有没有硫酸安普霉素测含量或效价的方法,谢谢!
(1)求助吉他霉素、硫酸粘菌素兽药典2010版标准,手上有的版友,能不能麻烦把这两个扫描一下或者传真一下也行,需要传真操作的版友麻烦站短联系,我告知传真号码。(2)吉他霉素对照品进液相色谱是只有一个A5峰,还是有多个色谱峰, 对照品的纯度是多少?
有关物质 取西索米星、小诺霉素标准品各适量,精密称定,用流动相制成每1ml中约含西索米星和小诺霉素各25μg、50μg和250μg的溶液作为标准品溶液(1)、(2)、(3)。照庆大霉素C组分项下色谱条件试验,取上述三种溶液各20µl,分别注入液相色谱仪,记录色谱图,计算标准品溶液浓度的对数值与相应的主峰面积对数值的回归方程,相关系数(r)应不小于0.99;另取本品适量,精密称定,用流动相制成每1ml中约含庆大霉素2.5mg的溶液,同法测定,供试品色谱图中如有西索米星、小诺霉素峰,用相应的回归方程计算西索米星、小诺霉素的含量。含西索米星不得过2.0%,小诺霉素不得过3.0%。除硫酸峰外,其他杂质按小诺霉素回归方程计算,单个杂质不得过2.0%,总杂质不得过5.0%西索米星标准品(标签上的) 每毫克相当于548单位小诺霉素标准品(标签上的) 每毫克相当于574单位组分:55.4%(供硫酸庆大霉素C组分测定及硫酸小诺霉素制剂组分测定用)组分(为理论值):84.8%(供硫酸小诺霉素组分测定用)取西索米星、小诺霉素标准品各适量,精密称定,用流动相制成每1ml中约含西索米星和小诺霉素各25μg、50μg和250μg的溶液作为标准品溶液(1)、(2)、(3)。欲配制100ml约含西索米星和小诺霉素各250微克的溶液,应该称取的西索米星标准品和硫酸小诺霉素标准品各多少?

【中文名称】硫酸多粘霉素E;硫酸抗敌素;硫酸粘菌素;硫酸粘杆霉素【英文名称】colistin sulfate;colimycin;polymyxine Multimycine【结构或分子式】 http://ng1.17img.cn/bbsfiles/images/2012/03/201203161918_355269_1855403_3.jpg【毒性LD50(mg/kg)】 饲料添加时常用粗制品,它对大鼠和小鼠的口服LD50均大于12g/kg。【性状】 白色粉末,有吸湿性。【溶解情况】 易溶于水。【用途】 硫酸多粘菌素E对革兰氏阴性菌有强大的抑菌作用。它可治疗志贺氏痢疾杆菌、大肠杆菌、绿浓杆菌、沙门氏杆菌和普通变形杆菌引起的感染。在动物体内不会产生耐药菌株,与其他抗生素不产生交叉耐药。一般都制成预混剂使用。【制备或来源】 本品系1950年小山康夫在日本福岛县分离出的多粘芽孢杆菌变种粘菌素(Bacilluspolmyxa var. cotistinus),在其培养液中提取的多粘菌素E,通常制成硫酸盐使用,它是由A、B、C三组分组成的。【生产单位】略
有关物质 取西索米星、小诺霉素标准品各适量,精密称定,用流动相制成每1ml中约含西索米星和小诺霉素各25µg、50µg和100µg的溶液作为标准品溶液(1)、(2)、(3)。照庆大霉素C组分项下色谱条件试验,取上述三种溶液各20µl,分别注入液相色谱仪,记录色谱图,计算标准品溶液浓度的对数值与相应的主峰面积对数值的回归方程,相关系数(r)应不小于0.99;另取本品适量,精密称定,用流动相制成每1ml中约含庆大霉素2.5mg的溶液,同法测定,供试品色谱图中如有西索米星、小诺霉素峰,用相应的回归方程计算西索米星、小诺霉素的含量。含西索米星不得过2.0%,小诺霉素不得过3.0%。除硫酸峰外,其他杂质按小诺霉素回归方程计算,单个杂质不得过2.0%,总杂质不得过5.0%问题1,每1ml中约含西索米星和小诺霉素各25µg、50µg和100µg的溶液作为标准品溶液(1)、(2)、(3)。如何称量?
请问谁有硫酸链霉素的含量分析方法(非效价法)?谢谢!
谁知道硫酸双氢链霉素的红外啊?我只查到了硫酸链霉素的光谱。大家帮帮忙,谢谢了。
大家好!我是个新手,有谁知道这个问题请联系我!硫酸新霉素的硫酸盐的含量测定原理,怎么计算?
我们在做阿奇霉素的释放度的检测,具体的方法是0.1mol/L的盐酸2小时,然后再加入0.2mol/L的磷酸盐,调节Ph至6.8,45分钟,去溶液适量,滤过,去续滤液1ml,加0.1mol/L的盐酸4ml,加硫酸(75→100)5ml,显色,放冷后在测紫外!我们遇到的问题是,加硫酸后不显色!!后来也确定了是磷酸盐的问题,我想问的是磷酸盐是怎么对显色反应起到干扰作用的,(有问题的磷酸盐是能够把药品完全溶解的),同时还想问的是:加入硫酸显色的原理是怎样的!
成分分析中硫酸法测定棉和聚酯纤维的含量时,标准要求1克样品200ML硫酸,这量比较大,大家有用100ML硫酸做过实验吗?
各位,请问用0.2mol/L的三氟乙酸去测定硫酸庆大霉素的时候,0.2mol/L的三氟乙酸pH0.58左右,而我用的C18柱子最低能耐受pH为1,请问各位0.2mol/L的三氟乙酸需要调节pH吗,如果需要用什么调节
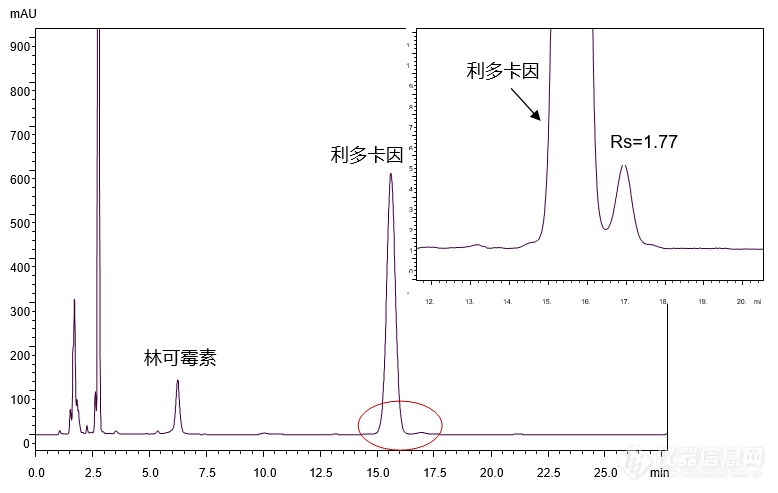
[align=center][b]【国家药品标准】林可霉素利多卡因凝胶的分析[/b][/align][align=center][b][/b][/align][align=right][b]——依据国家药品标准WS-10001-(HD-0140)-2002方法[/b][/align][b]林可霉素利多卡因凝胶[/b]为复方制剂,每克含林可霉素5毫克,利多卡因4毫克。适应症为用于轻度烧伤、创伤及蚊虫叮咬引起的各种皮肤感染。 [img=,193,127]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834522166_2994_2222981_3.gif!w193x127.jpg[/img] [img=,140,64]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260834520028_3541_2222981_3.gif!w140x64.jpg[/img] 林可霉素 利多卡因 Lincomycin Lidocaine M.W.: 406.54 M.W.: 234.34客户提供林可霉素利多卡因凝胶样品,希望本实验室帮忙通过筛选色谱柱及调节分析条件,依据[color=#ff0000][b]国家药品标准WS-10001-(HD-0140)-2002[/b][/color]方法,实现林可霉素利多卡因凝胶样品的良好分析。首先,使用能在纯水条件下稳定使用的高极性色谱柱[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ S5 4.6 mm i.d. × 150 mm[/b][/color],对林可霉素利多卡因凝胶样品进行分析,结果如图1所示,[color=#330099]利多卡因与其峰后杂质之间分离度为1.77[/color]。[align=center][img=,690,437]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858200006_8607_2222981_3.png!w690x437.jpg[/img][/align][align=center]图1 CAPCELL PAK C[sub]18 [/sub]AQ分析所得色谱图[/align]注:峰上标数字为分离度。[img=,528,205]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260858202566_2695_2222981_3.png!w528x205.jpg[/img]为进一步提高利多卡因与其峰后杂质之间的分离度,在原条件基础上将柱温由30℃降低至25℃,并分别使用 CAPCELL PAK C[sub]18[/sub] AQ、CAPCELL PAK C[sub]18[/sub] MG及高含碳量ODS色谱柱SUPERIOREX ODS进行分析,结果如图2所示。[align=center][img=,690,490]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859201516_7229_2222981_3.png!w690x490.jpg[/img][/align][align=center]图2 25℃条件下不同色谱柱分析结果对比[/align]注:峰上标数字为分离度。[img=,637,223]http://ng1.17img.cn/bbsfiles/images/2018/07/201807260859204236_7198_2222981_3.png!w637x223.jpg[/img]如图2所示,在柱温25℃条件下使用三款色谱柱进行分析,其中,[color=#ff0000][b]CAPCELL PAK C[sub]18[/sub] AQ色谱柱分析结果最好,利多卡因与其峰后杂质分离得到最佳分离,分离度为4.23[/b][/color];[color=#330099][b]使用CAPCELL PAK C[sub]18[/sub] MG色谱柱进行分析时,利多卡因与其峰后杂质分离度为3.27[/b][/color];而使用SUPERIOREX ODS色谱柱分析时,利多卡因与其峰后杂质未得到有效分离。综上,在国家药品标准WS-10001-(HD-0140)-2002方法基础上,将色谱柱柱温由30℃降低至25℃,使用高极性色谱柱CAPCELL PAK C[sub]18[/sub] AQ及中等极性色谱柱CAPCELL PAK C[sub]18[/sub] MG进行分析,均可在25 min内完成林可霉素利多卡因凝胶样品的分析,并得到利多卡因与其峰后杂质之间的良好分离结果。[align=right][/align][align=right][/align][align=right] [/align][align=right]三耀精细化工品销售(中国)有限公司[/align][align=right]技术开发部[/align][align=right]地址:北京经济技术开发区宏达南路5号[/align][align=right]宏达利德工业园1栋418室[/align][align=right]邮编:100176[/align]
跪求有没有人有“硫酸银、碳酸银”分析标准啊?
现我室来了批含量约30% (w/w)的硫酸氢钠NaHSO4试样,请教各位专业人士怎么分析其含量呀?或者谁能提供工业硫酸氢钠的执行标准?小弟先谢谢过!硫酸氢钠。。。。。。ZB G 12002-1987
[align=center][img=,450,798]https://ng1.17img.cn/bbsfiles/images/2019/02/201902281250168777_532_960_3.jpg!w677x1202.jpg[/img][/align][align=center][b]安谱实验带你打通关[/b][/align] 展青霉素,别名棒曲霉素,广泛存在于烂苹果中,具有很强的毒性,对人体有极强的损伤,在2019年的国抽检测项目中榜上有名。国标GB 2761—2011规定了展青霉素的限量值为50μg/kg。 目前食品中展青霉素的检测标准是国标《GB 5009.185-2016 食品安全国家标准 食品中展青霉素的测定》,其方法是:用MAX小柱净化,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]方法检测。安谱实验依据国标方法,运用CNW Poly-Sery MAX小柱(SBEQ-CA3381)和Athena UHPLC C18色谱柱,用[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]的方法对苹果汁中的展青霉素进行测定,结果稳定可靠,回收率高。[align=center][b]全部装备任你挑[/b][/align][align=center][/align][align=center]一、样品前处理[/align] 取2g苹果汁(如果是浑浊未过滤果汁,7000r/min离心10min去除悬浮物),加入标品(上机浓度50ppb),待净化。[align=center]二、SPE操作过程[/align] 活化:6mL甲醇 平衡:6ml水 上样:待净化液 淋洗:3mL乙酸铵溶液,3mL水,抽干 洗脱:4mL甲醇 浓缩:在洗脱液中加入20 μL乙酸,置40℃下用氮气缓缓吹至近干,用乙酸溶液定容至1.0mL,涡旋30s溶解残留物,0.22 μm 滤膜过滤,收集滤液于进样瓶中以备进样。[align=center]三、色谱条件[/align] [b] 液相条件:[/b] 色谱柱:Athena UHPLC C18(2.1*50mm,5μm); 流动相:乙腈:水=5:95,等度洗脱; 流速:300μL/min; 柱温:30oC; 进样量:10μL; [b]质谱参数:[/b] Ionization Mode: ESI- Capillary Voltage: 4000V Gas Temp:350oC Gas flow:8 L/min Nebulizer:35 psi[align=center]四、实验谱图[/align][align=center][img=,600,208]https://ng1.17img.cn/bbsfiles/images/2019/02/201902281258193638_3095_960_3.png!w807x281.jpg[/img][/align][align=center]▲50ppb标准品谱图(基质配标)[/align][align=center][/align][align=center][img=,600,213]https://ng1.17img.cn/bbsfiles/images/2019/02/201902281258451974_9937_960_3.png!w800x285.jpg[/img][/align][align=center]▲苹果汁谱图(空白)[/align][align=center][/align][align=center][img=,600,211]https://ng1.17img.cn/bbsfiles/images/2019/02/201902281259199259_8621_960_3.png!w802x283.jpg[/img][/align][align=center]▲苹果汁谱图(加标 50ppb)[/align][align=center]五、实验数据[/align][align=center][img=,445,46]https://ng1.17img.cn/bbsfiles/images/2019/02/201902281300177816_24_960_3.png!w445x46.jpg[/img][/align][b]实验结论:[/b]苹果汁中含有1.6μg/kg的展青霉素,符合国标限量标准(50μg/kg)。[align=center]六、实验耗材[/align][align=center][img=,527,813]https://ng1.17img.cn/bbsfiles/images/2019/02/201902281301189169_8438_960_3.png!w527x813.jpg[/img][/align]
各位,请问用0.2mol/L的三氟乙酸去测定硫酸庆大霉素的时候,0.2mol/L的三氟乙酸pH0.58左右,而我用的C18柱子最低能耐受pH为1,请问各位0.2mol/L的三氟乙酸需要调节pH吗,如果需要用什么调节
请教大家:这个硫酸新霉素鉴别的主斑点的颜色?是否是白色的?
进口兽药质量标准硫酸头孢喹肟注射液Liusuan Toubaokuiwo ZhusheyeCefquinome sulfate Injection本品为硫酸头孢喹肟与油酸乙酯等配制而成的混悬注射液。含头孢喹肟(C23H24N6O5S2)应为标示量的90.0%~105.0%。【性状】 本品为类白色至浅褐色混悬液体;久置分层。【鉴别】(1)含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品峰的保留时间一致。(2)取摇匀后的供试品2 ml,加水5 ml,稀盐酸1 ml,混匀,置超声浴中超声10分钟,弃去有机层,溶液显硫酸盐的鉴别反应(附录15页)。【检查】有关物质 照含量测定项下的方法。取摇匀后的供试品1.0 ml,加入流动相25.0 ml,置超声浴中超声5分钟,弃去有机层,取水层滤过,取续滤液10µ l,注入液相色谱仪,记录色谱图,2,3-环己基吡啶与头孢喹肟相对保留时间为0.20。按峰面积归一化法计算,2,3-环己基吡啶应不得过3.0%,其他单一杂质应不得过0.50%,杂质总量应不得过4.0%。水分 取本品,照水分测定法(附录58页,第一法)检查,含水分不得过0.2%。细菌内毒素 取摇匀后的供试品2 ml与细菌内毒素检查用水3 ml混匀,分成2等份,振摇30秒,离心15分钟(2000g),吸取水层1 ml,加1 mol/L氢氧化钠溶液0.06 ml调节pH值至6.5~7.5。用细菌内毒素检查用水按1:10稀释后,照细菌内毒素检查法(附录73页)检查,每1 mg头孢喹肟中含细菌内毒素的量应小于0.1 EU。无菌 取供试品8瓶,混合均匀,加入含6%吐温-80的蛋白胨缓冲液(1g/L)400ml,混匀,加入800×106单位青霉素酶(每1ml供试品溶液,加2×106单位青霉素酶),充分振摇,将供试品倒置,在37℃放置4小时;取供试品溶液,依法检查(附录79页,直接接种法),应符合规定。分散性 取本品1瓶,振摇30秒,将供试品转移置玻璃容器中,不得观察到结块或沉淀物。沉降 取本品1瓶,振摇30秒,取供试品10 ml置刻度试管中(内径1.0~1.5 cm),10分钟内不得沉淀。粒度 取摇匀后的供试品,置显微镜下检查,颗粒直径在5µ m以下应不得少于80%,10µ m以下不得少于90%,20µ m以下不得少于95%,50µ m以下不得少于100%。装量 按最低装量检查法(附录67页)检查,应符合规定。【含量测定】 照高效液相色谱法(附录24页)测定。色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;取一水合高氯酸钠3.45g溶于1000 ml水中,加磷酸12 ml和乙腈90 ml,用三乙胺调节pH至3.6为流动相;检测波长为270 nm。取头孢噻肟约25 mg,溶于100.0 ml流动相中,另取头孢喹肟约25 mg,置25 ml量瓶中,精密加入上述头孢噻肟溶液1 ml,用流动相稀释至刻度。精密量取10µ l注入液相色谱仪,记录色谱图;计算头孢喹肟与头孢噻肟的分离度,应大于1.0。
β-Glucuronidase/aryl sulfatase β-葡萄糖醛酸酶\芳香基硫酸酯酶( 葡萄糖苷酸酶/硫酸芳酯酶 ) 标准品——检瘦肉精等用的除了北京希凯创新科技有限公司提供,还可以从哪里购买么?
大家好。本人现正进行硫酸新霉素在鱼体的药物残留动力学检测,了解的液相检测方法有茚三酮衍生化、氯甲酸芴甲酯衍生和国标OPA衍生检测方法,都是用UV检测。本人已经试验过茚三酮衍生化方法,感觉效果不太理想,而国标OPA衍生法据说衍生化试剂及衍生物稳定性比较差,不知大家对于检测方面有什么建议!!!不胜感谢。
请问哪位有硫酸锰、硫酸镍、硫酸钴的国家标准或行业标准,最好带分析资料,谢谢!

一、背景1.1莫能菌素﹝Monensin﹞是聚醚类离子载体抗生素,是一种[url=http://baike.baidu.com/subview/138504/138504.htm][color=windowtext]反刍动物[/color][/url]中运用较广泛的饲料添加剂;原为[url=http://baike.baidu.com/subview/150635/150635.htm][color=windowtext]链霉菌[/color][/url]所分泌的一种物质,具有控制[url=http://baike.baidu.com/subview/39929/39929.htm][color=windowtext]瘤胃[/color][/url]中[url=http://baike.baidu.com/subview/3852509/3852509.htm][color=windowtext]挥发性脂肪酸[/color][/url]比例,减少瘤胃中蛋白质的降解,降低饲料干物质消耗,改善营养物质利用率和提高动物能量利用率等作用。1.2莫能菌素为百盛客户对肉类原料中兽药残留的监控项目,为了应对客户要求,满足实验室检测要求,对莫能菌素进行新项目技术开发。盐霉素为原有分析项目,此次技术开发对盐霉素前处理及仪器分析条件重新优化,与莫能菌素合并同为聚醚类抗生素检测检测方法。[img=,490,100]http://ng1.17img.cn/bbsfiles/images/2017/08/201708141014_01_3081717_3.png[/img]二、前处理流程2.1提取称取(2.00±0.02)g样品,加入10mL乙腈混匀,再加入3.00g无水硫酸钠,震荡混匀,超声提取10min;离心取上清液于50mL离心管中,残渣加入5mL乙腈重复提取,合并两次上清液,定容至20mL,加入5mL乙腈饱和正己烷,震荡离心。2.2净化取下层(乙腈层)5mL于圆底烧瓶中,旋转蒸发至1mL,氮气吹干。普通肉类基质:圆底烧瓶中加入1mL乙腈超声溶解。内脏类基质:圆底烧瓶中加入3mL甲醇+水(1+1)超声溶解,过Waters HLB柱(waters oasis HLB 6cc/200mg依次用5mL甲醇 5mL水活化),用5mL水淋洗,5mL甲醇洗脱于100mL圆底烧瓶中,40℃减压蒸干,加入1mL乙腈超声溶解。三、仪器分析条件([url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]MS8050)3.1质谱参数:离子源ESI源 Nebulizing Gas Flow: 3 L/min HeatingGas Flow: 10 L/min Interface Temperature 300 ℃;DLTemperature 250 ℃;Heating Block Temperature 400℃ Drying Gas Flow: 10 L/min [table=595][tr][td=1,2]化合物名称[/td][td=1,2]Precursor m/z[/td][td]Product[/td][td]Dwell Time[/td][td]Q1 Pre[/td][td=1,2]CE[/td][td]Q3 Pre[/td][/tr][tr][td]m/z[/td][td](msec)[/td][td]Bias(V)[/td][td]Bias(V) 1[/td][/tr][tr][td=1,3]莫能菌素[/td][td]688.6[/td][td]635.50*[/td][td]100[/td][td]-26[/td][td]-18[/td][td]-28[/td][/tr][tr][td]688.6[/td][td]461.35[/td][td]100[/td][td]-26[/td][td]-26[/td][td]-30[/td][/tr][tr][td]688.6[/td][td]617.5[/td][td]100[/td][td]-26[/td][td]-24[/td][td]-28[/td][/tr][tr][td=1,3]盐霉素[/td][td]773.1[/td][td]431.20*[/td][td]100[/td][td]-28[/td][td]-53[/td][td]-28[/td][/tr][tr][td]773.1[/td][td]531.35[/td][td]100[/td][td]-28[/td][td]-46[/td][td]-36[/td][/tr][tr][td]773.1[/td][td]413.2[/td][td]100[/td][td]-28[/td][td]-53[/td][td]-27[/td][/tr][/table]3.2液相参数: 流动相组成:A: 0.1%甲酸 B: 乙腈;流速:0.35Ml/min;A -10% B -90%恒流分析;进样量:2uL;色谱柱型号:ODS-III 1.6μm四、实验结果及分析4.1线性配制盐霉素、莫能菌素混标(稀释溶剂:乙腈),0.1ug/kg、1ug/kg、5ug/kg、10ug/kg、20ug/kg五个浓度点,仪器分析线性如下:[img=,490,157]http://ng1.17img.cn/bbsfiles/images/2017/08/201708141016_02_3081717_3.png[/img][img=,490,173]http://ng1.17img.cn/bbsfiles/images/2017/08/201708141017_01_3081717_3.png[/img]以上实验中,盐霉素、莫能菌素标准曲线R[sup]2[/sup]均大于0.99,各浓度点精密度良好;仪器分析标准品线性、稳定性符合实验要求。4.2选择性选取牛肉样品进行空白、添加回收实验,实验谱图如下:[img=,490,173]http://ng1.17img.cn/bbsfiles/images/2017/08/201708141019_01_3081717_3.png[/img][img=,490,173]http://ng1.17img.cn/bbsfiles/images/2017/08/201708141019_02_3081717_3.png[/img]4.3真度对空白牛肉进行前处理,采用空白样品萃取液与溶剂稀释统一标准品标准品,同一浓度下两者峰面积比值的百分比作为真度评价参数,实验结果及谱图如下: [table=386][tr][td]基质类型[/td][td]化合物名称[/td][td]基质稀释标准品[/td][td]溶剂稀释标准品[/td][td]真度(%)[/td][/tr][tr][td=1,2]牛肉[/td][td]盐霉素[/td][td]116,440[/td][td]107,537[/td][td]106.4%[/td][/tr][tr][td]莫能菌素[/td][td]9,845[/td][td]13,312[/td][td]74.0%[/td][/tr][/table]以上实验中牛肉基质对盐霉素无明显基质效应,对莫能菌素产生一定的基质效应,基质效应在70%左右,在可接受范围之内,日常分析准确定量可以做spiked校正。6实验总结本次试验,从前处理、仪器分析、实验分析的真度、精密度、准确度等多项参数验证了此方法的适用性及准确性,可初步满足试验要求,后续继续多基体验证。
本人有一个样品,只知道是阿奇霉素的一种,但不知道具体是磷酸二氢钠阿奇霉素、硫酸阿奇霉素、盐酸阿奇霉素.....等中的哪一种,想要分析出来它的具体种类及纯度。不知道哪位老师那里可以分析,或者有什么办法能分析出来。
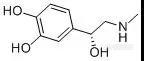
复方盐酸阿替卡因注射液为复方制剂,是盐酸阿替卡因与肾上腺素的灭菌水溶液,作为口腔用局部麻醉剂,适用于涉及切骨术及粘膜切开的外科手术过程。[img=,144,61]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191545418661_4518_2222981_3.jpg!w144x61.jpg[/img][color=black] [/color][color=#3e3e3e]肾上腺素 L(-)-Epinephrine M.W. : 183.2 [/color][b]在国家药品标准(YBH17082004-2015Z)[/b]中,在对复方盐酸阿替卡因注射液中肾上腺素进行分析时,使用[b]甲醇-水[/b]进行梯度洗脱,但由于[b]肾上腺素极性较强[/b],即使初始梯度为纯水相条件,肾上腺素仍紧邻死时间出峰,[b]保留不佳,易受到溶剂峰干扰,无法进行准确定量。[/b]我们分别尝试使用反相柱CAPCELL PAK C18 MGII加离子对试剂,以及直接使用离子交换色谱柱CAPCELL PAK SCX UG80两种方式,对复方盐酸阿替卡因注射液中肾上腺素和硫酸肾上腺素进行保留分析(复方盐酸阿替卡因注射液由客户提供)。CAPCELL PAK C18 MGII液相色谱柱,其采用高纯度硅胶作为基质,通过减少硅胶微细孔的数量来增大有效比表面积;并且采用新包被技术Ultimate Polymer Coating,实现了对硅醇基极大程度的封锁,兼具分离性能和普适性能,通用性非常好。CAPCELL PAK SCX UG80是强阳离子交换柱,使用高纯度硅胶,填料中金属杂质很少,使配位化合物的吸附得到了极大程度抑制,兼具聚合物和硅胶填料的优点。[b][color=#0070c0]实验方法[/color][color=#0070c0]方法一[/color][color=#0070c0]使用[/color][color=#0070c0]CAPCELL PAK C18MGII[/color][color=#0070c0]色谱柱[/color][color=#0070c0]+[/color][color=#0070c0]离子对试剂[/color][/b]如图1,对肾上腺素对照品溶液进行分析,肾上腺素主峰保留时间为5.69 min,拖尾因子为1.19,理论塔板数为12538。在相同色谱条件下,尝试对亚硫酸肾上腺素标准品及注射液中的亚硫酸肾上腺素进行分析。如图3,亚硫酸肾上腺素标准品溶液能够得到良好分析结果,注射液(客户提供的样品)中未明显见亚硫酸肾上腺素出峰,保留时间为3.55min,拖尾因子为1.14,理论塔板数为14955。[b][color=#0070c0]方法二[/color][color=#0070c0] CAPCELL PAK SCX UG80[/color][color=#0070c0]色谱柱[/color][/b][color=#000000]考虑到使用离子对试剂的流动相条件具有流动相配制麻烦、有损色谱柱寿命、平衡时间长等缺点,我们也尝试使用键合磺酸基团的强阳离子交换柱 ——CAPCELL PAK SCX UG80进行分析。[/color][color=#000000]如图4,在流动相中添加磷酸二氢铵,通过对盐浓度进行调整,在5 mmol/L磷酸二氢铵(磷酸调pH=2.5)条件下,亚硫酸肾上腺素保留时间为3.32 min,然而出现峰形拖尾现象,拖尾因子为2.0,不如CAPCELL PAK C18 MGII色谱柱添加离子对试剂所得分析结果好。[/color][align=center][/align][align=left][img=,400,284]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547381421_926_2222981_3.jpg!w584x416.jpg[/img] [img=,400,276]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548310561_8067_2222981_3.jpg!w572x395.jpg[/img][/align][align=left][img=,400,166]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191547576748_522_2222981_3.jpg!w612x254.jpg[/img] [img=,400,167]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191548525761_4184_2222981_3.jpg!w624x262.jpg[/img][/align][align=left]图1 MGII分析肾上腺素对照品溶液结果(离子对条件) 图2 MGII分析注射液结果(离子对条件)[/align][align=center][/align][img=,400,258]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191550354951_2539_2222981_3.jpg!w644x416.jpg[/img] [img=,400,250]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191555486850_3396_2222981_3.jpg!w644x403.jpg[/img][img=,400,147]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191551260881_9828_2222981_3.jpg!w696x256.jpg[/img] [img=,400,164]https://ng1.17img.cn/bbsfiles/images/2019/03/201903191556163846_9739_2222981_3.jpg!w632x260.jpg[/img][align=left]图3 MGII分析亚硫酸肾上腺素标准品和注射液结果(离子对条件) 图4 SCX UG80分析亚硫酸肾上腺素对照品溶液和供试品溶液[/align][align=left][/align][align=left]综上实验结果,使用中等极性色谱柱CAPCELL PAK C18 MGII S5 4.6 mm i.d. × 250 mm,在流动相中添加5 mM辛烷磺酸钠、30°C柱温条件下进行梯度洗脱,能够实现复方盐酸阿替卡因注射液中肾上腺素和亚硫酸肾上腺素的良好保留与分析。[/align][b][color=#0070c0][/color][/b][align=left][b][color=#0070c0] [/color][color=#0070c0] [/color][/b][/align]
(一)食品中氯霉素的限量1999年9月我国农业部发布了《动物性食品中兽药最高残留限量》,规定了氯霉素在所有食品动物的可食用组织中不得检出。2002年3月被我国农业部关于发布《食品动物禁用的兽药及其化合物清单》列为禁止使用的抗生素。欧盟、美国等国家规定动物源性食品中氯霉素的残留限量标准为“零容许量”,即不得检出。(二)测定氯霉素残留的方法①酶联免疫法。采用间接竞争ELISA筛选法,在酶标板微孔条上包被偶联抗原,样本中残留的氯霉素和微孑L条上包被的偶联抗原竞争抗氯霉素抗体,加入酶标二抗后,加入底物显色,样本吸光度值与其残留物氯霉素的含量成负相关,与标准曲线比较再乘以其对应的稀释倍数,即可得出样品中氯霉素的含量。该方法测定低限为氯霉素0.05/g/kg。⑦GB/T 22338--2008动物源性食品中氯霉素类药物残留量测定——气相色谱一质谱法和液相色谱一质谱法测定水产品、畜禽产品和畜禽副产品中氯霉素、氟甲砜霉素和甲砜霉素残留量。气相色谱一质谱法是样品用乙酸乙酯提取,4%氯化钠溶液和正己烷溶液液分配净化,再经弗罗里硅土(Florisil)柱净化后,以甲苯为反应介质,用N,O-I双(三甲基硅烷)三氟乙酰胺一三甲基氯硅烷(BSTFA+TMCS,99+1)于70℃硅烷化,用气相色谱/负化学电离源质谱测定,内标工作曲线法定量。该方法测定低限为氯霉素0.1/μg/kg,氟甲砜霉素和甲砜霉素0.5μg/kg。液相色谱一质谱法是针对不同动物源性食品中氯霉素、氟甲砜霉素和甲砜霉素残留量,分别采用乙腈、乙酸乙酯一乙醚或乙酸乙酯提取,提取液用固相萃取柱进行净化,液相色谱一质谱/质谱仪进行测定,氯霉素采用内标法定量,氟甲砜霉素和甲砜霉素采用外标法定量。该方法对氯霉素的测定低限为0.1μg/kg;氟甲砜霉素和甲砜霉素为O.1/μg/kg。
2011年2月15日,新西兰食品安全局(NZFSA)公布了2011年食品农化物最大残留限量标准。此次涉及的农化物包括阿维菌素、乙酰甲胺磷、阿苯达唑、氯氨吡啶酸、双甲脒、氨基三唑、阿莫西林、氨苄青霉素、安普罗铵、阿泊拉霉素、艾维激素、阿扎康唑、甲基谷硫磷、三唑锡、嘧菌酯、巴喹普林等256种化学物质,涵盖的食品范围包括鳄梨、猕猴桃、梨果、草莓;牛脂肪、牛肝脏、牛肉、绵羊脂肪、绵羊肾、绵羊肝、绵羊肉等126种,其中涉及最多的三类食品依次为梨果(43种),葡萄(34种),马铃薯(32种)。此次规定了食品中氯霉素的最大残留限量标准为0.0003mg/kg,是被允许的最大残留限量值中最低的。
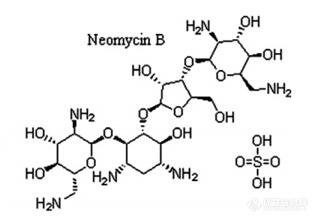
检测对象:硫酸新霉素,结构式如下[align=left][img=,319,223]http://ng1.17img.cn/bbsfiles/images/2017/12/201712191628_6456_3237657_3.jpg!w319x223.jpg[/img]检测方法:UP[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]LC-MS[/color][/url]/MS检测样品:生物组织(肌肉、皮脂、肝脏和肾脏)提取液:10mM的磷酸二氢钾溶液(1mL)+10%三氯乙酸(含0.4mM的EDTA)(5mL)上样液:过固相萃取柱的样品溶液 [b]遇到的问题:[/b]1、文献报道这个药物的PKa=8.8,如果在水溶液中要将该药物调成非离子状态,pH该怎么调?2、用水溶解硫酸新霉素对照品,并稀释成500ppb,过HLB萃取柱,回收率达90%以上;将标准品+提取液混合后,过HLB萃取柱,不调pH时回收率能达90%以上,但按文献报道的调pH6.0~8.0,回收率只有20%左右。将标准品加入至空白组织,混匀后再加相同的提取液,不调pH过HLB萃取柱,回收率在20%多,但上样液调pH至6.0后回收率能达到50%左右。完全相反的两种状态,这是什么原因?3、将标准品加入至空白组织,混匀后再加提取液,上样液调pH至6.0,过HLB萃取柱,回收率达50%。将空白组织加相同体积的提取液,上样液调pH至6.0,再加标准溶液混合,过HLB萃取柱,回收率只有8%左右。这个结果能说明什么问题?HLB不合适吗?还是上样液的pH调的不合适?4、将标准品加入至空白组织,混匀后再加提取液,上样液调系列pH(从2.0-10.0),不同pH值的上样液过HLB萃取柱,比较回收率差异,结果显示数值几乎没有变化。上样液的pH值不影响药物在HLB上的回收?5、大多数文献报道,这个药物用WCX或者MCX萃取柱净化,但实验结果表明,我用的Waters Oasis 的HLB跟MCX的提取回收率几乎一致,都在50%左右。用WCX提取效果更差。不知道原因何在,请赐教!谢谢![/align]
有谁做过硫酸新霉素的HPLC液相方法,能提供一下检测条件吗?
GB 29685-2013 食品安全国家标准 动物性食品中林可霉素、克林霉素和大观霉素多残留的测定 气相色谱—质谱法
请问硫酸庆大霉素是什么东西,庆大霉素B、C跟它又是什么关系?http://simg.instrument.com.cn/bbs/images/brow/em09512.gif







 我要推广仪器
我要推广仪器
 下载APP
下载APP