二胺系列产品,质量按Q/320801GHD001-2004标准执行、含量min.99.%
目前所做项目需要液相检测联苯乙腈(原料)和联苯乙酸(产物),但两者出峰时间一致,已尝试多种方法进行混合样分离,但始终只有一个尖峰显示,没有任何分离趋势,目前试过的方法有:甲醇:0.05%磷酸水=70:30(C-18长柱);乙腈:水=70:30;乙腈:0.1%氨水水溶液=70:30(NX-C18);甲醇:0.1%醋酸水=55:45(C18短柱)。求助是否有可行的方法能够较好的分离两种物质。谢谢。
[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#333333]4-羟基苯乙酸酯[/color][/size][/font],[font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]CAS No.:[b]58556-55-1[/b][/color][/size][/font][font=Helvetica Neue,Helvetica,PingFang SC,Tahoma,Arial,sans-serif][size=14px][color=#999999]分子式:C[sub]1[/sub][sub]0[/sub]H[sub]1[/sub][sub]2[/sub]O[sub]3[/sub][/color][/size][/font]
哪位高人有甲苯、苯、甲醇、乙酸乙酯、N-甲基吡咯烷酮的检测标准,望分享,不胜感激!
我用的是据二甲基硅氧烷的柱子,30m*0.25mm*0.25um,溶剂是乙酸乙酯,溶质是苯,两者出峰时间太接近,分离效果很不好进样口:200度检测器:240度程序升温:35度恒温15min各位大大有什么好的方法呢?
请问从哪儿能查到海水中下面的杂质含量测定的方法:甲醇氯化石蜡-52乙二醇二乙二醇二甲基甲酰胺苯乙烯丙酮冰醋酸氯仿甲苯二甲苯1,4-丁二醇苯酚环氧乙烷
食品中二甲基黄的测定解决方案二甲基黄属于亲脂性偶氮染料,用作显色剂可测定硒、钌、碘、砷、铜和溴酸根等。工业上常用于油漆、鞋油和纺织品等的染色。因具神经及生殖毒性,长期摄取二甲基黄会增加罹患肝癌、肺癌、膀胱癌风险,国际癌症研究署(IARC)已将其列为2B等级的致癌物,禁止作为食品添加剂使用。但是,我国台湾地区不断报道出在豆干中检出了二甲基黄。目前二甲基黄的测定方法主要有气相色谱串联质谱法,固相萃取-超高效液相串联质谱。我们采用的是固相萃取-超高效液相串联质谱。方法优势:迪马科技开发的《食品中二甲基黄的测定》采用固相萃取-超高效液相串联质谱测定食品中的二甲基黄,以乙酸乙酯和水为提取液,采用ProElutDMY固相萃取柱净化样品,通过UPLC检测;本方案前处理步骤简便、操作简单、净化效果好,重现性好,回收率高。以下为详细解决方案,敬请参考!食品中二甲基黄的测定1、适用范围 适用于豆干、糕点和饼干中二甲基黄的检测,方法检出限是0.03μg/kg,定量限是0.1 μg/kg。2、提取取1.0 g样品(易乳化的样品需加1.0 g氯化钠),加入2 mL水,涡旋混匀,加入5 mL乙酸乙酯,振荡5 min,6000 rpm下离心2 min,精密量取2.5 mL上清液待净化。3、净化——ProElut DMY 3 mL(Cat.#65914) a活 化:3 mL乙酸乙酯活化;b上 样:c 淋 洗:加入待净化液,弃去流出液;加入3 mL乙酸乙酯,弃去流出液(推干小柱);d洗 脱:加入4 mL10%氨水甲醇,收集流出液;e重新溶解:将流出液在50 ℃下氮吹至干,用流动相定容至1 mL,过0.22 μm微孔滤膜,供LC-MS分析。4、色谱条件4.1UPLC 条件:色谱柱:Endeavosil C18,100× 2.1 mm,1.8 μm (Cat#:87003)流 速:0.2 mL/min进样量:5 μL柱 温:35 ℃流动相: A:0.1%甲酸水 B:乙腈 A:B=20:804.2质谱条件:电离模式:ESI 扫描方式:正离子扫描检测方式:多反应监测 电喷雾电压:5500 V雾化气压力:50 psi 辅助气压力:50 psi气帘气压力:20 psi 离子源温度:500 ℃定性离子对、定量离子对、碰撞气能量及去簇电压见下表 目标物 定性离子对 定量离子对 碰撞气能量/eV 去簇电压/ V (m/z) (m/z) (母离子/子离子) (母离子/子离子) 二甲基黄 226.3/77.2 226.2/77.2 30 74 226.3/134.1 30 5、添加回收结果食品中二甲基黄的LC-MS检测添加回收结果 基质 添加水平(μg/kg) 回收率(%) 豆干 1.0 100.08 糕点 1.0 90.75 饼干 1.0 108.63 http://ng1.17img.cn/bbsfiles/images/2016/02/201602021751_584170_708_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602021752_584171_708_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602021752_584172_708_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/02/201602021752_584173_708_3.png食品中二甲基黄的测定相关产品信息: 货号 名称 规格 样品前处理 65914 ProElut DMY 3 mL, 50/pk 244358 12管防交叉污染真空SPE萃取装置 12位 4803 1,3,6mL柱管通用连接器 15/pk 4806 考克(控制流量) [/
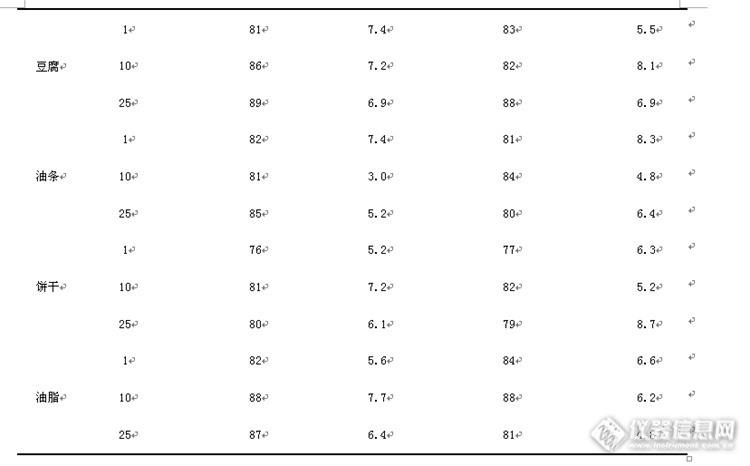
2014年底台湾惊爆二甲基黄食品安全事件,不法商贩将非食用色素二甲基黄添加到豆制品中着色牟利。二甲基黄及其同系物二乙基黄属于亲脂偶氮性染料,这种偶氮类物质多含有R-N=N-R键和其他芳香环或其衍生物的结构,被人体食用后易在肠道还原或分解为易致癌的芳香胺类,在此次台湾食品安全事件发生之前,偶氮染料如苏丹红、甲苯胺红、对位红等早已被禁止添加到食品中。除了豆制品外,粮食、油脂、油炸食品及饼干糕点都有可能为了追求色泽和降低成本而在生产过程中非法添加这两种偶氮染料。因此,如何在各类食品中有效、准确地检出这类非食用色素也成为当前的食品安全热点之一。本文尝试建立气相色谱质谱联用法对大米、豆腐、油条、饼干以及油脂中的二甲基黄和二乙基黄进行检测,在实现化合物有效分离的基础上,提高检测效率,为食品安全风险监测提供有效技术支撑。1 材料与方法1.1材料与试剂二甲基黄(≥98.5%,Dr.EhrenstorferGmbH)、二乙基黄(≥98.5%,Dr.EhrenstorferGmbH)、乙腈(色谱纯,Merck公司)、氯化钠(分析纯,国药集团化学试剂有限公司)、实验用水超纯水1.2 仪器与设备气相色谱-质谱联用仪:GCMS-QP2010, 日本岛津公司离心机:Centrifuge 5804R,德国Eppendorf公司超声波清洗器:KQ-500B型,昆山市超声仪器有限公司旋转蒸发仪:RE-2000A,上海亚荣生化仪器厂分析天平:BS224s,北京赛多利斯仪器系统有限公司涡混振荡仪:CM-1000,东京理化器械株式会社有油基质玻璃萃取管:上海安谱科学仪器有限公司1.3 方法1.3.1 色谱条件色谱柱:HP-5 MS,色谱条件:柱温: 40 ℃用于1分钟,30 ℃ /min升至180 ℃ (保持 3min),5 ℃ /min升至250 ℃,保持6min进样口:220 ℃分流方式:不分流1.3.2 质谱条件离子源为电子轰击离子(EI)源,电子轰击能量为70eV,离子源温度为230℃,四极杆温度为150℃,分别采用全扫描SCAN和选择离子SIM模式,溶剂延迟时间为5min。二甲基黄选择离子:77,105,120,225,二乙基黄选择离子:253,238,148,133。1.3.3 样品前处理大米、豆腐:称取4g,视含水量酌情加入少量去离子水后静置10min,加入2-3g NaCl后以10mL1%醋酸的乙腈提取并超声20min。振荡10min后以2500r/min离心5min。提取上清液,重复一次提取过程后收集上清液于45℃下浓缩至近干,并以1mL乙腈定容后上机。植物油:称取0.5g油脂,将其放入有油基质玻璃萃取管中,加入2mL 1%醋酸的乙腈涡混振荡2min后离心,将上清液上机。油条、饼干:称取4g样品,视含水量酌情加入少量去离子水后静置10min,加入少量NaCl后以10mL1%醋酸的乙腈提取并超声20min。振荡10min后以2500r/min离心5min。提取上清液,重复一次提取过程后收集上清液于45℃下浓缩至近干,以乙腈定容至2mL,转移至有油基质玻璃萃取管中,涡混2min后离心取上清液上机。2 结果与分析2.1 样品提取溶剂的选择在提取过程中,提取溶剂的选择对二甲基黄、二乙基黄的回收率有很大影响。根据二甲基黄的油溶性,分别选择丙酮、乙酸乙酯和乙腈为提取溶剂,通过比较回收率考察提取溶剂的合适程度。由下图可得,在样品含水的情况下,通常不使用与水混溶的提取溶剂,提取液中会含有大量水分,而影响浓缩效果。乙酸乙酯和丙酮的提取效率较好,但其易提取样品中的大分子物质,提取出的杂质较多,且丙酮不易与水分开,不易用盐析出其中的水分。乙酸乙酯在提取油性样品时,将一些非极性亲脂性干扰物质同时提取出来,杂质较多。乙腈不溶于油,能沉淀蛋白质,且提取的脂肪少,与样品混合匀浆后,虽然提取液中可能有水分,但较易用盐析出。而加入1%醋酸的乙腈其回收率高,且稳定。这可能是由于二甲基黄、二乙基黄是酸性化合物,低pH的环境可使得它不会发生离解和溶剂化作用,保持其稳定性。故本实验采用1%醋酸的乙腈作为提取溶剂。http://ng1.17img.cn/bbsfiles/images/2016/12/201612281034_01_2238288_3.png2.2 净化方法的选择二甲基黄和二乙基黄为阴离子酸性化合物,宜用反相SPE小柱对其进行萃取。当样品为大米及豆腐时,其基质简单,由于在前处理过程中每多增一步骤即有可能伴随目标物损失,故应在保证回收率的前提下尽量简化净化步骤。实验中加入少量去离子水后可活化分子状态,便于溶剂与微细试样反复接触萃取。加入NaCl促进两相分配,有效降低待测物对水相的亲和力。对于含油量较多的饼干、油条及油脂,净化的重点集中在油脂的去除。我们分别对低温冷冻法、PSA基质分散固相萃取以及氨基小柱固相萃取三种净化模式进行考察。结果表明,经过氨基小柱的净化效果略差,低温冷冻法和PSA基质分散固相萃取的净化效果相似,但耗时长,不利于风险监测时效性的提高。因此净化方式选择PSA基质分散固相萃取,在实验中我们选用上海安谱科学仪器有限公司的有油基质玻璃萃取管。这种萃取管最初用于邻苯二甲酸酯类的检测,除油效果较好。2.3 色谱分离条件选择根据文献,采用HP-5MS作为分离色谱柱。由于二甲基黄出峰较晚,优化仪器条件时将前面的升温速率提高,并放缓第二段升温速率。进行样品测定时,如满足以下条件则判断样品为阳性结果:1、色谱峰的保留时间与标准样品色谱峰的保留时间一致,且偏差在±2.5%之内,2、所选择的监测离子均出现,3、离子丰度比符合下表要求。http://ng1.17img.cn/bbsfiles/images/2016/12/201612281036_01_2238288_3.png在提取过程中,提取溶剂的选择对二甲基黄、二乙基黄的回收率有很大影响。根据二甲基黄的油溶性,分别选择丙酮、乙酸乙酯和乙腈为提取溶剂,通过比较回收率考察提取溶剂的合适程度。由下图可得,在样品含水的情况下,通常不使用与水混溶的提取溶剂,提取液中会含有大量水分,而影响浓缩效果。乙酸乙酯和丙酮的提取效率较好,但其易提取样品中的大分子物质,提取出的杂质较多,且丙酮不易与水分开,不易用盐析出其中的水分。乙酸乙酯在提取油性样品时,将一些非极性亲脂性干扰物质同时提取出来,杂质较多。乙腈不溶于油,能沉淀蛋白质,且提取的脂肪少,与样品混合匀浆后,虽然提取液中可能有水分,但较易用盐析出。而加入1%醋酸的乙腈其回收率高,且稳定。这可能是由于二甲基黄、二乙基黄是酸性化合物,低pH的环境可使得它不会发生离解和溶剂化作用,保持其稳定性。故本实验采用1%醋酸的乙腈作为提取溶剂。得到的标准物质及加标样品的TIC图及SIM图如下所示:http://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_01_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_02_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_03_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_04_2238288_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/12/201612281042_05_2238288_3.png2.4 线性范围和检出限以2种色素的质量浓度为横坐标,色谱峰面积为纵坐标绘制标准工作曲线,二甲基黄的线性方程为y=1.257*105-5.579*104,二乙基黄的线性方程为y=1.444*105-5.651*104。结果表明在0.125-25ug/mL范围内,标准曲线线性关系良好,相关系数均大于0.99。以3倍信噪比计算检出限,二甲基黄的检出限为0.002mg/kg,二乙基黄的检出限为0.001mg/kg。2.5 回收率和精密度由于二甲基黄及二乙基黄浓度低于1ug/mL时即接近无色,实验中称样量为2-5g,故将加标浓度设为25mg/kg,10mg/kg和1mg/kg。加标方式:由于二甲基黄与二乙基黄是脂溶性物质,采用乙醇为油性模拟介质。将二甲基黄与二乙基黄分别溶于乙醇后,将已知浓度的溶液浸泡于大米和豆腐制品,超声并过夜。油脂类则直接称量一定质量的标准物质并超声溶于油脂。由下表可见,各组分测定结果的相对标准偏差在2.8%-10.1%,平均空白加标回收率为74%-93%,满足GB/T 27404-2008的相关要求。http://ng1.17img.cn/bbsfiles/images/2016/12/201612281044_01_2238288_3.pnghttp://ng
请问DMBC Acetate乙酸二甲基苄基原醇酯的质谱图是怎样的?CAS000151-05-3
完全按GB29708-2013的方法处理,衍生化后质谱检不出五氯苯乙酸酯,不知道哪里有问题!请做过这个项目的指点。
[color=#444444]求助一下啊!!!![/color][color=#444444]我做的羟基苯乙酸去送样,结果GC分析人员做出了两个峰,做了两次都是这样。[/color][color=#444444]我的样品应该是纯的,是分析方法不对,还是其它什么原因啊?有没有哪个遇到这个的情况呢?求助哈[/color]
氧化器主要含异丙苯,测定其中所含杂质含量,主要杂质有二甲基苯甲醇,苯乙酮和过氧化氢异丙苯等,选用何种色谱柱?如何选择操作条件,我们要使用安捷伦1200液相色谱仪,
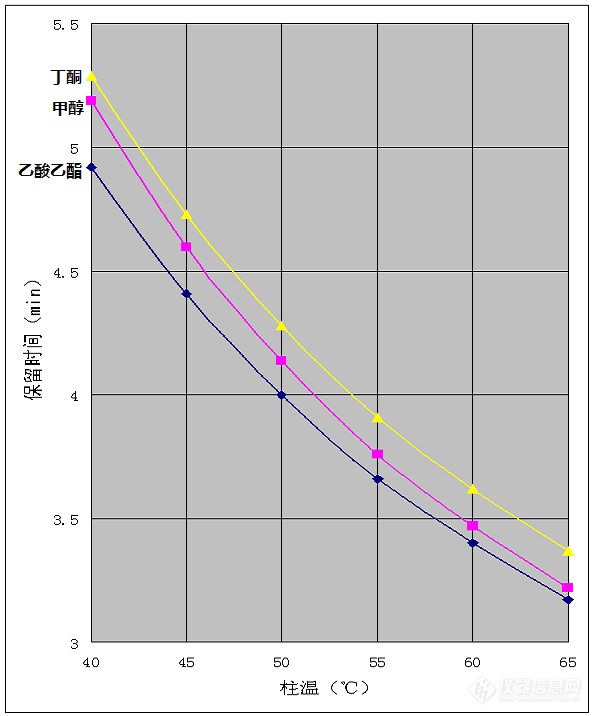
溶剂残留分析是[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的重要应用之一,在药品、食品、包装等领域都是必测的项目。常见溶剂中涉及到的检测目标物经常有乙酸乙酯、甲醇、丁酮,以及二甲苯异构体这几项。最近看到 @m3091333、@p3109800、@Insm_c1196d2b 等多人发帖子讨论相关问题,我从原理上进行了一些解释,但终究纸上谈兵,于是找别的实验室要了这几种试剂,用实践检验了一下。首先,如果二甲苯异构体不要求分离,用624柱可以很容易的解决问题,这里就不讨论了。如果要求乙苯、对二甲苯、间二甲苯、邻二甲苯四种异构体分离,用624柱是无法完成的。因为二甲苯异构体色散力差异非常小,只能靠诱导力的差异分离,不同异构体在强极性柱上的极化率不同,乙苯极化率最低,其次是对二甲苯、间二甲苯,邻二甲苯极化率最大,出峰时间也随极化率的增加而延长。而624柱的极性比较弱,不能产生足够的极化作用,特别是对二甲苯与间二甲苯的极化差异非常小,无法实现分离。这个问题是由分子结构决定的,无论怎么调节色谱条件都不能解决。要想解决只能换强极性柱,常见的就是聚乙二醇柱,包括各种wax柱和FFAP柱等。三氟丙基柱也是强极性的,可以分离二甲苯异构体,但是这种柱很少使用。在聚乙二醇类的色谱柱上,乙酸乙酯、甲醇、丁酮三种目标物分离困难,各种类型的聚乙二醇柱选择性略有差异,但这三种物质都是较为接近的,想要分离是不太容易的。但是这三种物质与聚乙二醇固定相之间的作用力存在本质上的差异,因此通过调整柱温条件是可以分离的。下面三幅图是用60米*0.53mm*1um的INNOWAX柱分离乙酸乙酯、甲醇、丁酮的效果,柱温分别是40℃、50℃、60℃。[img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157168864_5041_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157170984_7926_2204387_3.png!w690x796.jpg[/img][img=,690,796]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022157172914_736_2204387_3.png!w690x796.jpg[/img]图中很明显,柱温低时甲醇与丁酮出峰时间接近分不开,高温时甲醇与乙酸乙酯出峰时间接近分不开,温度适中时三者可以实现分离。虽然未达到基线分离,但分离度都超过1,用来定量是完全可以的。这是找别人借的一根旧柱子,柱效只有4万塔板,如果是新柱子柱效应该能达到七八万塔板,分离度肯定更高,如果是0.32mm口径的柱子分离就更没问题了。要强调的是,能够实现分离的条件并不是完全靠盲目尝试获得的。我们看一看三种目标物的保留时间随柱温的变化就能发现其中的规律,见下图:[img=,594,716]http://ng1.17img.cn/bbsfiles/images/2018/08/201808022156374904_6999_2204387_3.png!w594x716.jpg[/img]图中可以看出,三种目标物的保留时间都是随温度升高而减小的,但是减小的幅度却并不相同。甲醇的保留时间随温度升高而减小的幅度明显大一些。这是因为甲醇具有羟基,与聚乙二醇固定相的相互作用力以氢键为主,氢键的强度随温度升高而迅速减弱。而乙酸乙酯、丁酮与聚乙二醇固定相的作用力都是以诱导力和取向力为主,这种力是由分子偶极矩决定的,受温度的影响要小一些。甲醇峰位置在乙酸乙酯与丁酮之间,温度升高时保留时间都减小,但甲醇减小更多,于是甲醇与乙酸乙酯靠的更近,与丁酮的分离度提高。温度降低时保留时间都增大,但甲醇增大更多,于是甲醇与丁酮靠的更近,与乙酸乙酯的分离度提高。用其他的柱子,如DB-wax或者FFAP时,各组分之间的相对位置会有差别,甚至有时出峰顺序都会变,但是保留时间随温度变化的这种规律仍然是适用的。所以遇到分不开的情况,一定不要盲目的乱试一通,也不用盲目的换柱子,一定要把问题想明白,有针对性的优化条件。最后要强调的是,这里虽然是以溶剂检测为例讨论了如何只用一根柱子就实现分离,但实际样品很复杂,并不是每次都能通过这种优化实现全部分离目的。所以色谱实验室配备多种不同极性的色谱柱是非常重要的。特别是做复杂样品时,即使谱图上看起来分离不错,最好也能用另外一种柱子进行一次验证,以免实际样品中有干扰物共流出,造成假阳性。
做药物的有机溶剂残留,用二甲基亚砜做溶解药品的溶剂,二甲基亚砜本身有好多小杂质峰干扰药物中的待测有机溶剂的测定,有谁用过二甲基亚砜做过溶解样品的溶剂的,在2-5min是不是也有好多小杂质峰干扰测定?换了好几个厂家的二甲基亚砜了,或多或少都有干扰,如何处理?还有什么其它溶剂可用没有,甲醇、甲苯、正丁醇、正己烷做为溶剂药物都不溶解(我测药物中乙醇,乙酸乙酯、二氯甲烷、四氢呋喃、NN-二甲基甲酰胺的残留)
谁告诉我下 巯基乙酸异辛酯 二甲基二氯锡 用GC什么检测器?什么柱子分析啊?
用二甲基亚砜做溶剂溶样,一般用什么类型的柱子不会出现干扰峰啊(测定乙酸乙酯和酒精),各位都说说啊,
2,4-二甲基苯胺和2,6-二甲基苯胺的鉴别2,4-二甲基苯胺和2,6-二甲基苯胺同属于国家强制标准GB18401-2003附录C中所列的还原条件下染料中不允许分解出的23种芳香胺之一,二者又属于同分异构体,沸点和极性都很接近,故在检测过程中很难鉴别。目前,对于两者的分离鉴别主要靠液相色谱来实现,而使用气-质联用仪来鉴别两者还没有很好的方法。而针对有害芳香胺的气相色谱-质谱检测方法,大多采用非极性或极性较弱的色谱柱,如HP-5MS,DB-5MS,DB-35MS,这些色谱柱普遍存在的缺点是对常见的芳香胺异构体不能很好的分离。由于2,4-二甲基苯胺和2,6-二甲基苯胺沸点太接近,单纯依靠两者的沸点差异来实现其分离鉴别是有一定难度的。于是,作者考虑采用中等极性色谱柱DB-17MS(固定相等同于50%苯甲基聚硅氧烷),除了利用2,4-二甲基苯胺和2,6-二甲基苯胺的沸点差异外,再利用中等极性柱对于二者的保留作用差异来研究二者的分离鉴别。通过改善优化色谱条件,作者使用中等极性色谱柱DB-17MS,同时使用三阶程序升温,实现了2,4-二甲基苯胺和2,6-二甲基苯胺的较好分离。1 试验1.1 仪器与试剂气相色谱-质谱联用仪(GC-MS):Agilent 7890A/5975C,美国Agilent公司毛细管柱:DB-17MS柱(30m×0.25mm×0.25μm)叔丁基甲醚 分析纯 国药集团化学试剂有限公司甲醇 色谱纯 美国Fisher公司旋转蒸发仪 上海亚荣生化仪器厂2,4-二甲基苯胺和2,6-二甲基苯胺均为德国Dr.Ehrenstorfer公司。1.2 试样的制备分别称取适量的2,4-二甲基苯胺和2,6-二甲基苯胺,以甲醇为溶剂分别配制适宜浓度的2,4-二甲基苯胺溶液、2,6-二甲基苯胺溶液和2,4-二甲基苯胺和2,6-二甲基苯胺混合溶液。1.3 仪器操作条件色谱柱:DB-17MS 30m×0.25mm×0.25μm;温度:进样口220℃ ;辅助器280℃;离子源230℃ ;四极杆温度:150℃;柱温:40℃保持2分钟,以15℃/分钟升温至[/font
有人有N,N-二甲基对苯二胺纯度测定方法或相关标准吗?谢谢了。
[font=&][size=18px]N,N-二甲基乙酰胺又称乙酰基二甲胺、乙酰二甲胺,简称DMAC,是一种非质子高极性溶剂,有微氨气味,溶解力很强,可溶解的物质范围很广,能与水、芳香族化合物、酯、酮、醇、醚、苯和三氯甲烷等任意混溶,且能使化合物分子活化,因此广泛用作溶剂及催化剂。在溶剂方面作为高沸点、高闪点、热稳定性高、化学性稳定的溶剂,可用于聚丙烯腈的抽丝溶剂、合成树脂及天然树脂、甲酸乙烯酯、乙烯基吡啶等共聚物及芳烃羧酸的溶剂;在催化剂方面可用于尿素加热制氰尿酸、卤代烷与金属氰化物反应制腈、乙炔钠与卤代烷反应制烷基炔、有机卤化物与氰酸盐反应制异氰酸酯等过程。N,N-二甲基乙酰胺还可用作电解溶剂及摄影用成色剂的溶剂、脱油漆剂、有机合成原料、农药及医药原料。从C8馏分中分离苯乙烯的萃取蒸馏溶剂等。[/size][/font]
工作场所空气有毒物质测定 饱和脂肪族酯类化合物 [url=http://www.stdbuy.cn/Standard/StdInfo.aspx?ca=99VLGLHxWzU=]GBZ/T 160.63-2007[/url] 是作废了吧?它被 工作场所空气有毒物质测定 第122部分:甲酸甲酯和甲酸乙酯和工作场所空气有毒物质测定 第126部分:硫酸二甲酯和三甲苯磷酸酯 部分代替。那么 原先 [url=http://www.stdbuy.cn/Standard/StdInfo.aspx?ca=99VLGLHxWzU=]GBZ/T 160.63-2007[/url] 中的乙酸乙酯 乙酸丙酯 现行的标准是什么?
GB 21027-2007胶粘剂中苯、甲苯、二甲苯含量的测定。标准里面说是测苯的话用N,N-二甲基甲酰胺溶剂胶粘剂;测甲苯和二甲苯用乙酸乙酯溶解。可是两种溶剂都溶解不了?各位有做过这个实验吗?是怎么溶解胶粘剂的?
[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]质谱做邻苯二甲酸酯类(二丁酯、二乙基己酯),进溶剂空白(甲醇、乙腈、二氯甲烷、乙酸乙酯,均为色谱纯或农残级),空白均有很高响应,以致标准曲线都无法做,选择离子扫描,149出很大峰,希望各位专家能有解决办法,谢谢!急!







 我要推广仪器
我要推广仪器
 下载APP
下载APP