百灵威权威提供“地沟油”检测标样
&ldquo 地沟油&rdquo 是y个泛指概念,是对各类劣质油的统称,y般包括潲水油、煎炸废油、食品及相关企业产生的废弃油脂等。&ldquo 地沟油&rdquo 对人体的危害j大,长期食用可能会引发癌症。 虽然g家明令禁止将废弃油脂再加工进行使用或者销售,但出于利益驱使,个别不法企业或个人仍冒天下之大不韪,造成每年几百万吨的&ldquo 地沟油&rdquo 流向餐桌,给民众食品安全带来严重威胁。目前,我g还没有专门针对&ldquo 地沟油&rdquo 的检测标准。据了解,北京市食品安全监控中心在筛查了&ldquo 地沟油&rdquo 可能涉及的80多个技术检验项目后,已经找到了包括多环芳烃(PAHs)、胆固醇、电导率和特定基因组成等4类能够排查&ldquo 地沟油&rdquo 的有效指标,初步建立了&ldquo 地沟油&rdquo 检测的指标体系。百灵威作为分析化学l域的引l者,以维护民众的生命安全为己任,整合全球优秀产品资源,提供专业的、品种齐全的检测标样,为&ldquo 地沟油&rdquo 的检测保驾护航!针对性强、价格低廉、具有溯源性纯品、液标等多种规格液标具有多种溶剂、多种浓度产品经过ISO 9001:2000、ISO 17025:1999质量认证产品经过了NIST、NVLAP和EPA认证订购标样附带质检报告、材料安全数据卡■ 混标产品名称:PAHs Solution Mix(多环芳烃混标)产品编号:Z-013-17溶剂:0.2 mg/mL in CH2Cl2 : MeOH(1:1)包装:1 mL组分数量:16种编号CAS英文名称中文名称浓度(mg/mL)156-55-31,2-Benzanthracene苯并(a)蒽0.2283-32-9Acenaphthene二氢苊0.23208-96-8Acenaphthylene苊0.24120-12-7Anthracene蒽0.2550-32-8Benzo(a)pyrene苯并(a)芘0.26205-99-2Benzo(b)fluoranthene苯并(b)荧蒽0.27191-24-2Benzo(g,h,i)perylene苯并(g,h,i)二萘嵌苯0.28207-08-9Benzo(k)fluoranthene苯并(k)荧蒽0.29218-01-9Chrysene屈0.21053-70-3Dibenz(a,h)anthracene二苯并(a,h)蒽0.211206-44-0Fluoranthene荧蒽0.21286-73-7Fluorene芴0.213193-39-5Indeno(1,2,3-cd)pyrene茚并(1,2,3-cd)芘0.21491-20-3Naphthalene萘0.21585-01-8Phenanthrene菲0.216129-00-0Pyrene芘0.2※若需要混标中的具体单标请致电400-666-7788垂询!■ 单标■ 氘代单标CAS英文名称中文名称浓度包装1718-53-21,2-Benz(a)anthracene D12氘代苯并(a)蒽2.0 mg/mL in CH2Cl21 mL15067-26-2Acenaphthene D10氘代苊4.0 mg/mL in CH2Cl21 mL93951-97-4Acenaphthylene D8氘代苊烯10 ng/&mu L10 mL1719-06-8Anthracene D10氘代蒽2.0 mg/mL in CH2Cl21 mL93951-66-7Benzo(g,h,i)perylene D12氘代苯并(g,h,i)苝10 ng/&mu L1 mL1719-03-5Chrysene D12氘代屈4.0 mg/mL in CH2Cl21 mL13250-98-1Dibenzo(a,h)anthracene D14氘代二苯并(a,h)蒽10 ng/&mu L10 mL93951-69-0Fluoranthene D10氘代荧蒽ampule of 50 mg1 EA81103-79-9Fluorene D10氘代芴10 ng/&mu L10 mL1146-65-2Naphthalene D8氘代萘4.0 mg/mL in CH2Cl21 mL1517-22-2Phenanthrene D10氘代菲0.2 mg/mL in CH2Cl21 mL4.0 mg/mL in CH2Cl21 mL1718-52-1Pyrene D10氘代芘0.5 mg/mL in Acetone1 mL※更多氘代单标请致电400-666-7788垂询!■ 氟代单标CAS英文名称中文名称浓度包装17521-01-65-Fluoroacenaphthylene5-氟代苊烯10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL113600-15-09-Fluorobenzo[k]Fluoranthene9-氟代苯并(k)荧蒽10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mLN/A1-Fluorochrysene1-氟代屈10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL36288-22-93-Fluorochrysene3-氟代屈10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL1691-66-33-Fluorofluoranthene3-氟代荧蒽10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL343-43-12-Fluorofluorene2-氟代芴10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL321-38-01-Fluoronaphthalene1-氟代萘0.1 mg/mL in Acetone1 mL523-41-12-Fluorophenanthrene2-氟代菲10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL440-40-43-Fluorophenanthrene3-氟代菲10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL1691-65-21-Fluoropyrene1-氟代芘10 &mu g/mL in Toluene1 mL100 &mu g/mL in Toluene1 mL※更多氟代单标请致电400-666-7788垂询!■ 黄曲霉毒素类、胆固醇CAS英文名称中文名称备注包装1162-65-8Aflatoxin B1黄曲霉毒素 B1定性用对照品5 mgAflatoxin B1 solution黄曲霉毒素 B1 (液标)标样20 &mu g/mL in methanol1 U7220-81-7Aflatoxin B2黄曲霉毒素 B2定性用对照品2 mgAflatoxin B2 solution黄曲霉毒素 B2 (液标)标样3 &mu g/mL in benzene:acetonitrile (98:2)1 U1165-39-5Aflatoxin G1黄曲霉毒素 G1定性用对照品2 mgAflatoxin G1 solution黄曲霉毒素 G1 (液标)标样3 &mu g/mL in benzene:acetonitrile (98:2)1 U7241-98-7Aflatoxin G2黄曲霉毒素 G2定性用对照品1 mgAflatoxin G2 solution黄曲霉毒素 G2 (液标)标样3 &mu g/mL in benzene:acetonitrile (98:2)1 U6795-23-9Aflatoxin M1黄曲霉毒素 M1定性用对照品0.25 mgAflatoxin M1 solution黄曲霉毒素 M1 (液标)标样10 &mu g/mL in acetonitrile1 U6885-57-0Aflatoxin M2黄曲霉毒素 M2定性用对照品0.25 mg57-88-5Cholesterol胆固醇标样0.25 g※更多产品欢迎致电400-666-7788垂询!■ 配套溶剂■ 色谱溶剂高纯度:HPLC分析中无干扰峰低含水量:避免了正相色谱柱的失活低 UV 背景吸收:避免了鬼峰及得出错误的结论优异的批次稳定性:更换批次时无需更改 HPLC 标准方法低挥发、低残留:使用前无需过滤,减少了色谱柱的污染并防止了系统堵塞■ 产品列表(以下产品可提供20 L / 200 L包装)CAS产品编号英文名称中文名称包装67-56-1116481Methanol, 99.9% [HPLC/ACS]甲醇1 L / 4 L982150Methanol, 99.8% [HPLC/PREP]甲醇(制备j)4 L/20 L/200 L75-05-8134752Acetonitrile, 99.9% [HPLC/ACS]乙腈1 L / 4 L925301Acetonitrile, 99.9% [HPLC/PREP]乙腈(制备j)4 L/20 L/200 L110-54-3133516Hexane, 95% [HPLC/ACS]正己烷1 L / 4 L141-78-6300999Ethyl acetate, 99.9% [HPLC/ACS]乙酸乙酯1 L / 4 L67-66-3508435Chloroform, 99.9% [HPLC/ACS]氯仿1 L / 4 L109-99-9990407Tetrahydrofuran, 99.8% [HPLC/ACS]四氢呋喃4 L※更多色谱溶剂欢迎致电400-666-7788垂询!■ 离子对试剂离子对试剂是高效液相色谱专用试剂,y般是将离子性化合物添加到流动相中以促使离子与带电荷分析物形成配对离子,达到可靠的分析效果。百灵威不仅可提供系列化磺酸类(酸性)或铵盐类(碱性)离子对试剂,而且可以根据实验要求,定制从5 g 至1 kg多种包装。CAS产品编号英文名称产品名称包装207605-40-12568821-Pentanesulfonic acid sodium salt monohydrate, 98% [HPLC grade]戊烷磺酸钠y水合物5 g/25 g/100 g/500 g207300-91-22389191-Hexanesulfonic acid sodium salt monohydrate, 98% [HPLC grade]己烷磺酸钠y水合物5 g/25 g/100 g/500 g207300-90-12353851-Heptanesulfonic acid sodium salt monohydrate, 98% [HPLC grade]庚烷磺酸钠y水合物5 g/25 g/100 g/500 g207596-29-01653021-Octanesulfonic acid sodium salt monohydrate, 98% [HPLC grade]辛烷磺酸钠y水合物5 g/25 g/100 g/500 g22767-49-33587891-Pentanesulfonic acid sodium salt,99% [HPLC grade]戊烷磺酸钠5 g/25 g/100 g2832-45-35738321-Hexanesulfonic acid sodium salt monohydrate, 98% [HPLC grade]己烷磺酸钠5 g/25 g/100 g22767-50-61491161-Heptanesulfonic acid sodium salt monohydrate, 98% [HPLC grade]庚烷磺酸钠25 g/100 g5324-84-51945001-Octanesulfonic acid sodium salt, 99.5% [HPLC grade]辛烷磺酸钠5 g/25 g/100 g※更多离子对试剂欢迎致电400-666-7788垂询!■ 配套仪器耗材■ 液相色谱柱高度的柱间重现性高度可控的单分子层形成和封尾技术高选择性,提高了分离效率适合分离酸性、中性和碱性化合物,以及多肽和蛋白等产品编号产品名称适用pH范围特征包装S02001C18液相色谱柱柱长:150× 外径4.6 mm填料直径:5 µ mpH 2-7★ 母体为高纯度(99.999%)硅胶;★ 均y且完全呈球状的硅胶粒径,可以在低压力下使用;★有理想的端基封尾处理,既不会有碱性化合物吸附问题,也不会有酸性化合物吸附问题;★ 即使是在酸性条件下,也有着较高的耐受性。1 PakS02302C18液相色谱柱柱长:250× 外径4.6 mm填料直径:5 µ mpH 2-71 PakS02303C18 WpH液相色谱柱 柱长:150× 外径4.6 mm填料直径:5 µ mpH 1-10★ 保留能力强,与母体成分的分离更容易;★ 均y且完全呈球状的硅胶粒径,使用压力小,给泵带来的负担更小;★ 高惰性,不论酸性化合物还是碱性化合物,都能得到尖锐的峰型;★ 硅胶纯度高,可用于分析金属配合物;★ pH1-10,即使使用强碱性溶离液也能维持高性能。 1 PakS02304C18 WpH液相色谱柱柱长:250× 外径4.6 mm填料直径:5 µ mpH 1-101 Pak※更多液相色谱柱欢迎致电400-666-7788垂询!■ J&K-Abel气相色谱柱高性能:低流失、独特的去活技术高惰性:能得到更尖锐的锋形高选择性:更高的信噪比高的柱间稳定性:提高了分离效率,保证了结果的重现性创新型设计:保证更长的色谱柱使用寿命产品类型:聚硅氧烷色谱柱聚合乙二醇(PEG)色谱柱PLOT色谱柱熔融石英管产品编号型号规格耐受温度S010125-3002AB-1, 30 m × 0.25 mm × 0.25 &mu m-60 to 325/350 19091Z-433S011125-3002AB-1MS, 30 m × 0.25 mm × 0.25 &mu m-60 to 325/350 19091S-933S010525-3002AB-5, 30 m × 0.25 mm × 0.25 &mu m-60 to 325/350 19091J-433S011525-3002AB-5MS, 30 m × 0.25 mm × 0.25 &mu m-60 to 325/350 19091S-433S016125-3002AB-1701, 30 m × 0.25 mm × 0.25 &mu m-20 to 280/300 122-0732S016132-3002AB-1701, 30 m × 0.32 mm × 0.25 &mu m-20 to 280/300 123-0732S016225-3014AB-624, 30 m × 0.25 mm × 1.40 &mu m-20 to 260 122-1334S016253-3030AB-624, 30 m × 0.53 mm × 3.00 &mu m-20 to 260 125-1334S012025-3002AB-INOWAX, 30 m × 0.25 mm × 0.25 &mu m40 to 260/280 19091N-133S018653-3030AB-PLOT Q, 30 m × 0.53 mm × 30.0 &mu m-80 to 280/290 19095P-QO4S011125-3002-G5AB-1MS Builtin-Guard 30 m,0.25 mm,0.25 &mu m with 5 m Guard Column-60 to 325/350※更多气相色谱柱欢迎致电400-666-7788垂询!■ 其它配套仪器耗材产品编号产品名称包装3581025加热磁力搅拌器1台3810025RCT 基本型磁力搅拌器1台1572500磁力搅拌子1PKE03935569手动单道可调式移液枪,1000-5000 µ L1支E02901275瓶口分液器,5-50 mL1个WX-7009-0020-18247 R95 有机蒸气异味防护口罩,120个/箱1箱5982-3236SCX Polymer - Box, 50 x 3 mL tubes, 60 mg50支/盒959741-902Eclipse Plus C18, 2.1 x 50 mm,1.8 µ m, 600 bar1支BR36849100 mL, DURAN, NS 14/23, -stoer1套5182-0714Screw cap vials, clear 100/PK 透明螺口2 mL样品瓶1盒WKLM-2.1微孔滤膜Ф50 0.2 &mu (水)混合纤维素100片/包WKLM-4.1微孔滤膜Ф50 0.2 &mu (有机)尼龙6100片/包RJGL1L-C溶剂过滤器(1 L) 杯300 mL 瓶1000 mL,PTFE滤板1套5982-911012 Port Vacuum Extraction Manifold Assy1套※更多产品欢迎致电400-666-7788垂询!








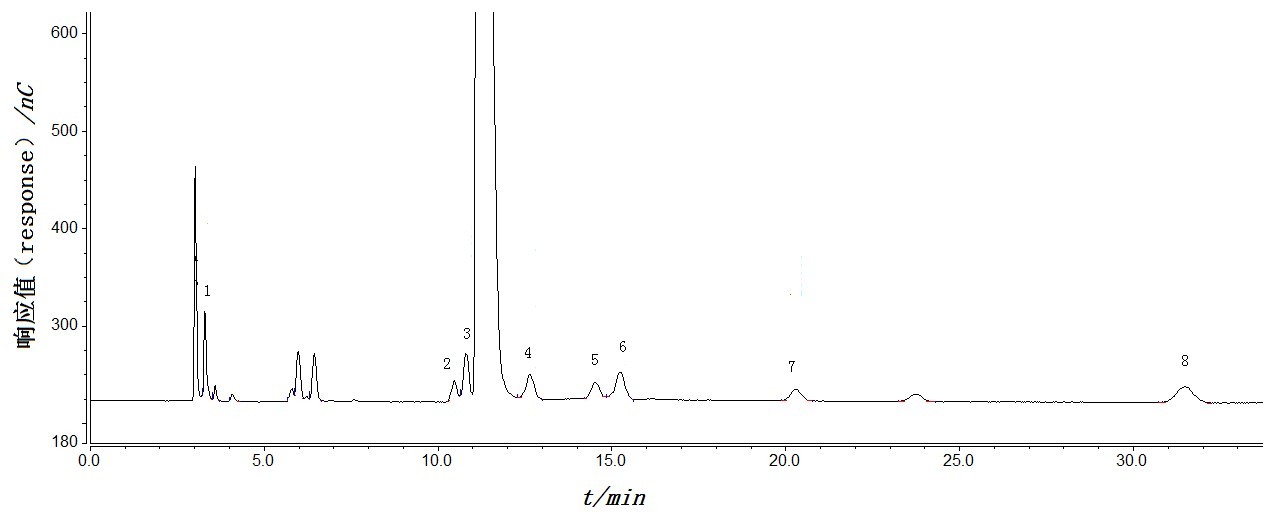





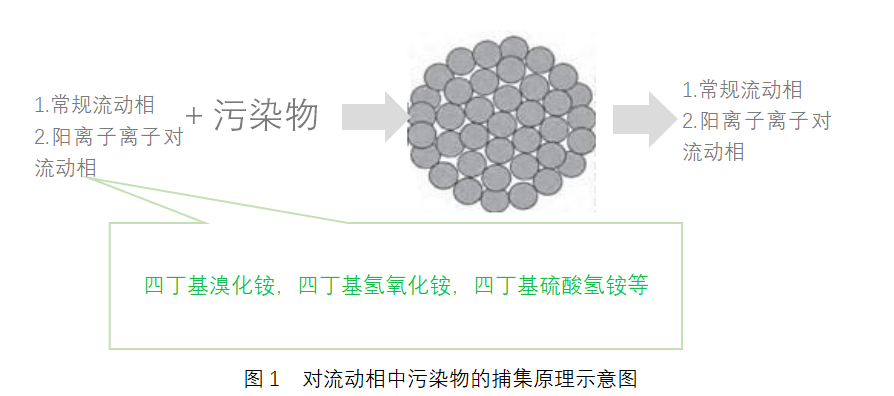










 我要推广仪器
我要推广仪器
 下载APP
下载APP